1. Introduction
The maintenance of reactive oxygen species (ROS) homeostasis is a critical determinant of cell survival. In proliferating tissues, a decreased supply of oxygen and nutrients and deregulated metabolic pathways usually pose a threat to the redox balance [
1]. The role played by ROS in tumor initiation and development is controversial and has been shown to depend on many factors, including tumor origin, developmental stage, and tissue microenvironment. Moreover, an increasing body of evidence indicates that cancer cells adapt to an imbalanced redox status by developing alternative metabolic reactions or by strengthening existing ones. This allows them to balance ROS according to their needs and renders them insensitive to further stress inducers such as chemotherapy and radiation [
1].
The TP53-induced glycolysis and apoptosis regulator (
TIGAR) is an important gene coding for a regulatory enzyme of glycolysis and ROS detoxification [
2]. The activity of this enzyme that has attracted most research interest is the dephosphorylation of fructose-2,6-bisphosphate (Fru-2,6-P
2), the most potent allosteric activator of 6-phosphofructo-1-kinase (PFK-1) and an inhibitor of fructose 1,6-bisphosphatase [
3,
4]. In so doing, TIGAR increases the flux from fructose-6-phosphate to glucose-6-phosphate, fueling the pentose phosphate pathway (PPP). Therefore, TIGAR contributes to the production of nicotinamide adenine dinucleotide phosphate (NADPH) for glutathione (GSH) regeneration. Importantly, the capacity of TIGAR to increase the NADPH/NADP+ ratio and confer protection from ROS-induced apoptosis was found to be dependent on the activity of glucose-6-phosphate dehydrogenase (G6PD) [
2]. This indicates that TIGAR constitutes a link between glycolysis and the PPP. In physiological conditions, TIGAR expression confers cytoprotection and can prevent tumor development, as shown in mouse intestinal epithelia [
5]. However, in a neoplastic context, TIGAR helps increase the antioxidant capacity of malignant cells and favors tumor growth. This phenomenon has been described in different cancer cell lines and tumor types [
4,
6,
7]. Consequently, the downregulation of TIGAR in tumors has been proposed as an appropriate strategy to increase the effectiveness of antineoplastic therapies based on ROS-mediated cell death [
8,
9]. Recently, the cytotoxic effects of the DNA methyltransferase inhibitor decitabine and the inhibitor of heme-oxygenase tin mesoporphyrin have been linked to decreased expression of TIGAR and reduced PPP in myeloid leukemia and non-small cell lung carcinoma (NSCLC), respectively [
10,
11].
One of the most relevant players in the cellular antioxidant response is the transcription factor nuclear factor (erythroid-derived 2)-like 2 (NRF2), encoded by the gene
NFE2L2 [
12]. Through the activation of a wide range of detoxifying enzymes and metabolic effectors, NRF2 maintains ROS homeostasis and tissue integrity [
13]. Under normal conditions, NRF2 is subjected to rapid turnover in the cytoplasm due to its binding to the ubiquitin ligase Kelch-like ECH-associated protein 1 (KEAP1) and Cullin 3 (CUL3). CUL3 ubiquitinates NRF2 and promotes its proteasomal degradation in the cytoplasm [
13]. Upon exposure to electrophilic molecules or ROS, cysteine residues in KEAP1 are oxidized resulting in a conformational change, which disrupts the KEAP1-NRF2 binding and renders NRF2 free to translocate to the nucleus [
13]. NRF2 forms heterodimers with small musculoaponeurotic fibrosarcoma oncogene homologue (sMaf) proteins, which are primarily located in the nucleus. The NRF2-sMaf heterodimer binds to antioxidant response elements (AREs) in the promoter of different genes thereby enhancing transcription [
14]. AREs are typically sequences of 5’-TGAC/GnnnGC-3’ that function as cis-regulatory elements or enhancers [
15]. Constitutive activation of NRF2 is common in malignant cells regardless of ROS levels [
16,
17]. NRF2 target genes include enzymes with a wide range of functions: ROS detoxification such as NAD(P)H quinone dehydrogenase 1 (NQO1) and thioredoxin reductase 1 (TXNRD1), NADPH synthesis such as G6PD or malic enzyme 1 (ME-1), and glutathione metabolism such as the glutamate-cysteine ligase catalytic (GCLC) subunit, among others [
18]. Far from having a restricted role as antioxidant, the pleiotropy of NRF2 makes it a key transcription factor in processes related to each of the hallmarks of cancer [
19]. Importantly, analysis of genetic alterations from different cancer genome sequencing studies using cBioportal [
20,
21] revealed that
KEAP1/
NFE2L2 mutations are highly frequent in NSCLC, endometrial cancer, ovarian cancer, nerve sheath tumor and various forms of squamous cell carcinoma including cervical cancer. Abnormalities of
KEAP1/
NFE2L2 in NSCLC and are associated with poor clinical outcome [
22]. In cervical carcinoma,
KEAP1 expression is significantly decreased in the cytoplasm of cancer cells compared with that of normal cervical epithelial cells, which increases NRF2 activity. Constitutive NRF2 activation is associated with advanced stages of this type of tumors [
23].
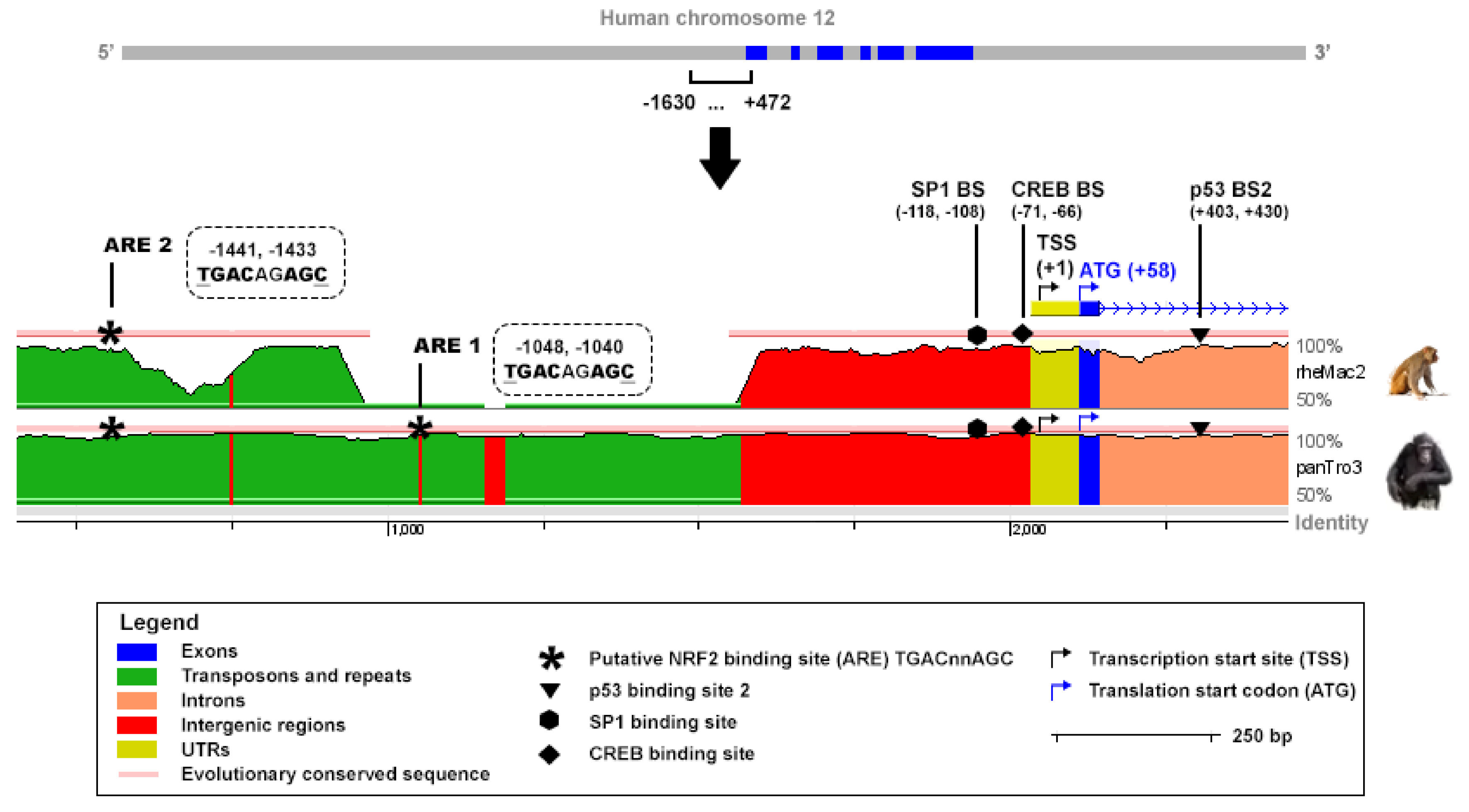
TIGAR expression can be regulated by p53 through a binding site in the proximal region of the first intron of the gene [
2]. However, TIGAR has been found overexpressed in cell lines and tumors in which
TP53 has been either lost or mutated [
4,
5]. In fact, the expression of mouse
Tigar is independent of p53 and p73 [
24]. Considering that
TP53 is mutated in more than 50% of tumors [
25], there is a clear need to find other transcriptional mechanisms which can account for TIGAR modulation. To date, only the specificity protein 1 (SP1) and the cAMP response element-binding protein 1 (CREB1) have been described to bind to the promoter of
TIGAR [
26,
27]. However, neither of them is intrinsically involved in the antioxidant program of cells. Recently, it has been shown that TIGAR expression is dynamically regulated during the development of pancreatic ductal adenocarcinoma, and that together with NRF2 it has a key role in balancing ROS levels during the carcinogenic process [
28].
