
mTOR Signaling Components in Tumor Mechanobiology


 ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tumor Mechanobiology
The Role of ECM
3. mTOR Biology
3.1. mTOR Structure and Functions
3.2. mTOR in Cancer Development and Progression
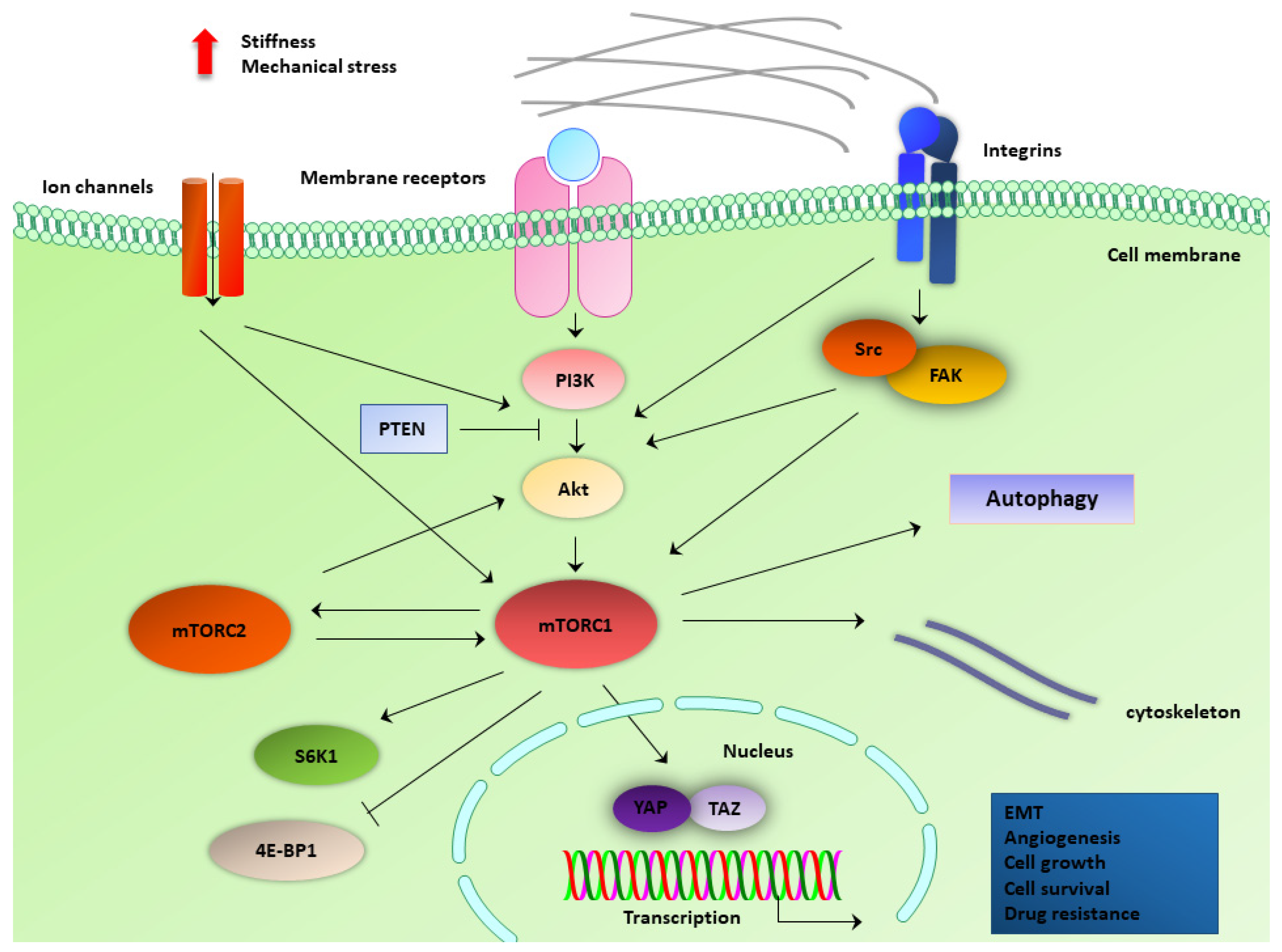
4. mTOR Components Involved in Mechanotransduction in Cancer Cells
4.1. Force-Regulated Autophagy
4.2. PI3K/Akt Pathway
4.3. ECM Stiffness-Induced Mechanisms
4.4. Mechanosensitive Membrane Protein Complexes and Ligands
4.5. Mechanosensitive Ion Channels and Cytoskeletal Components
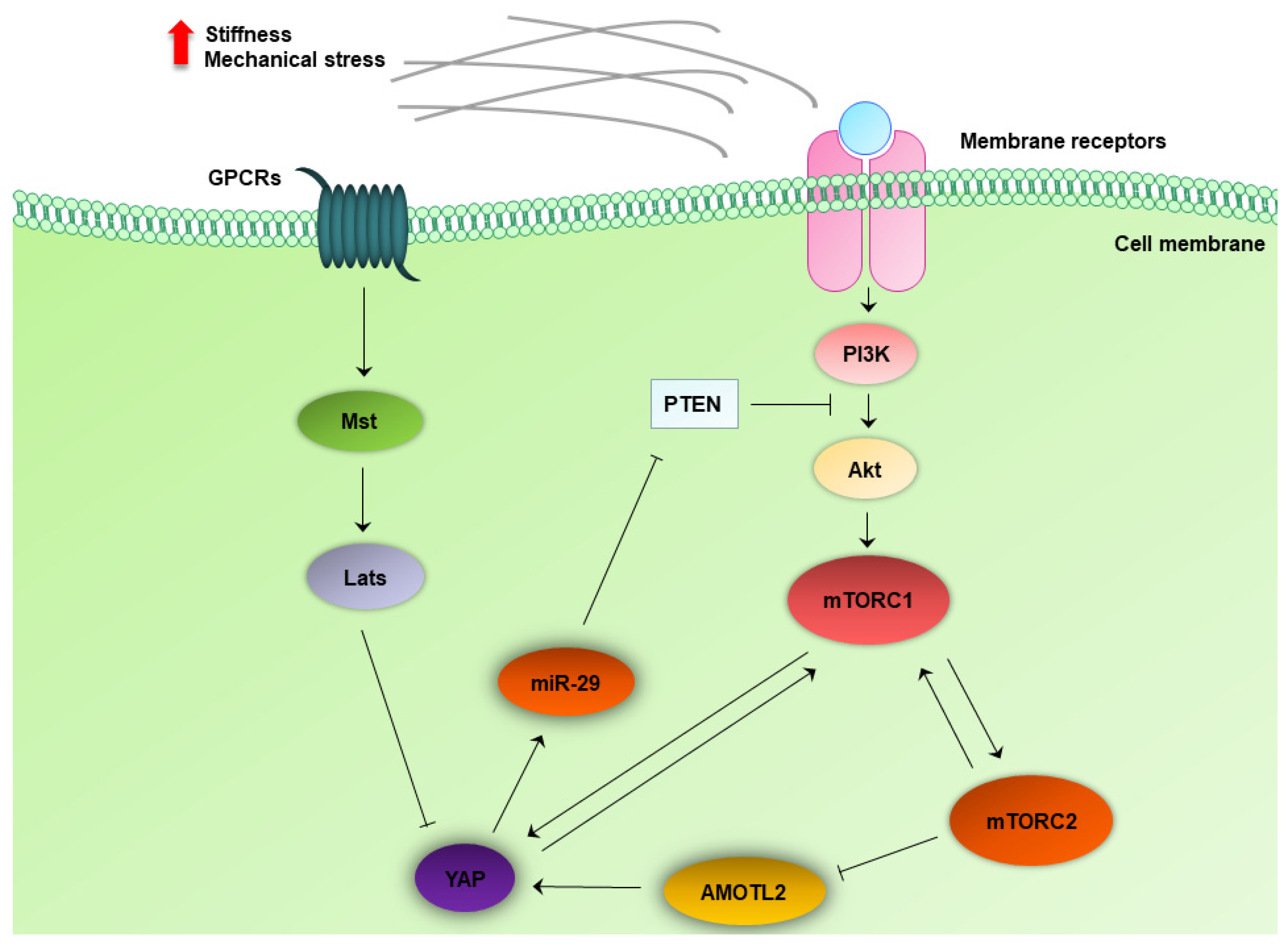
4.6. Crosstalk between the MTOR and Hippo Pathway
5. mTOR Targeting and Mechanotransduction Interplay in Anticancer Treatments
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dumont, F.J.; Su, Q. Mechanism of action of the immunosuppressant rapamycin. Life Sci. 1996, 58, 373–395. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, P.K.; Low, B.C.; Lim, C.T. Mechanobiology of Tumor Growth. Chem. Rev. 2018, 118, 6499–6515. [Google Scholar] [CrossRef] [PubMed]
- Gargalionis, A.N.; Basdra, E.K.; Papavassiliou, A.G. Tumor mechanosensing and its therapeutic potential. J. Cell. Biochem. 2018, 119, 4304–4308. [Google Scholar] [CrossRef]
- Gargalionis, A.N.; Basdra, E.K.; Papavassiliou, A.G. Cancer mechanobiology: Effects and therapeutic perspectives. Int. J. Cancer 2018, 142, 1298–1299. [Google Scholar] [CrossRef] [Green Version]
- Di-Luoffo, M.; Ben-Meriem, Z.; Lefebvre, P.; Delarue, M.; Guillermet-Guibert, J. PI3K functions as a hub in mechanotransduction. Trends Biochem. Sci. 2021, 46, 878–888. [Google Scholar] [CrossRef]
- King, J.S.; Veltman, D.M.; Insall, R.H. The induction of autophagy by mechanical stress. Autophagy 2011, 7, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Nazemi, M.; Rainero, E. Cross-Talk between the Tumor Microenvironment, Extracellular Matrix, and Cell Metabolism in Cancer. Front. Oncol. 2020, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Jansen, K.A.; Donato, D.M.; Balcioglu, H.E.; Schmidt, T.; Danen, E.H.; Koenderink, G.H. A guide to mechanobiology: Where biology and physics meet. Biochim. Biophys. Acta 2015, 1853, 3043–3052. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, V.; Gloerich, M. Decoding mechanical cues by molecular mechanotransduction. Curr. Opin. Cell Biol. 2021, 72, 72–80. [Google Scholar] [CrossRef]
- Xia, Y.; Pfeifer, C.R.; Cho, S.; Discher, D.E.; Irianto, J. Nuclear mechanosensing. Emerg. Top. Life Sci. 2018, 2, 713–725. [Google Scholar] [CrossRef]
- Vogel, V. Unraveling the Mechanobiology of Extracellular Matrix. Annu. Rev. Physiol. 2018, 80, 353–387. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.; Shi, L.; Wolfenson, H. The ‘Yin and Yang’ of Cancer Cell Growth and Mechanosensing. Cancers 2021, 13, 4754. [Google Scholar] [CrossRef] [PubMed]