The main aim of the present study was to determine whether NRF2 can transcriptionally regulate the expression of TIGAR in human carcinoma cells.
3. Discussion
Several studies have shown that expression levels of
TIGAR are high in tumor samples from different origin [
4]. The Gene Expression Profiling Interactive Analysis (GEPIA) [
35], which collects data from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx), shows that higher expression of
TIGAR is related to lower overall survival in different cancer types, including cervical squamous cell carcinoma. According to cBioportal [
20,
21], the most common genetic alteration in
TIGAR is amplification, but even in the tumor types in which the
TIGAR DNA sequence is most altered, that is, uterine carcinosarcoma, genomic alterations are found in only 7% of the patients. Therefore, understanding the mechanisms that drive
TIGAR overexpression in cancer cells is crucial to unveil how the antioxidant potential of this enzyme can be blocked. In this study, we have characterized the transcriptional regulation of
TIGAR by the pleiotropic antioxidant factor NRF2 in cell lines and primary cells.
TIGAR was initially identified as a p53-induced gene coding for a bisphosphatase enzyme that has activity on Fru-2,6-P
2 [
2]. Thereafter, most studies involving TIGAR have characterized the capacity of this enzyme to redirect the glycolytic flux to the PPP and increase the NADPH/NADP+ ratio [
4]. NRF2 orchestrates an antioxidant and metabolic program within the cell that has been related to the resistance of ROS-induced cell death and adaptation to nutritional stress, and which has an impact on all the defined hallmarks of cancer [
13,
19]. We show here that NRF2 activation increases the expression of
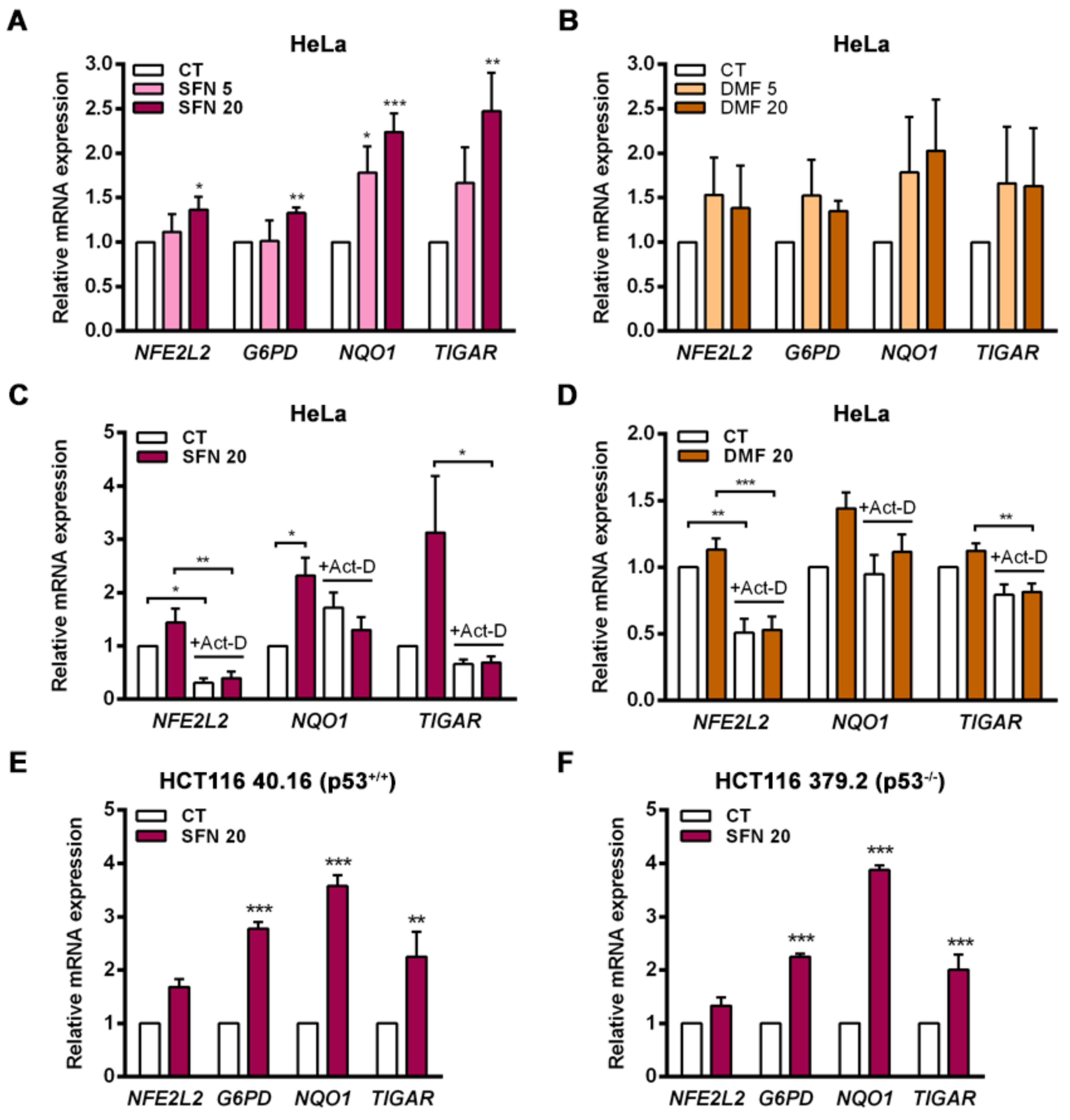
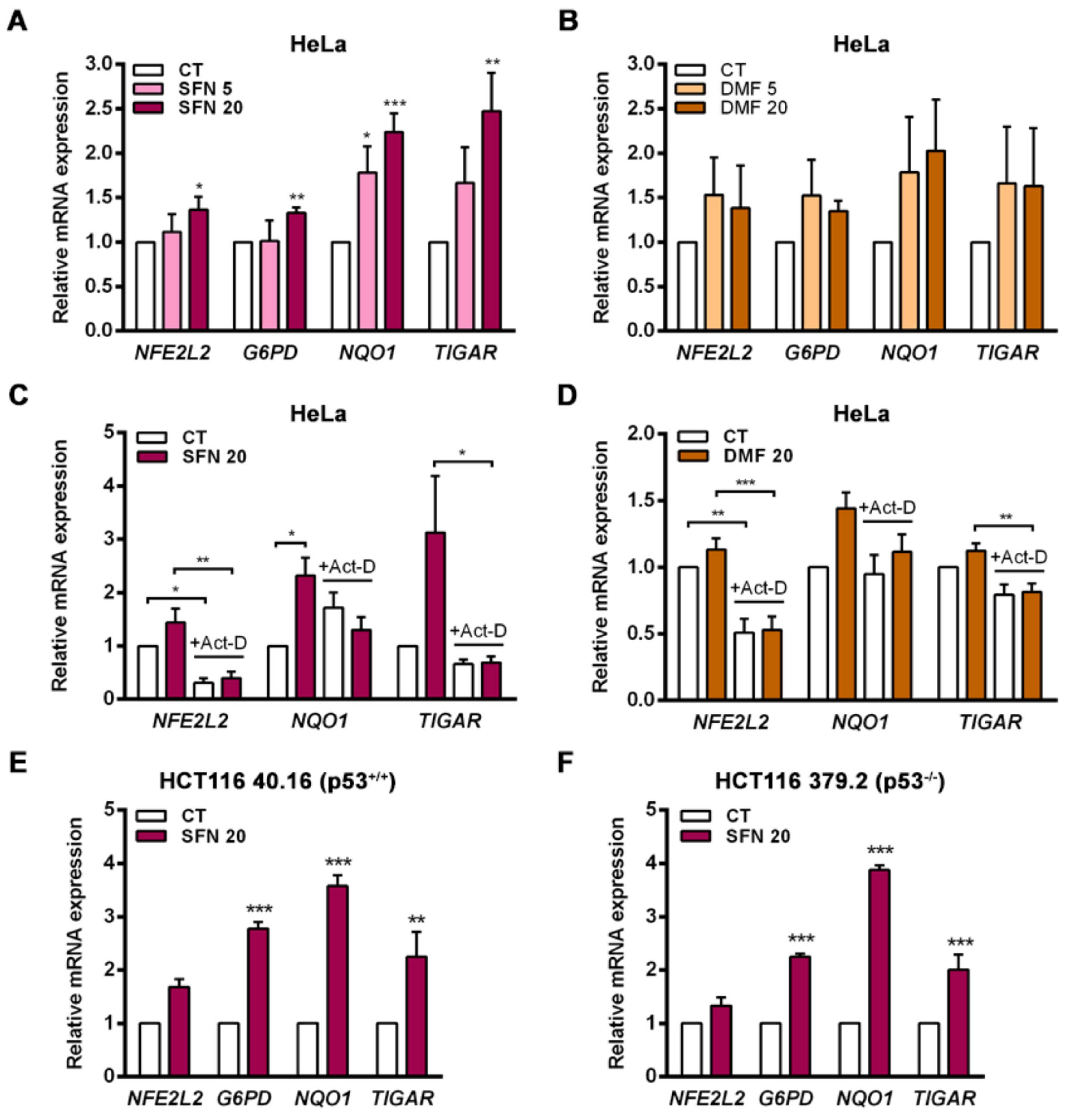
TIGAR in the cervical cancer HeLa cell line both in response to NRF2 overexpression and following exposure to the electrophilic molecules SFN and DMF, which induce the dissociation of NRF2 from KEAP1. Enhancement of TIGAR expression by SFN was also observed in the human colorectal carcinoma cell lines HCT116 40.16 and HCT116 379.2, which bear wild-type and null p53, respectively. These results demonstrate that
TIGAR can be regulated by NRF2 in different cellular contexts regardless of p53 expression, despite being initially described as a p53 response gene [
2]. This finding is in line with previous studies that have reported p53-independent expression of
TIGAR [
5]. NRF2 can be an important regulator of
TIGAR in these models.
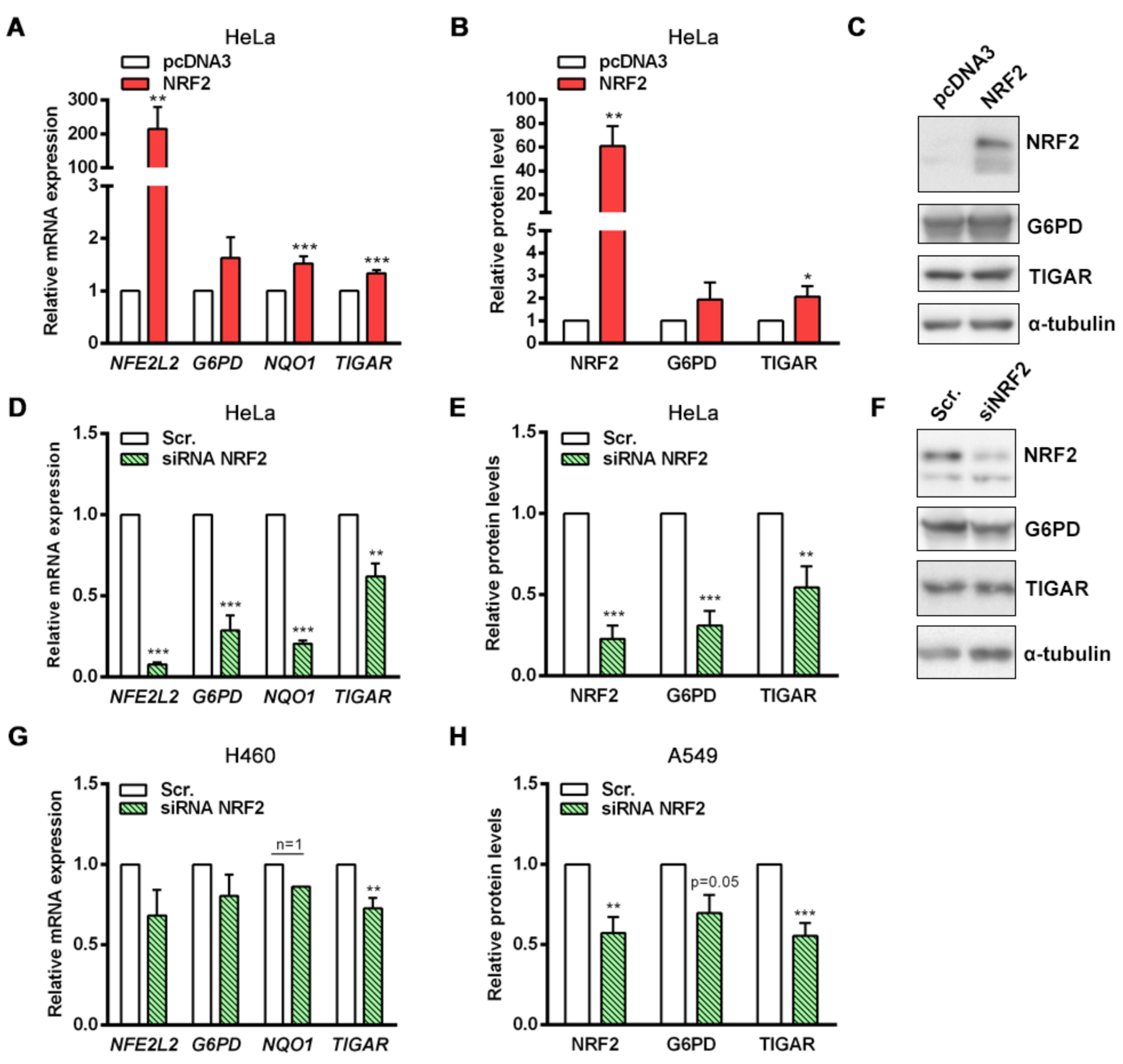
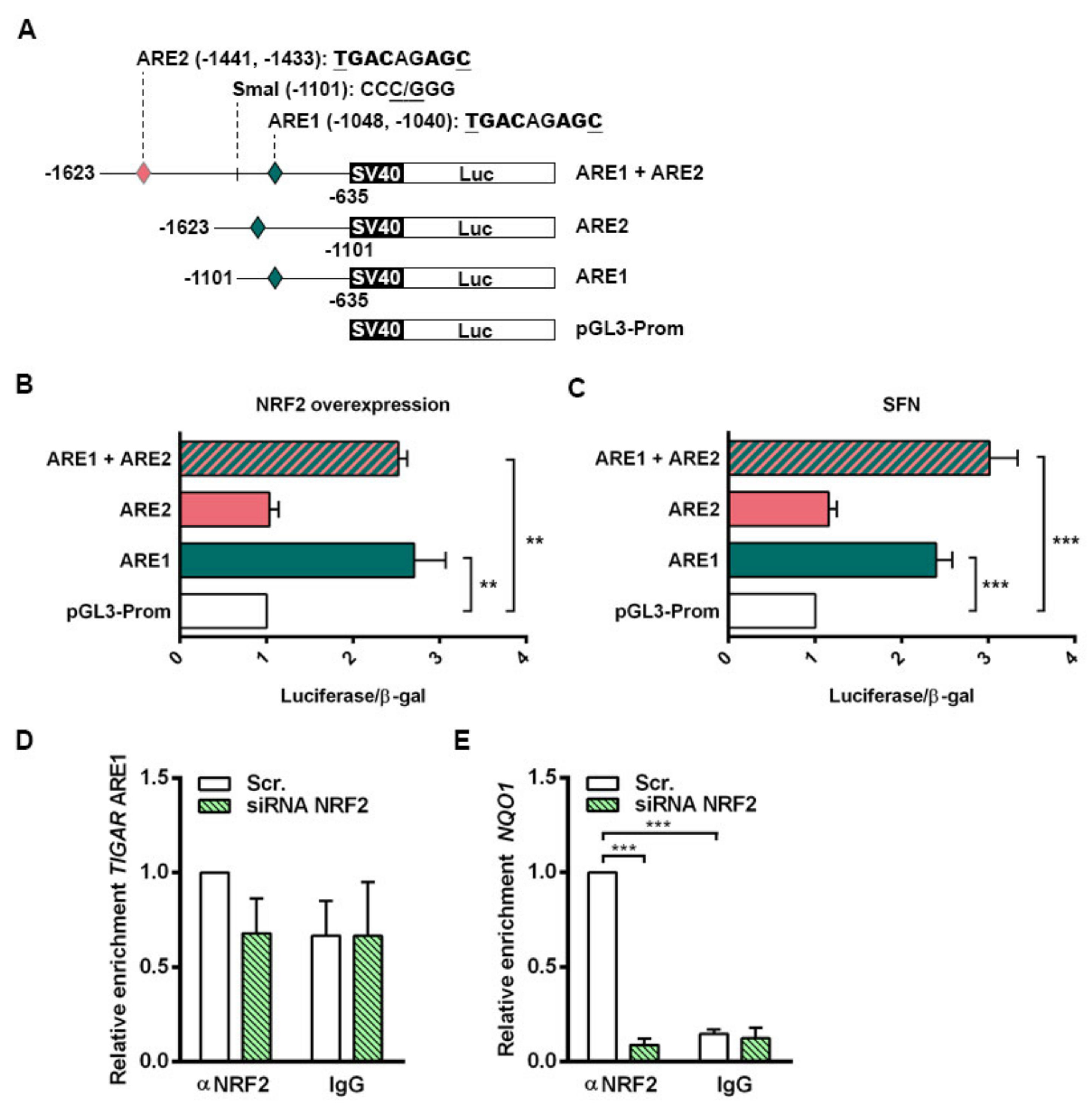
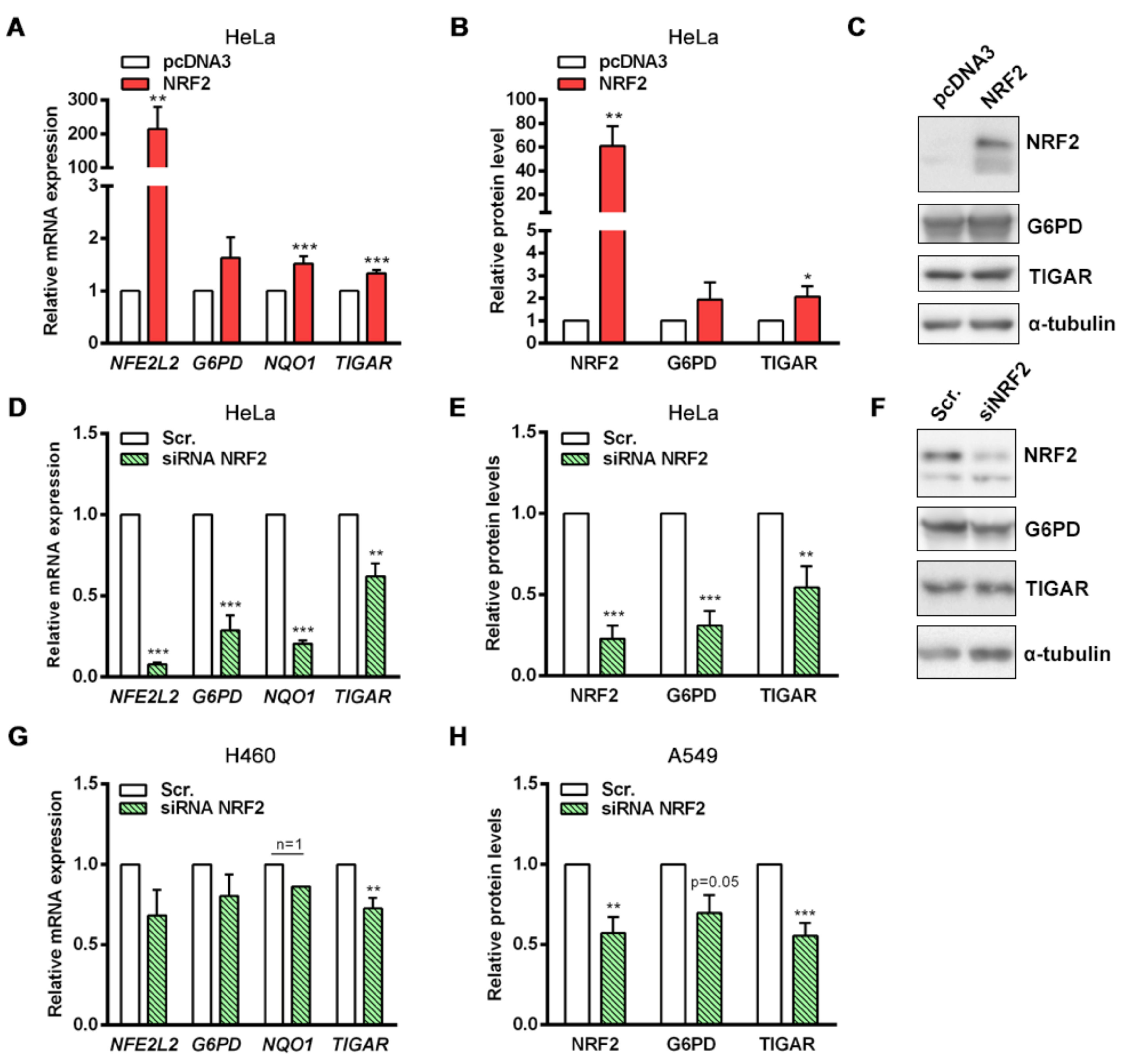
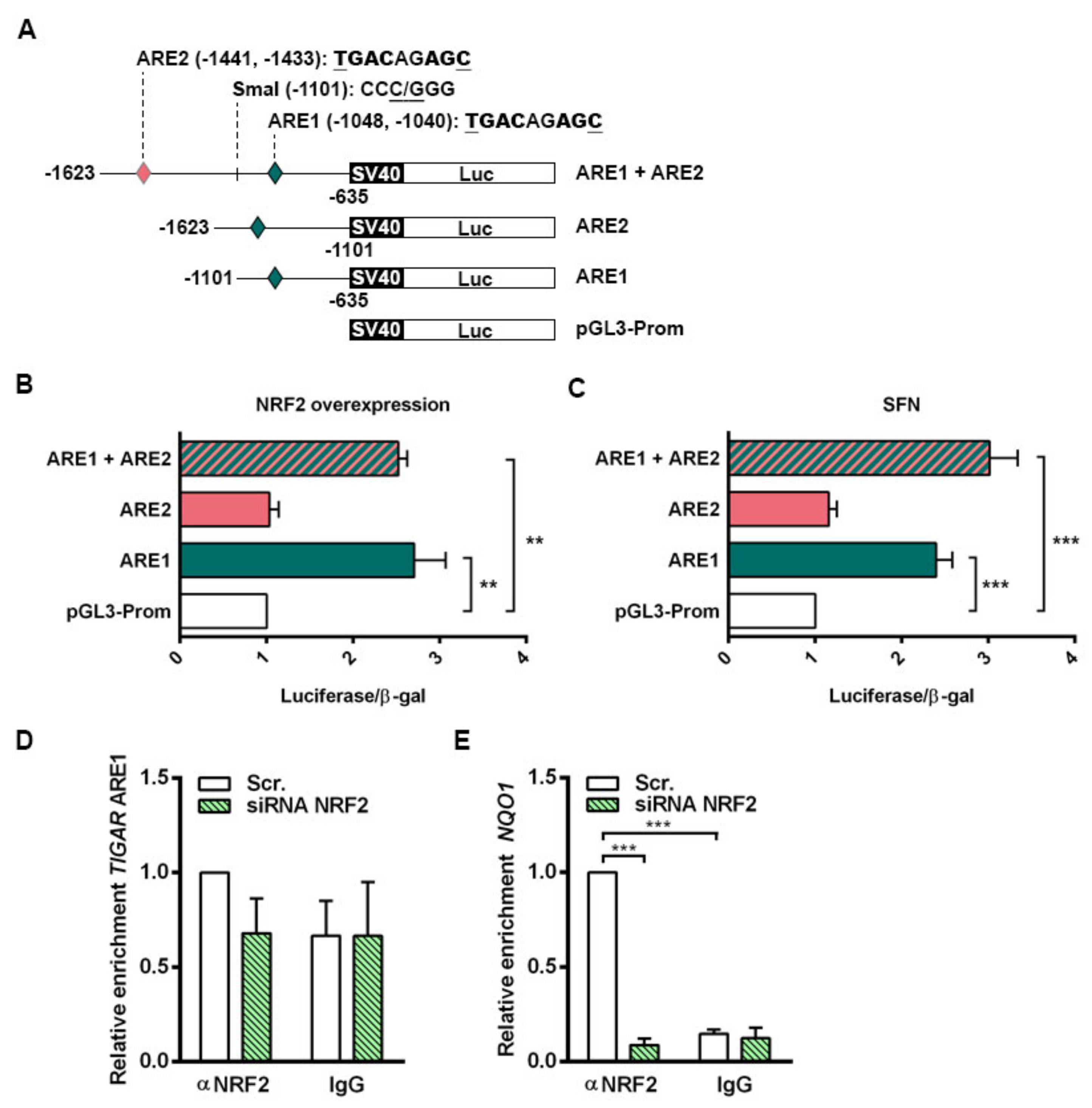
NRF2 downregulation by siRNA resulted in decreased TIGAR levels in HeLa, H460 and A549 cells. Accordingly, we have shown that NRF2 enhances the transcription of human
TIGAR through an ARE located on its promoter at −1048 from the TSS, and that the amount of chromatin immunoprecipitated from this specific region is decreased in NRF2-depleted cells. However, the effect of NRF2 depletion on ChIP assays is more striking in the
NQO1 ARE than in
TIGAR ARE1, suggesting that other motifs might also be involved in the regulation of
TIGAR by NRF2. These results are consistent with the data obtained by RT-qPCR, which showed that the decrease in
NQO1 expression upon NRF2 depletion is more pronounced than that of
TIGAR. Plausible explanations for that could be differential recruitment of sMaf proteins to the promoter of NRF2 target genes, or different chromatin availability throughout the genomic regions where these target genes are located due to epigenetic marks. In a previous study, these features were reported to compromise NRF2 activity on its target genes [
14]. The participation of other antioxidant response elements, such as the distal motif involved in the control of mouse
Tigar, or other transcription factors in the modulation of human
TIGAR in response to NRF2 activation cannot be ruled out. Our results indicate that NRF2 can function as an enhancer of
TIGAR expression under certain cellular conditions, but it is not responsible for the maintenance of the basal expression of this gene. The transcription factor SP1 was described as binding to the proximal promoter of
TIGAR [
26], in a region that is highly conserved between species, as evidenced in the in-silico analysis with the ECR Browser. Therefore, SP1 might account for the maintenance of
TIGAR basal expression levels.