- Wullkopf, L.; West, A.V.; Leijnse, N.; Cox, T.R.; Madsen, C.D.; Oddershede, L.B.; Erler, J.T. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol. Biol. Cell 2018, 29, 2378–2385. [Google Scholar] [CrossRef]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Sun, Q.; Li, X.; Feng, J.; Ao, Z.; Li, X.; Wang, J. Substrate Stiffness Modulates the Growth, Phenotype, and Chemoresistance of Ovarian Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 718834. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.; Sligar, A.D.; Chavarria, D.; Lee, J.; Choksi, D.; Patil, N.P.; Lee, H.; Veith, A.P.; Riley, W.J.; Desai, S.; et al. Biomechanical regulation of breast cancer metastasis and progression. Sci. Rep. 2021, 11, 9838. [Google Scholar] [CrossRef]
- Baker, E.L.; Bonnecaze, R.T.; Zaman, M.H. Extracellular matrix stiffness and architecture govern intracellular rheology in cancer. Biophys. J. 2009, 97, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Gensbittel, V.; Krater, M.; Harlepp, S.; Busnelli, I.; Guck, J.; Goetz, J.G. Mechanical Adaptability of Tumor Cells in Metastasis. Dev. Cell 2021, 56, 164–179. [Google Scholar] [CrossRef]
- Moose, D.L.; Henry, M.D. Survival of the resilient: Mechano-adaptation of circulating tumor cells to fluid shear stress. Mol. Cell Oncol. 2020, 7, 1766338. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341, 1236566. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.X.; Guan, K.L. The SIN1-PH Domain Connects mTORC2 to PI3K. Cancer Discov. 2015, 5, 1127–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, J.T.; Ray, C.; Fox, A.L.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. Mammalian EAK-7 activates alternative mTOR signaling to regulate cell proliferation and migration. Sci. Adv. 2018, 4, eaao5838. [Google Scholar] [CrossRef] [Green Version]
- Mendonca, D.B.; Nguyen, J.T.; Haidar, F.; Fox, A.L.; Ray, C.; Amatullah, H.; Liu, F.; Kim, J.K.; Krebsbach, P.H. MicroRNA-1911-3p targets mEAK-7 to suppress mTOR signaling in human lung cancer cells. Heliyon 2020, 6, e05734. [Google Scholar] [CrossRef]
- Nguyen, J.T.; Haidar, F.S.; Fox, A.L.; Ray, C.; Mendonca, D.B.; Kim, J.K.; Krebsbach, P.H. mEAK-7 Forms an Alternative mTOR Complex with DNA-PKcs in Human Cancer. iScience 2019, 17, 190–207. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27 (Suppl. 2), S43–S51. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.J.; Crowe, P.; Yang, J.L. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 671–689. [Google Scholar] [CrossRef]
- Nathan, N.; Keppler-Noreuil, K.M.; Biesecker, L.G.; Moss, J.; Darling, T.N. Mosaic Disorders of the PI3K/PTEN/AKT/TSC/mTORC1 Signaling Pathway. Dermatol. Clin. 2017, 35, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilagan, E.; Manning, B.D. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer 2016, 2, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalski, J.R.; Bhaduri, A.; Zehnder, A.M.; Neela, P.H.; Che, Y.; Wozniak, G.G.; Khavari, P.A. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol. Cell 2019, 73, 830–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison Joly, M.; Williams, M.M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Young, C.D.; Sarbassov, D.D.; Muller, W.J.; Brantley-Sieders, D.; Cook, R.S. Two distinct mTORC2-dependent pathways converge on Rac1 to drive breast cancer metastasis. Breast Cancer Res. 2017, 19, 74. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.M.; Dietrich, P.; Hackl, C.; Guenzle, J.; Bronsert, P.; Wagner, C.; Fichtner-Feigl, S.; Schlitt, H.J.; Geissler, E.K.; Hellerbrand, C.; et al. Inhibition of mTORC2/RICTOR Impairs Melanoma Hepatic Metastasis. Neoplasia 2018, 20, 1198–1208. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- Hornberger, T.A. Mechanotransduction and the regulation of mTORC1 signaling in skeletal muscle. Int. J. Biochem. Cell Biol. 2011, 43, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, B.L.; Goodman, C.A.; Hornberger, T.A. The mechanical activation of mTOR signaling: An emerging role for late endosome/lysosomal targeting. J. Muscle Res. Cell Motil. 2014, 35, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.S.; Liu, Y.W. Mechanical Stretch Induces mTOR Recruitment and Activation at the Phosphatidic Acid-Enriched Macropinosome in Muscle Cell. Front. Cell Dev. Biol. 2019, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Caceres, M.P.; Munoz, L.; Pradenas, J.M.; Pena, F.; Lagos, P.; Aceiton, P.; Owen, G.I.; Morselli, E.; Criollo, A.; Ravasio, A.; et al. Mechanobiology of Autophagy: The Unexplored Side of Cancer. Front. Oncol. 2021, 11, 632956. [Google Scholar] [CrossRef]
- Phillip, J.M.; Aifuwa, I.; Walston, J.; Wirtz, D. The Mechanobiology of Aging. Annu. Rev. Biomed. Eng. 2015, 17, 113–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Liu, C.; Zhang, D.; Men, H.; Huo, L.; Geng, Q.; Wang, S.; Gao, Y.; Zhang, W.; Zhang, Y.; et al. Mechanosensitive ion channel Piezo1 promotes prostate cancer development through the activation of the Akt/mTOR pathway and acceleration of cell cycle. Int. J. Oncol. 2019, 55, 629–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 14, 1110–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathage, B.; Gehlert, S.; Ulbricht, A.; Ludecke, L.; Tapia, V.E.; Orfanos, Z.; Wenzel, D.; Bloch, W.; Volkmer, R.; Fleischmann, B.K.; et al. The cochaperone BAG3 coordinates protein synthesis and autophagy under mechanical strain through spatial regulation of mTORC1. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 62–75. [Google Scholar] [CrossRef]
- Blawat, K.; Mayr, A.; Hardt, M.; Kirschneck, C.; Nokhbehsaim, M.; Behl, C.; Deschner, J.; Jager, A.; Memmert, S. Regulation of Autophagic Signaling by Mechanical Loading and Inflammation in Human PDL Fibroblasts. Int. J. Mol. Sci. 2020, 21, 9446. [Google Scholar] [CrossRef]