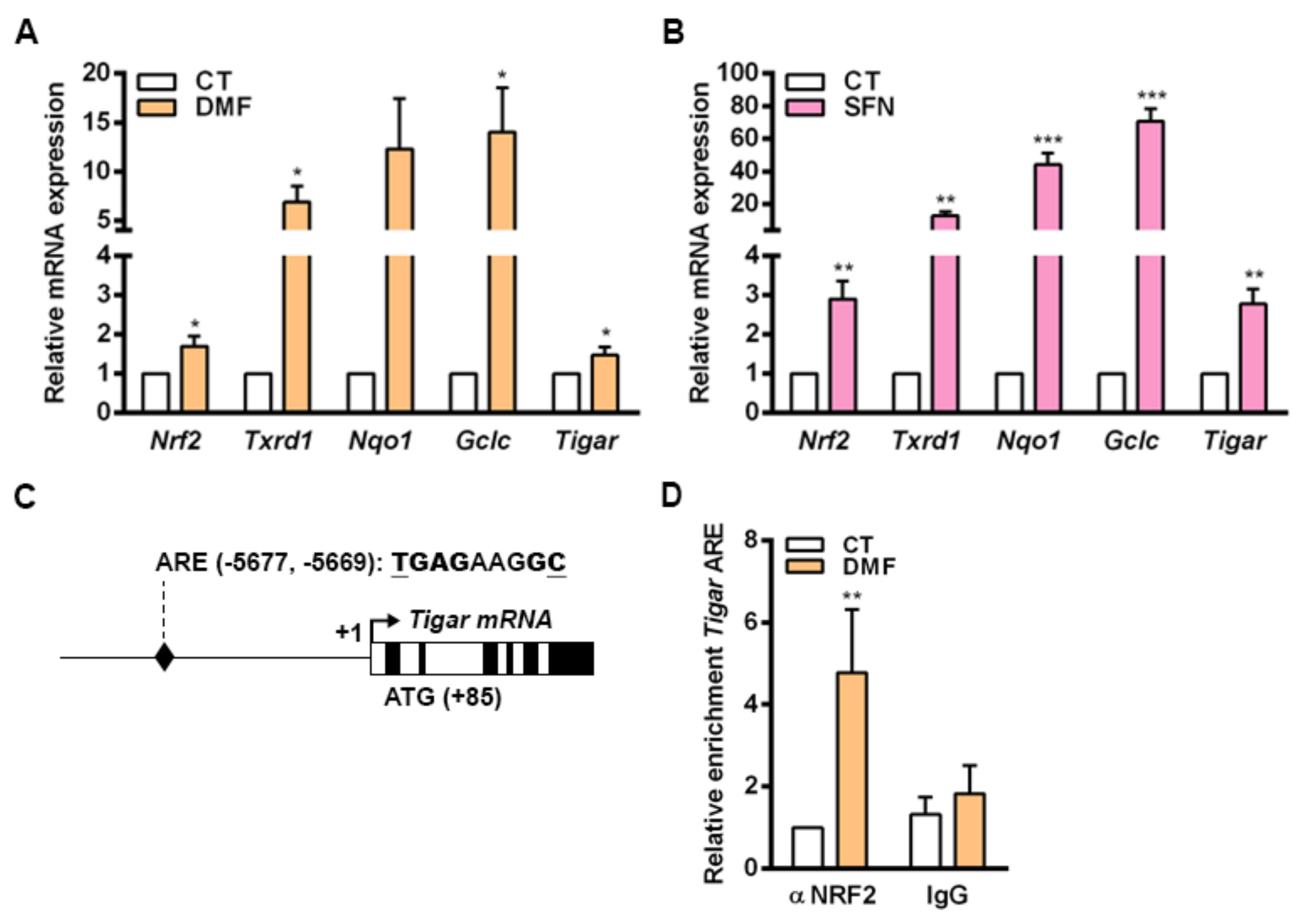
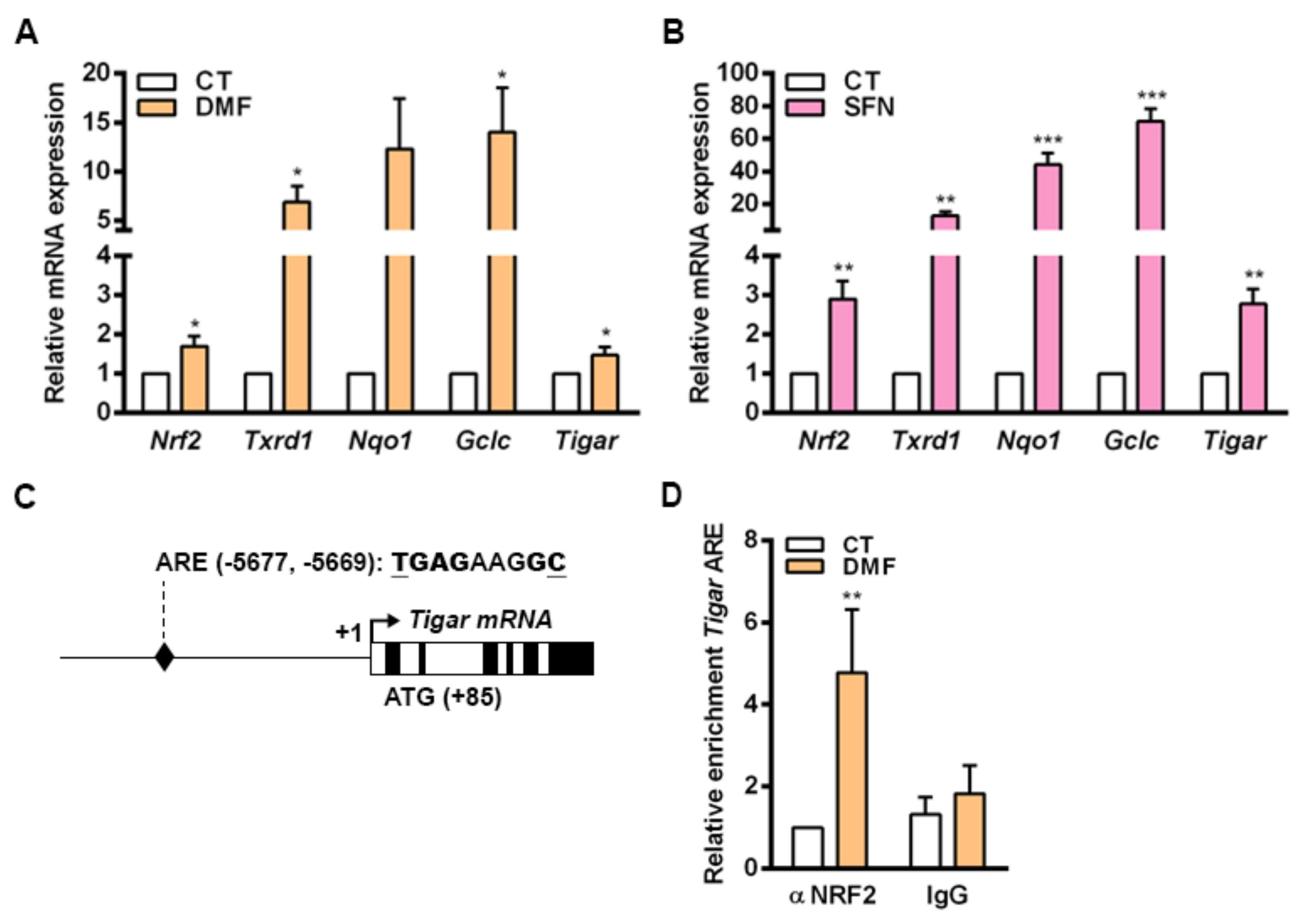
In mouse primary cells cultured in vitro, the transcriptional regulation of
Tigar by NRF2 involves an ARE located at −5.677 bp from the TSS. Increased binding of NRF2 to this ARE was observed in parallel to enhanced
Tigar expression in response to the NRF2 inducers DMF and SFN in mouse osteoblasts. The process of differentiation of osteocytes in the bone is linked to a shift of the metabolic machinery from glycolysis towards oxidative phosphorylation [
36]. Recently, increased mitochondrial biogenesis has been described as increasing ROS levels and NRF2 activity during osteocytogenesis [
34]. The induction of Tigar by NRF2 in mouse osteoblasts might also play a role in the promotion of mitochondrial metabolism in this cell type. In breast carcinoma cells,
TIGAR overexpression leads to increased oxygen consumption and ATP production in parallel to enhanced levels of the translocase of the outer mitochondrial membrane 20 (TOM20) [
6].