- Das, J.; Agarwal, T.; Chakraborty, S.; Maiti, T.K. Compressive stress-induced autophagy promotes invasion of HeLa cells by facilitating protein turnover in vitro. Exp. Cell Res. 2019, 381, 201–207. [Google Scholar] [CrossRef]
- Das, J.; Maji, S.; Agarwal, T.; Chakraborty, S.; Maiti, T.K. Hemodynamic shear stress induces protective autophagy in HeLa cells through lipid raft-mediated mechanotransduction. Clin. Exp. Metastasis 2018, 35, 135–148. [Google Scholar] [CrossRef]
- Danciu, T.E.; Adam, R.M.; Naruse, K.; Freeman, M.R.; Hauschka, P.V. Calcium regulates the PI3K-Akt pathway in stretched osteoblasts. FEBS Lett. 2003, 536, 193–197. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.; Yashiro, M.; Nishioka, N.; Hirakawa, K.; Olden, K.; Roberts, J.D. PI3K/Akt signalling is required for the attachment and spreading, and growth in vivo of metastatic scirrhous gastric carcinoma. Br. J. Cancer 2012, 106, 1535–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubashkin, M.G.; Cassereau, L.; Bainer, R.; DuFort, C.C.; Yui, Y.; Ou, G.; Paszek, M.J.; Davidson, M.W.; Chen, Y.Y.; Weaver, V.M. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Res. 2014, 74, 4597–4611. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Montminy, T.; Azad, T.; Lightbody, E.; Hao, Y.; SenGupta, S.; Asselin, E.; Nicol, C.; Yang, X. PI3K Positively Regulates YAP and TAZ in Mammary Tumorigenesis Through Multiple Signaling Pathways. Mol. Cancer Res. 2018, 16, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Kalli, M.; Minia, A.; Pliaka, V.; Fotis, C.; Alexopoulos, L.G.; Stylianopoulos, T. Solid stress-induced migration is mediated by GDF15 through Akt pathway activation in pancreatic cancer cells. Sci. Rep. 2019, 9, 978. [Google Scholar] [CrossRef] [PubMed]
- Kalli, M.; Li, R.; Mills, G.B.; Stylianopoulos, T.; Zervantonakis, I.K. Mechanical stress in pancreatic cancer: Signaling pathway adaptation activates cytoskeletal remodeling and enhances cell migration. bioRxiv 2021. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell. Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef]
- Kim, B.G.; Sung, J.S.; Jang, Y.; Cha, Y.J.; Kang, S.; Han, H.H.; Lee, J.H.; Cho, N.H. Compression-induced expression of glycolysis genes in CAFs correlates with EMT and angiogenesis gene expression in breast cancer. Commun. Biol. 2019, 2, 313. [Google Scholar] [CrossRef] [Green Version]
- Frith, J.E.; Kusuma, G.D.; Carthew, J.; Li, F.; Cloonan, N.; Gomez, G.A.; Cooper-White, J.J. Mechanically-sensitive miRNAs bias human mesenchymal stem cell fate via mTOR signalling. Nat. Commun. 2018, 9, 257. [Google Scholar] [CrossRef] [Green Version]
- Rainero, E.; Howe, J.D.; Caswell, P.T.; Jamieson, N.B.; Anderson, K.; Critchley, D.R.; Machesky, L.; Norman, J.C. Ligand-Occupied Integrin Internalization Links Nutrient Signaling to Invasive Migration. Cell Rep. 2015, 10, 398–413. [Google Scholar] [CrossRef]
- Husain, A.; Khadka, A.; Ehrlicher, A.; Saint-Geniez, M.; Krishnan, R. Substrate stiffening promotes VEGF-A functions via the PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2022, 586, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Shea, M.P.; O’Leary, K.A.; Wegner, K.A.; Vezina, C.M.; Schuler, L.A. High collagen density augments mTOR-dependent cancer stem cells in ERalpha+ mammary carcinomas, and increases mTOR-independent lung metastases. Cancer Lett. 2018, 433, 1–9. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix stiffness-mediated effects on stemness characteristics occurring in HCC cells. Oncotarget 2016, 7, 32221–32231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.; Qiao, X.; Xing, X.; Huang, J.; Qian, J.; Wang, Y.; Zhang, Y.; Zhang, X.; Li, M.; Cui, J.; et al. Matrix Stiffness-Upregulated MicroRNA-17-5p Attenuates the Intervention Effects of Metformin on HCC Invasion and Metastasis by Targeting the PTEN/PI3K/Akt Pathway. Front. Oncol. 2020, 10, 1563. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wang, J.; Sun, Q.; Li, F.; Gao, H.; Xu, L.; Zhang, J.; Sun, X.; Tian, Y.; Zhao, Q.; et al. Interleukin-8 promotes integrin beta3 upregulation and cell invasion through PI3K/Akt pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 449. [Google Scholar] [CrossRef]
- Colombero, C.; Remy, D.; Antoine-Bally, S.; Mace, A.S.; Monteiro, P.; ElKhatib, N.; Fournier, M.; Dahmani, A.; Montaudon, E.; Montagnac, G.; et al. mTOR Repression in Response to Amino Acid Starvation Promotes ECM Degradation Through MT1-MMP Endocytosis Arrest. Adv. Sci. 2021, 8, e2101614. [Google Scholar] [CrossRef]
- Lee, F.Y.; Zhen, Y.Y.; Yuen, C.M.; Fan, R.; Chen, Y.T.; Sheu, J.J.; Chen, Y.L.; Wang, C.J.; Sun, C.K.; Yip, H.K. The mTOR-FAK mechanotransduction signaling axis for focal adhesion maturation and cell proliferation. Am. J. Transl. Res. 2017, 9, 1603–1617. [Google Scholar]
- Wu, C.; You, J.; Fu, J.; Wang, X.; Zhang, Y. Phosphatidylinositol 3-Kinase/Akt Mediates Integrin Signaling To Control RNA Polymerase I Transcriptional Activity. Mol. Cell. Biol. 2016, 36, 1555–1568. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Guan, L.; Li, S.; Jiang, Y.; Xiong, N.; Li, L.; Wu, C.; Zeng, H.; Liu, Y. Mechanosensitive caveolin-1 activation-induced PI3K/Akt/mTOR signaling pathway promotes breast cancer motility, invadopodia formation and metastasis in vivo. Oncotarget 2016, 7, 16227–16247. [Google Scholar] [CrossRef] [Green Version]
- Rosselli-Murai, L.K.; Almeida, L.O.; Zagni, C.; Galindo-Moreno, P.; Padial-Molina, M.; Volk, S.L.; Murai, M.J.; Rios, H.F.; Squarize, C.H.; Castilho, R.M. Periostin responds to mechanical stress and tension by activating the MTOR signaling pathway. PLoS ONE 2013, 8, e83580. [Google Scholar] [CrossRef]
- Jia, Y.Y.; Yu, Y.; Li, H.J. POSTN promotes proliferation and epithelial-mesenchymal transition in renal cell carcinoma through ILK/AKT/mTOR pathway. J. Cancer 2021, 12, 4183–4195. [Google Scholar] [CrossRef]