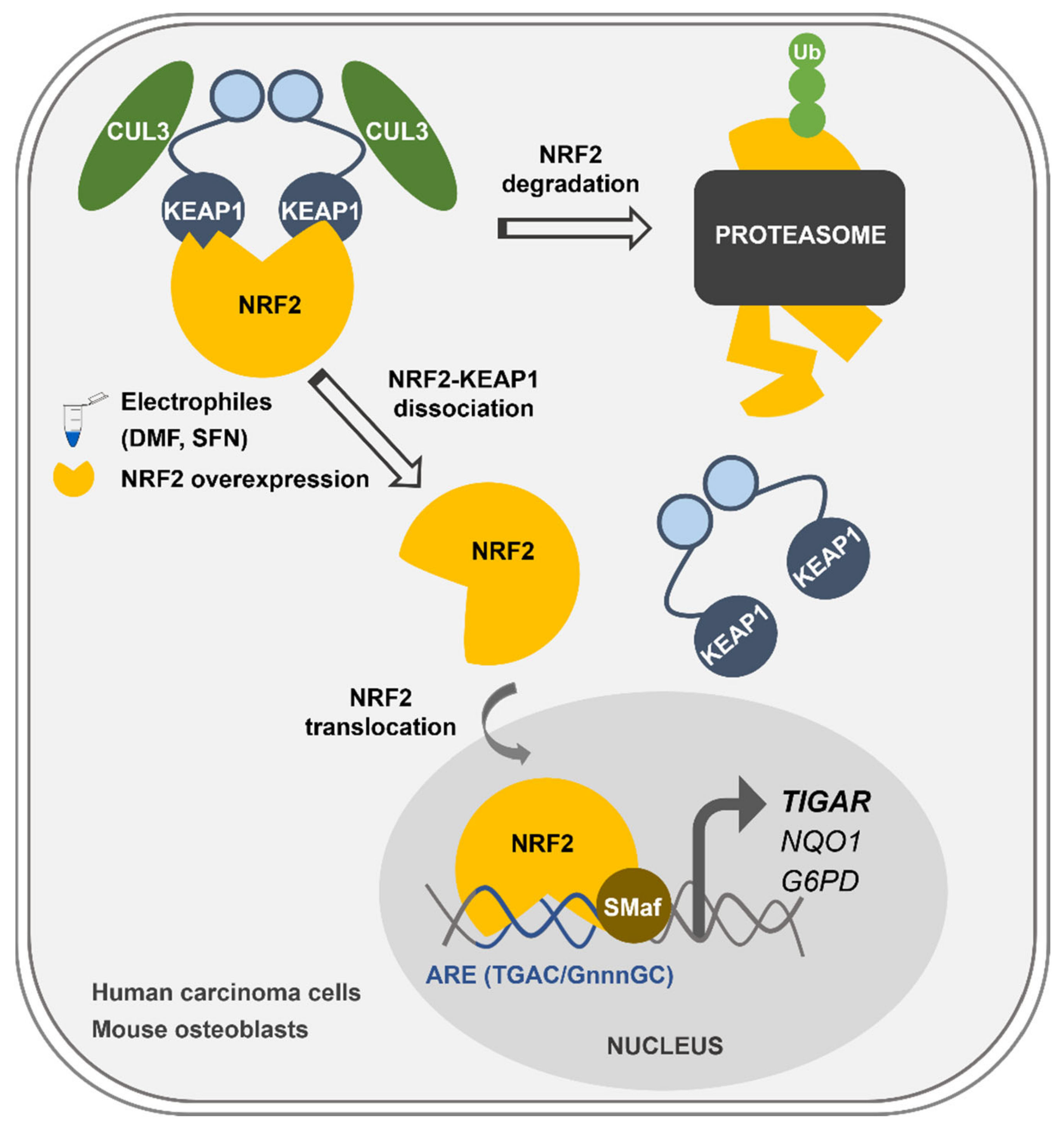
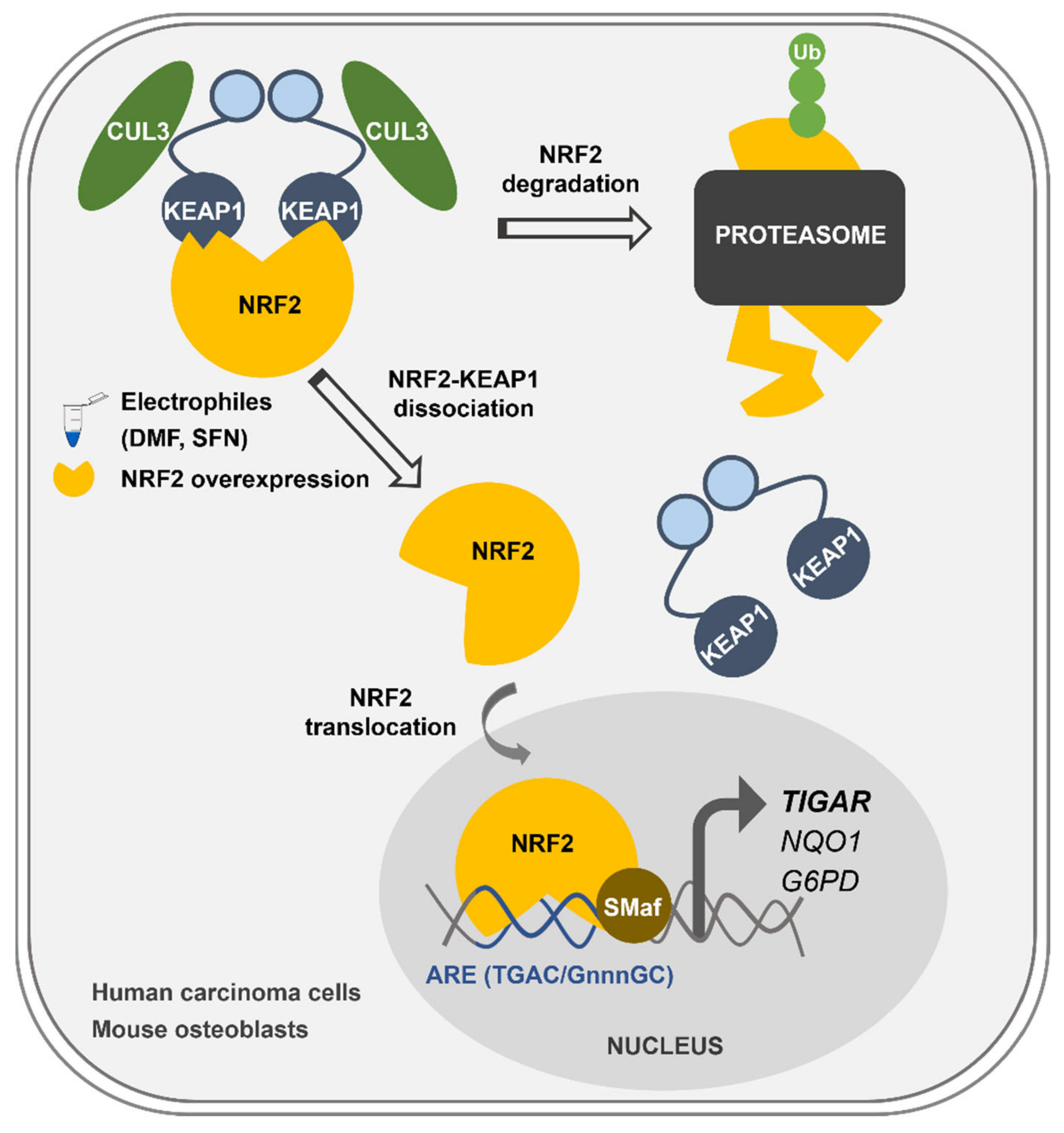
Overall, we describe here the mechanism of control of
TIGAR expression by NRF2, which can explain how
TIGAR is upregulated in response to oxidative stress and electrophilic molecules in cancer cell lines and healthy primary cells regardless of the expression of p53 (
Figure 6). This newly described mechanism of
TIGAR regulation by NRF2 can be of special interest in human cell lines lacking p53, or in other species where
TIGAR expression is independent of p53, such as mice [
24].
The
KEAP1/
NFE2L2 mutational status has been directly linked to the responsiveness of certain cancer types to radio- and chemotherapy [
22]. Besides, NRF2 addiction has been shown to define the metabolic signature of specific types of tumors. For example, glutamine addiction and induction of the PPP in NSCLS [
17,
18,
37]. Recently, it has been observed that TIGAR and NRF2 can fulfil similar functions in the initiation and dissemination of mouse pancreatic ductal adenocarcinoma. Expression of NRF2 and TIGAR is required for tumor initiation and for the development of established metastases, however loss of either one of these genes leads to increased ROS and metastatic potential [
28]. Targeting the NRF2 axis has shown promising preclinical results in terms of sensitivity to therapy [
18]. It would be interesting to analyze whether NRF2 inhibition is also beneficial for those cancer types in which TIGAR abrogation has been shown to increase sensibility to radio- and chemotherapy, the case, for example, of glioblastoma [
8,
38]. Whereas pharmacological inhibitors of the KEAP1-NRF2-sMAF pathway are currently being developed [
18,
39] and constitute potential antineoplastic drugs [
40], no molecules have been proposed as potential inhibitors of TIGAR to date. The specific effects of NRF2 inhibitors that might be attributed to the downregulation of TIGAR have yet to be deciphered. Heterogenous modulation of NRF2 target genes should also be considered, as experiments performed by our group indicate that downregulation of NRF2 in cell lines of different origin does not always have the same effects at the transcriptional level. Thus, strategies based on the disruption of the genetic expression or the pharmacological inhibition of the KEAP1-NRF2 axis should first evaluate the consequences of this inhibition on NRF2 targets. The expression of certain NRF2 downstream effectors might be compensated by alternative mechanisms that, ultimately, could render cancer cells even more aggressive or resistant to therapies.
The establishment of a direct link between NRF2 and TIGAR is of relevance as it should help in anticipating the coordinated functions of these two proteins while, at the same time, provide additional information as to how metabolic and redox processes are interconnected. As it has become evident, metabolism is no longer seen as the group of reactions that provide cellular energy; rather, metabolism is also at the heart of ROS homeostasis, anabolism, and cellular division. The most frequently studied role of TIGAR is its antioxidant capacity due to the bisphosphatase activity on Fru-2,6-P
2; however, reported activity of this enzyme on other glycolytic intermediates [
41] and in mitochondrial function [
6] should not be overlooked. The branching of glycolytic metabolites to anabolic pathways such as amino acid or nucleotide synthesis and the regulation of mitochondrial metabolism are important processes in tumor development, and NRF2 has been shown to control key enzymes in these pathways. Our work situates TIGAR in the wide landscape of NRF2-regulated pathways by demonstrating that NRF2 functions as an enhancer in the transcriptional control of human and mouse
TIGAR gene.
4. Materials and Methods
4.1. Human Cell Lines
HeLa (cervical carcinoma), HCT116 40.16 and HCT116 379.2 (colorectal carcinoma) cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). Lung adenocarcinoma cell lines H460 and A549 were kindly provided by the laboratories of Dr. Ricardo Pérez-Tomás and Dr. Jose Luis Rosa, respectively. All cells were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Biological Industries, Kibbutz Beit-Haemek, Israel, 01-055-1A-24) supplemented with 10% foetal bovine serum (FBS; Biological Industries, 04-007-1A), 2 mM glutamine (Biological Industries, 03-020-1B) and penicillin/streptomycin (100 U/mL and 100 μg/mL, respectively; Biological Industries, 03-031-1B). Elsewhere, this medium composition is referred to as complete DMEM. Cells were maintained at 37 °C, in a 5% CO2 atmosphere and a relative humidity of 70–80%.
4.2. Primary Mouse Osteoblasts
Primary osteoblasts were isolated from mice calvariae as previously described [
36]. All procedures were approved by the Ethics Committee for Animal Experimentation of the Generalitat de Catalunya. Briefly, the calvariae were dissected from P1–P4 pups, and soft tissue was discarded. A total of five to eight calvariae were pooled and digested in α-Minimum Essential Medium Eagle Alpha Modification (α-MEM; SIGMA, St. Louis, MO, USA, M8042) containing trypsin (0.025%)/collagenase II (1 mg/mL; Thermo Fisher Scientific, Waltham, MA, USA, 17101015). The product of the first 5 minutes of digestion was discarded, while the product of a double 20-minute digestion was centrifuged and seeded on 60 mm culture plates. Cells were used between passages 1 to 4. Osteoblasts were cultured at 37 ºC in osteogenic media ((α-MEM with 10% FBS (Biological Industries, 04-007-1A), 2 mM glutamine (Biological Industries, 03-020-1B), 1 mM pyruvate (Biological Industries, 03-042-1B), 100 U/mL penicillin and 0.1 mg/mL streptomycin (Biological Industries, 03-031-1B), 50 µg/mL ascorbic acid and 4 mM β-glycerophosphate).
4.3. Transient Overexpression of NRF2
HeLa cells were plated at a density of 30% in six-well plates and the transfection procedure was performed the following day using Lipofectamine LTX (Invitrogen, Carlsbad, CA, USA, 15338-100) according to the manufacturer’s indications in Opti-MEM (Thermo Fisher Scientific, 11058021). One microgram of NC16 pcDNA3.1 Flag NRF2 (Addgene, Watertown, MA, USA, 36971) or the corresponding pcDNA3.1 empty vector (Invitrogen) was transfected into each well. After 4 h of transfection, complete DMEM was added to each well. Cellular extracts were collected after 24 h of plasmid transfection.
4.4. Transient Downregulation of NRF2
HeLa, H460, A549, HCT116 40.16 or HCT116 379.2 cells were plated at a density of 15% in six-well plates and the transfection procedure was performed the following day using Oligofectamine (Invitrogen, 12252-011) according to the manufacturer’s indications in Opti-MEM (Thermo Fisher Scientific, 11058021). The small-interfering RNA (siRNA) HSS107130 (Thermo Fisher Scientific) targeting exon 5 of NFE2L2 (NM_006164.5) was used at 100 nM: 5’-CCAACCAGUUGACAGUGAACUCAUU-3’. Negative Stealth RNAi control, medium GC was used as negative control (Invitrogen, 12935300) and referred to as scrambled siRNA (Scr.). After 4 h of transfection, complete DMEM was added to each well. Cellular extracts were collected after 72 h of siRNA transfection.
4.5. NRF2 Inducers
Dimethylfumarate (DMF; SIGMA, 242926) and sulforaphane (SFN; SIGMA, S4441) were prepared with DMSO and used at a final concentration of 5 or 20 µM. Treatments were performed in the corresponding culture medium for each cell type. DMSO was used as vehicle control. DMF was freshly prepared for each experiment. Cells were plated at a density of 30% the day before treatment. Cell lines were treated for 24 h whereas primary osteoblasts were treated for 48 h with the indicated compounds.
4.6. Protein Extraction and Western Blot
Cells were lysed with whole cell lysis buffer containing 50 mM Tris–Cl, 1% SDS and 10% glycerol and protein concentration was determined with the Pierce BCA protein assay (Thermo Fisher Scientific, 23228 and 23224). Equal amounts of total protein extracts were analyzed in 12.5% (w/v) SDS–PAGE. Western blot was performed using specific antibodies for NRF2 (Santa Cruz Biotechnologies, Dallas, TX, USA, sc-722, 1:1000), G6PD (Abcam, Cambridge, UK, ab993, 1:500), TIGAR (Santa Cruz Biotechnologies, sc-67273, 1:1000) and α-tubulin (SIGMA, T8203, 1:4000). Peroxidase-conjugated secondary antibodies goat anti-rabbit and goat anti-α-mouse (Advansta, San Jose, CA, USA, R-050072-500 and R-050071-500, respectively) were used. Immunostaining was carried out using the ECL technique (Biological Industries, 20-500-500). Densitometric analysis was performed using Multi-Gauge v3.0 (FujiFilm Corporation, Tokyo, Japan, v. 2007) software. Protein levels were normalized to α-tubulin in all experiments.
4.7. RTqPCR Analysis
For RNA extraction, cells were maintained on ice and lysed with TRIsure (Bioline, London, UK, BIO-38033). Extraction was performed according to the manufacturer’s indications and RNA was quantified with NanoDrop One (Thermo Fischer Scientific) and reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Thermo Fischer Scientific, 4368813). mRNA expression analysis was performed through RT-qPCR using SensiFAST™ Probe Hi-ROX (Bioline, BIO-82005) and the following TaqMan Assays (Thermo Fisher Scientific): human NFE2L2 (Hs00975961_g1), G6PD (Hs00166169_m1), NQO1 (Hs01045993_g1), TIGAR (Hs00608644_m1) and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Hs99999905_m1); and mouse Nfe2l2 (Mm00477784_m1), Txnrd1 (Mm00443675_m1), Nqo1 (Mm01253561_m1), Gclc (Mm00802655_m1), Tigar (Mm01183137_m1) and TATA-box binding protein (Tbp) (Mm01277042_m1). The expression of each gene of interest was normalized to that of GAPDH in human cells and Tbp in mouse cells.