- Gargalionis, A.N.; Korkolopoulou, P.; Farmaki, E.; Piperi, C.; Dalagiorgou, G.; Adamopoulos, C.; Levidou, G.; Saetta, A.; Fragkou, P.; Tsioli, P.; et al. Polycystin-1 and polycystin-2 are involved in the acquisition of aggressive phenotypes in colorectal cancer. Int. J. Cancer 2015, 136, 1515–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombroski, J.A.; Hope, J.M.; Sarna, N.S.; King, M.R. Channeling the Force: Piezo1 Mechanotransduction in Cancer Metastasis. Cells 2021, 10, 2815. [Google Scholar] [CrossRef] [PubMed]
- Nam, S.; Gupta, V.K.; Lee, H.P.; Lee, J.Y.; Wisdom, K.M.; Varma, S.; Flaum, E.M.; Davis, C.; West, R.B.; Chaudhuri, O. Cell cycle progression in confining microenvironments is regulated by a growth-responsive TRPV4-PI3K/Akt-p27(Kip1) signaling axis. Sci. Adv. 2019, 5, eaaw6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, Z.S.; Hussain, F. Shear Stress Increases V-H + -ATPase and Acidic Vesicle Number Density, and p-mTORC2 Activation in Prostate Cancer Cells. Cell. Mol. Bioeng. 2020, 13, 591–604. [Google Scholar] [CrossRef]
- Chantaravisoot, N.; Wongkongkathep, P.; Loo, J.A.; Mischel, P.S.; Tamanoi, F. Significance of filamin A in mTORC2 function in glioblastoma. Mol. Cancer 2015, 14, 127. [Google Scholar] [CrossRef] [Green Version]
- Diz-Munoz, A.; Thurley, K.; Chintamen, S.; Altschuler, S.J.; Wu, L.F.; Fletcher, D.A.; Weiner, O.D. Membrane Tension Acts Through PLD2 and mTORC2 to Limit Actin Network Assembly During Neutrophil Migration. PLoS Biol. 2016, 14, e1002474. [Google Scholar] [CrossRef] [Green Version]
- Ibar, C.; Irvine, K.D. Integration of Hippo-YAP Signaling with Metabolism. Dev. Cell 2020, 54, 256–267. [Google Scholar] [CrossRef]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat. Cell Biol. 2012, 14, 1322–1329. [Google Scholar] [CrossRef]
- Csibi, A.; Blenis, J. Hippo-YAP and mTOR pathways collaborate to regulate organ size. Nat. Cell Biol. 2012, 14, 1244–1245. [Google Scholar] [CrossRef]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artinian, N.; Cloninger, C.; Holmes, B.; Benavides-Serrato, A.; Bashir, T.; Gera, J. Phosphorylation of the Hippo Pathway Component AMOTL2 by the mTORC2 Kinase Promotes YAP Signaling, Resulting in Enhanced Glioblastoma Growth and Invasiveness. J. Biol. Chem. 2015, 290, 19387–19401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Lin, Y.; Zhang, X.C.; Tan, Y.H.; Yao, Y.L.; Tan, J.; Zhang, X.; Cui, Y.H.; Liu, X.; Wang, Y.; et al. Phosphorylated mTOR and YAP serve as prognostic markers and therapeutic targets in gliomas. Lab. Invest. 2017, 97, 1354–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, C.G.; Ng, Y.L.; Lam, W.L.; Plouffe, S.W.; Guan, K.L. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015, 25, 1299–1313. [Google Scholar] [CrossRef]
- Liang, N.; Zhang, C.; Dill, P.; Panasyuk, G.; Pion, D.; Koka, V.; Gallazzini, M.; Olson, E.N.; Lam, H.; Henske, E.P.; et al. Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J. Exp. Med. 2014, 211, 2249–2263. [Google Scholar] [CrossRef]