4.8. In silico Analysis of Human and Mouse TIGAR Promoter
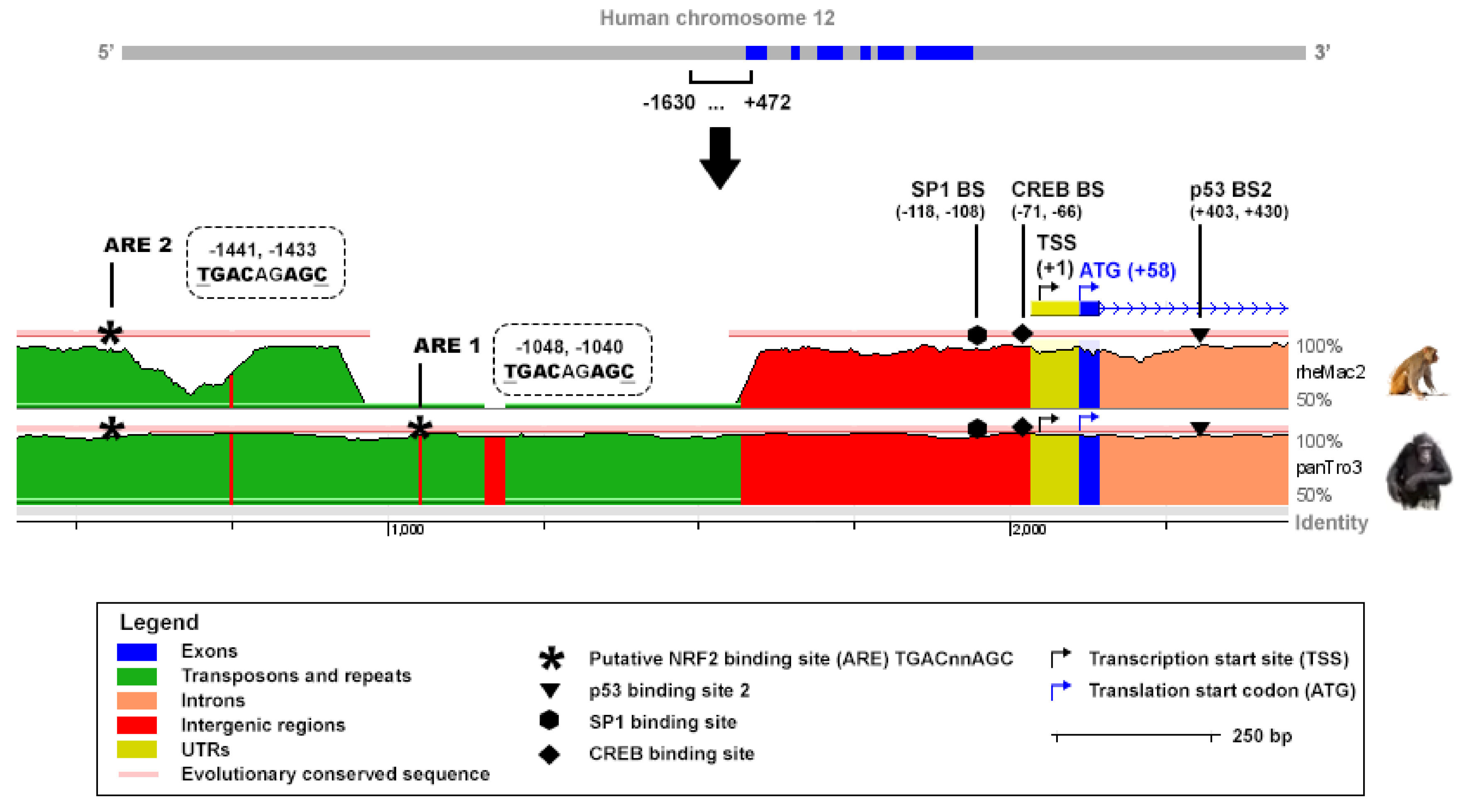
An in silico analysis was performed to identify putative AREs corresponding to the sequence TGAC/GnnnGC in the promoter of human and mouse
TIGAR. The sequences tracked covered −1630, +472 bp and −8000, +1 of the TSS of human and mouse TIGAR, respectively. The reference for the TSS was the NCBI (human TIGAR mRNA: NM_020375.3, mouse Tigar mRNA: NM_177003.5). The ECR Browser (available at
http://ecrbrowser.dcode.org, accessed on 10 January 2022) [
31] was used to analyse the degree of conservation of the AREs identified across species by comparing the genome sequences of Homo sapiens, Pan troglodytes (panTro3), Rhesus macaque (rheMac2) and Mus musculus (mm10).
4.9. Generation of TIGAR Promoter Constructs
Two bacterial artificial chromosomes (BACs) of human chromosome 12 (RP11-177D20 and RP11-74J21) were provided by the BACPAC Service of the Children’s Hospital Oakland Research Institute (ChORI, Oakland, CA, USA) as agar-stab culture. Specific information about the clones is available at bacpacresources.org. Bacteria were cultured and amplified in Luria Bertani (LB) medium and BACs were purified with a phenol/chloroform-based DNA extraction protocol. The region covering from −1623 to −635 from the TSS of TIGAR, containing the two AREs of interest, was amplified by PCR using MyFi™ DNA Polymerase (Bioline, BIO-21118) and the following primers FW: 5’-GCGTCCTTACAGATCTAGCATGG-3’ and RV: 5’-GCCCCTTGATAGCTAGCAAAGTTC-3’. The resulting 989 bp PCR product was cloned into the pCRR2.1-TOPO vector using the TOPO TA Cloning Kit (45-1641, Thermo Fisher Scientific) and then subcloned into the pGL3-Promoter Vector (Promega, Madison, WI, E1761), under the control of SV40 promoter, through double restriction enzyme digestion with SacI/XhoI (New England Biolabs, Ipswich, MA, USA, R0156S and R0146S, respectively). To generate the constructs containing each of the ARE, the pGL3-Promoter Vector containing the 989 bp construct was further digested with SmaI (New England Biolabs, R0141S)/SacI or SmaI/XhoI to generate a 467 bp construct containing only ARE1 (−1101, −635 bp) and a 522 bp construct containing only ARE2 (−1623, −1101 bp), respectively. The single ARE constructs were subcloned again in pGL3-Promoter Vectors. For all cloning procedures, JM109 competent cells (Promega, L2005) were used, cultured in LB agar plates and LB liquid culture containing 50 µg/mL ampicillin and purified with GeneElute Plasmid Miniprep or Maxiprep Kits (SIGMA, PLN350 and NA0410, respectively). Phosphatase FastAP (Thermo Fisher Scientific, EF0651) and T4 DNA Ligase (Thermo Fisher Scientific, EL0011) were used to dephosphorylate 5’-P termini of vectors and to perform ligations, respectively. Specific PCR, digestion and ligation conditions are available on request.
4.10. Luciferase Assays
HeLa cells were plated at a density of 30% in six-well plates and the following day 0.8 µg of the corresponding luciferase reporter construct was transfected to each well using Lipofectamine LTX (Invitrogen, 15338-100) according to the manufacturer’s indications in Opti-MEM (Thermo Fisher Scientific, 11058021). The constructs used were pGL3-Promoter Vector containing the 989 bp with both AREs of TIGAR promoter (ARE1 + ARE2), pGL3-Promoter Vector containing only ARE1 (ARE1), pGL3-Promoter Vector containing only ARE2 (ARE2) or the corresponding pGL3-Promoter empty vector (Promega, E1761). To assess the efficiency of the transfection and to normalize luciferase expression values, 0.5 µg of RSV-β-galactosidase plasmid was transfected into each well. After 4h of transfection, cells were trypsinized and split (each well of a six-well plate was split into three wells of a 12-well plate) and complete DMEM was added to each well. Luciferase experiments were performed in cells overexpressing NRF2 or in cells treated with SFN. For NRF2 overexpression, 1 µg of NC16 pcDNA3.1 Flag NRF2 (Addgene, 36971) was co-transfected with the luciferase and β-galactosidase plasmids. For SFN treatment, SFN was added to a final concentration of 20 µM when cells were split into 12-well plates. After 24 h of transfection, cells were washed twice in cold PBS and lysed in buffer of the Luciferase Assay System (Promega, E1500). Fifty microliters of cell lysate were used to measure luciferase activity. β-Galactosidase activity was determined in 30 μL of cell lysate using the luminescent β-galactosidase detection kit II (Takara Bio, Shiga, Japan, 631712). Both activities were measured in a TD 20/20 luminometer (Turner Designs, San Jose, CA, USA) in acquisition intervals of 10 and 5 s, respectively.
4.11. Chromatin Immunoprecipitation (ChIP) Assays
HeLa and osteoblasts cells were plated at a density of 30% in 10 cm plates and either transfected with 6 µg of NC16 pcDNA3.1 Flag NRF2 (Addgene, 36971) or treated with DMF the following day. After 24 h, cells were fixed with 1% formaldehyde for 10 min, and the reaction was stopped with 0.25 M glycine for 5 min. Cells were maintained on ice, lysed with a buffer containing 1% SDS, 10 mM EDTA and 50 mM Tris and sonicated to obtain chromatin fragments of 500–1000 bp in a Branson Sonifier 250 (Branson Ultrasonics, Brookfield, CT, USA) with six cycles of ten pulses at duty cycle 0.8 seconds and output 5. Two 10-cm plates were lysed for each experimental condition. ChIP was carried out using 7.6 µg of NRF2 antibody (Cell Signaling Technology, Danvers, MA, USA, 12721) or nonspecific rabbit IgG (Merck Millipore, Burlington, Massachusetts, 12-372) as a control and purified with 20 µL of Magna ChIP protein A+G magnetic beads (Merck Millipore, 16-663). The complexes were washed twice with four different buffers and eluted with a solution of 1% SDS and 0.1 M NaHCO3. Reversion of cross-linking was carried out by overnight incubation with 0.2 M NaCl at 65 °C, followed by treatment with proteinase K and RNase A. The DNA fragments were purified using the QIAquick gel extraction kit (Qiagen, Hilden, Germany, 28706) and analyzed by RT-qPCR. Input samples were prepared with 12.5% of the chromatin material used for an immunoprecipitation in each experimental condition. Inputs were analyzed in parallel and used to normalize chromatin enrichment. RTqPCR was performed with SYBR Select Master Mix (Thermo Fischer Scientific, 4472908) at 45 cycles with the following primers:
FW human TIGAR (5’-AATGGCGTGAACCCGGGAGGCG-3’)
RV human TIGAR (5’-GCCTTCAGAACGTTGAGGGAGTTGC-3’)
FW human NQO1 (5’- TGGCATGCACCCAGGGAAGTGTGT-3’)
RV human NQO1 (5’-AGCCGGATGCGGATTACTGTGGTGC-3’)
FW mouse Tigar (5’-AGTGACAGGCTAAACGGCCAGGCA-3’)
RV mouse Tigar (5’-AGCTGGGGGCGGAGGAAGATTGGTT-3’).
4.12. Statistical Analysis
Results are expressed as the mean ± SEM of independent experiments. Differences were analyzed with the corresponding statistical test, according to the nature of the data represented. The number of experiments as well as the statistical method applied are indicated in each figure. Significant differences at p < 0.05, 0.01, and 0.001 between conditions are indicated by ∗, ∗∗, and ∗∗∗, respectively. All calculations were performed using the GraphPad Prism version 4.00 for Windows (GraphPad Software, La Jolla, CA, USA).


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}