- Liu, P.; Calvisi, D.F.; Kiss, A.; Cigliano, A.; Schaff, Z.; Che, L.; Ribback, S.; Dombrowski, F.; Zhao, D.; Chen, X. Central role of mTORC1 downstream of YAP/TAZ in hepatoblastoma development. Oncotarget 2017, 8, 73433–73447. [Google Scholar] [CrossRef] [Green Version]
- Frtus, A.; Smolkova, B.; Uzhytchak, M.; Lunova, M.; Jirsa, M.; Hof, M.; Jurkiewicz, P.; Lozinsky, V.I.; Wolfova, L.; Petrenko, Y.; et al. Hepatic Tumor Cell Morphology Plasticity under Physical Constraints in 3D Cultures Driven by YAP-mTOR Axis. Pharmaceuticals 2020, 13, 430. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, M.; Li, Y.; Wang, Y.; Wang, K.; Chen, Q.; Zhang, R.; Song, W.; Huang, Q.; Zhao, W.; et al. YAP manipulates proliferation via PTEN/AKT/mTOR-mediated autophagy in lung adenocarcinomas. Cancer Cell Int. 2021, 21, 30. [Google Scholar] [CrossRef]
- Rizzuti, I.F.; Mascheroni, P.; Arcucci, S.; Ben-Meriem, Z.; Prunet, A.; Barentin, C.; Riviere, C.; Delanoe-Ayari, H.; Hatzikirou, H.; Guillermet-Guibert, J.; et al. Mechanical Control of Cell Proliferation Increases Resistance to Chemotherapeutic Agents. Phys. Rev. Lett. 2020, 125, 128103. [Google Scholar] [CrossRef]
- Muranen, T.; Selfors, L.M.; Worster, D.T.; Iwanicki, M.P.; Song, L.; Morales, F.C.; Gao, S.; Mills, G.B.; Brugge, J.S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012, 21, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Klapproth, E.; Dickreuter, E.; Zakrzewski, F.; Seifert, M.; Petzold, A.; Dahl, A.; Schrock, E.; Klink, B.; Cordes, N. Whole exome sequencing identifies mTOR and KEAP1 as potential targets for radiosensitization of HNSCC cells refractory to EGFR and beta1 integrin inhibition. Oncotarget 2018, 9, 18099–18114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, A.K.; Viloria-Petit, A.M. Targeting Mechanotransduction in Osteosarcoma: A Comparative Oncology Perspective. Int. J. Mol. Sci. 2020, 21, 7595. [Google Scholar] [CrossRef] [PubMed]
- Molina, E.R.; Chim, L.K.; Salazar, M.C.; Mehta, S.M.; Menegaz, B.A.; Lamhamedi-Cherradi, S.E.; Satish, T.; Mohiuddin, S.; McCall, D.; Zaske, A.M.; et al. Mechanically tunable coaxial electrospun models of YAP/TAZ mechanoresponse and IGF-1R activation in osteosarcoma. Acta Biomater. 2019, 100, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, D.; Zhang, Y.; Wen, Y.; Wang, Y. mTOR signal transduction pathways contribute to TN-C FNIII A1 overexpression by mechanical stress in osteosarcoma cells. Mol. Cells 2014, 37, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Estrada, M.V.; Bianchini, G.; Moore, P.D.; Zhao, J.; Cheng, F.; Koch, J.P.; Gianni, L.; Tyson, D.R.; Sanchez, V.; et al. Extracellular Matrix/Integrin Signaling Promotes Resistance to Combined Inhibition of HER2 and PI3K in HER2(+) Breast Cancer. Cancer Res. 2017, 77, 3280–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, R.; Kawahara, A.; Watari, K.; Murakami, Y.; Sonoda, K.; Maeda, M.; Fujita, H.; Kage, M.; Uramoto, H.; Costa, C.; et al. Erlotinib resistance in lung cancer cells mediated by integrin beta1/Src/Akt-driven bypass signaling. Cancer Res. 2013, 73, 6243–6253. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, S.; Higuchi, Y.; Ishibashi, M.; Hashimoto, H.; Yasunaga, M.; Matsumura, Y.; Tsuchihara, K.; Tsuboi, M.; Goto, K.; Ochiai, A.; et al. Collagen type I induces EGFR-TKI resistance in EGFR-mutated cancer cells by mTOR activation through Akt-independent pathway. Cancer Sci. 2018, 109, 2063–2073. [Google Scholar] [CrossRef]
- Petras, M.; Lajtos, T.; Friedlander, E.; Klekner, A.; Pintye, E.; Feuerstein, B.G.; Szollosi, J.; Vereb, G. Molecular interactions of ErbB1 (EGFR) and integrin-beta1 in astrocytoma frozen sections predict clinical outcome and correlate with Akt-mediated in vitro radioresistance. Neuro Oncol. 2013, 15, 1027–1040. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.F.; Li, K.S.; Shen, Y.H.; Gao, P.T.; Dong, Z.R.; Cai, J.B.; Zhang, C.; Huang, X.Y.; Tian, M.X.; Hu, Z.Q.; et al. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016, 7, e2201. [Google Scholar] [CrossRef]
- Yu, C.; Zhang, M.; Song, J.; Zheng, X.; Xu, G.; Bao, Y.; Lan, J.; Luo, D.; Hu, J.; Li, J.J.; et al. Integrin-Src-YAP1 signaling mediates the melanoma acquired resistance to MAPK and PI3K/mTOR dual targeted therapy. Mol. Biomed. 2020, 1, 12. [Google Scholar] [CrossRef]
- Lesovaya, E.A.; Savinkova, A.V.; Morozova, O.V.; Lylova, E.S.; Zhidkova, E.M.; Kulikov, E.P.; Kirsanov, K.I.; Klopot, A.; Baida, G.; Yakubovskaya, M.G.; et al. A novel approach to safer glucocorticoid receptor–targeted anti-lymphoma therapy via REDD1 (Regulated in Development and DNA Damage 1) inhibition. Mol. Cancer Ther. 2020, 19, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Moutsatsou, P.; Papavassiliou, A.G. The glucocorticoid receptor signalling in breast cancer. J. Cell. Mol. Med. 2008, 12, 145–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargalionis, A.N.; Papavassiliou, K.A.; Basdra, E.K.; Papavassiliou, A.G. mTOR Signaling Components in Tumor Mechanobiology. Int. J. Mol. Sci. 2022, 23, 1825. https://doi.org/10.3390/ijms23031825
Gargalionis AN, Papavassiliou KA, Basdra EK, Papavassiliou AG. mTOR Signaling Components in Tumor Mechanobiology. International Journal of Molecular Sciences. 2022; 23(3):1825. https://doi.org/10.3390/ijms23031825
Chicago/Turabian StyleGargalionis, Antonios N., Kostas A. Papavassiliou, Efthimia K. Basdra, and Athanasios G. Papavassiliou. 2022. "mTOR Signaling Components in Tumor Mechanobiology" International Journal of Molecular Sciences 23, no. 3: 1825. https://doi.org/10.3390/ijms23031825
APA StyleGargalionis, A. N., Papavassiliou, K. A., Basdra, E. K., & Papavassiliou, A. G. (2022). mTOR Signaling Components in Tumor Mechanobiology. International Journal of Molecular Sciences, 23(3), 1825. https://doi.org/10.3390/ijms23031825