
Hidden Information Revealed Using the Orthogonal System of Nucleic Acids


Abstract
1. Introduction
2. Results and Discussion
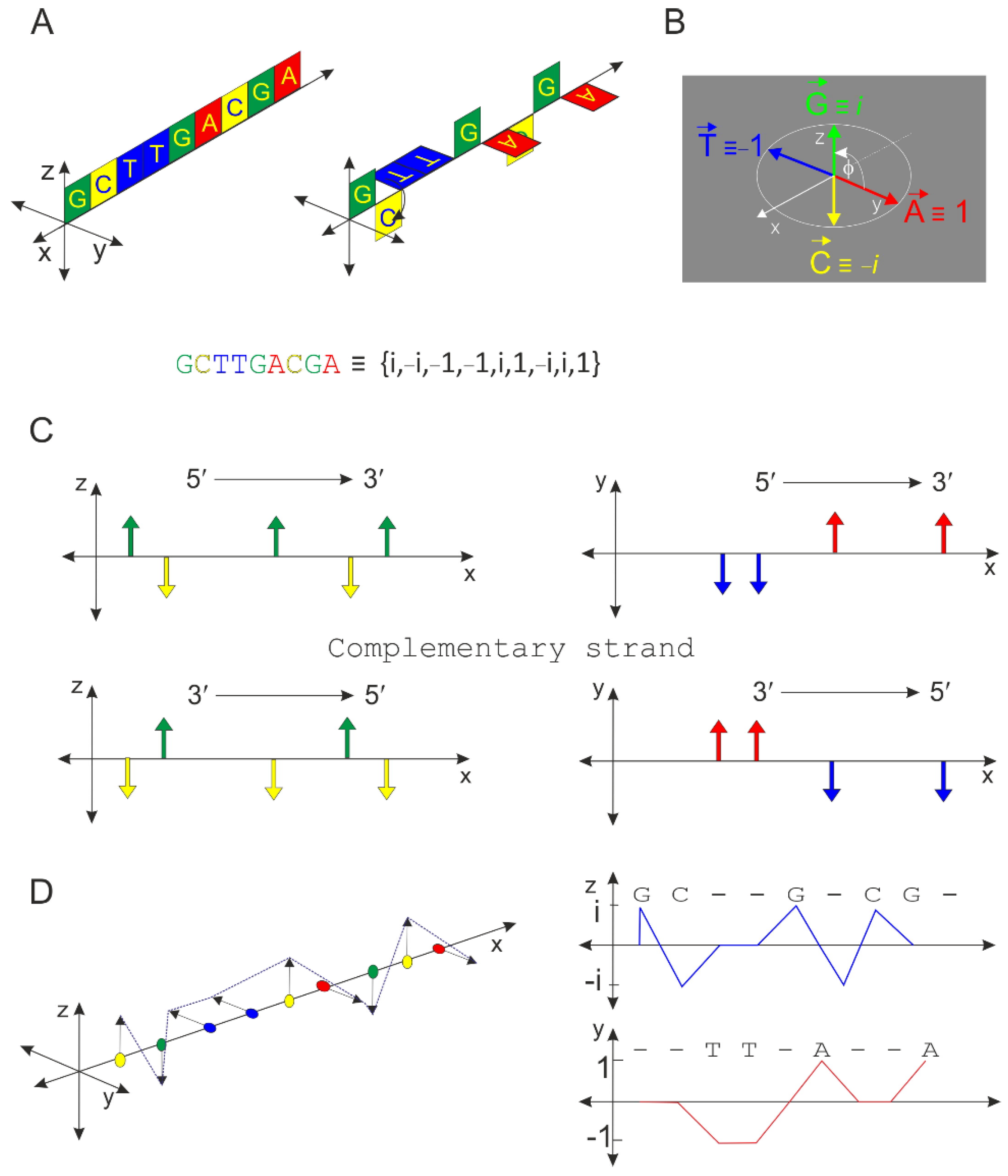
2.1. Principle of the Orthogonal Algorithm
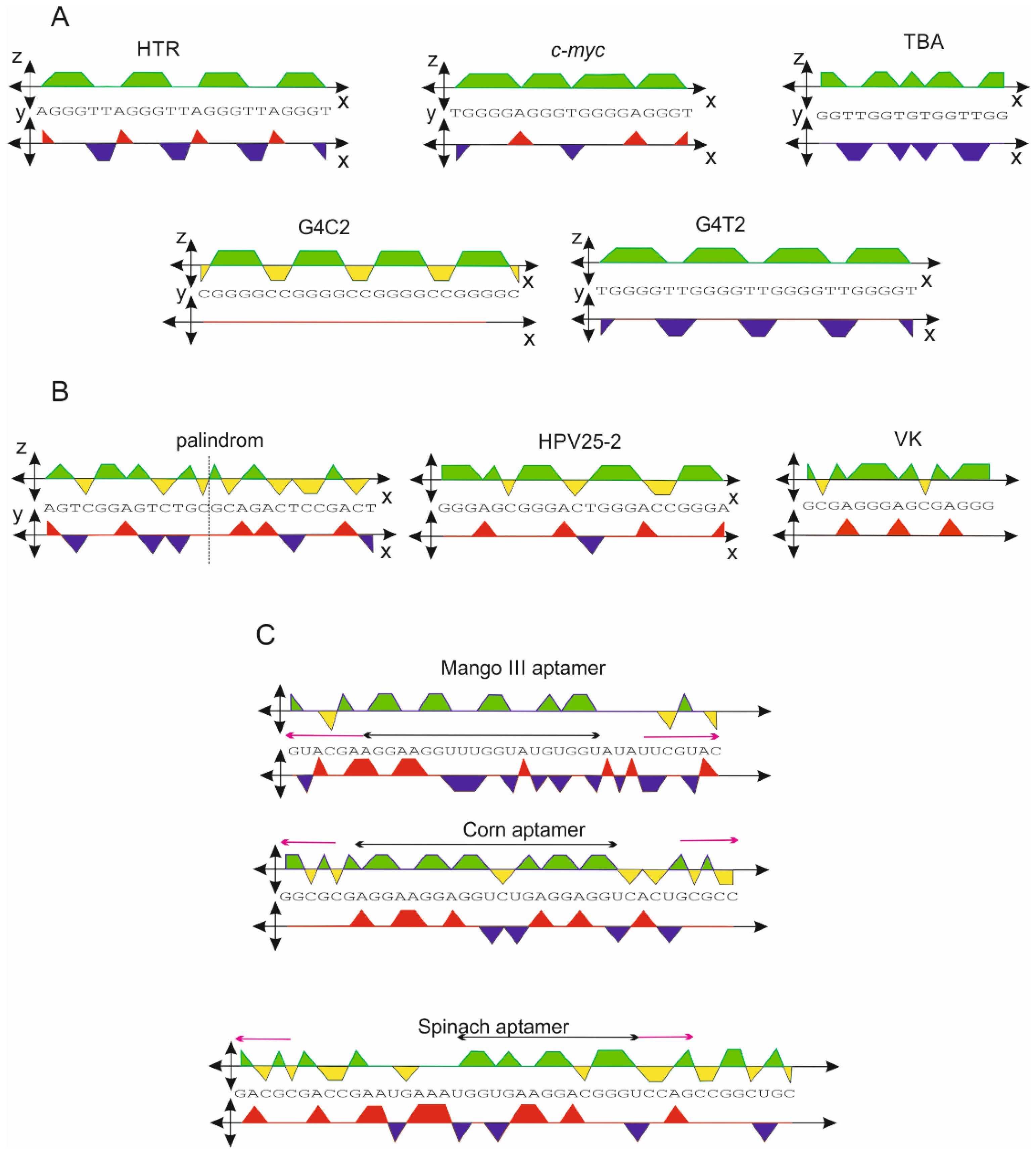
2.2. G-Quadruplex Forming Sequences and Non-Canonical Motifs
2.3. Genetic Code in Orthogonal Presentation
3. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, S.; Sugimoto, N. Stability prediction of canonical and non-canonical structures of nucleic acids in various molecular environments and cells. Chem. Soc. Rev. 2020, 49, 8439–8468. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Sugimoto, N. Roles of non-canonical structures of nucleic acids in cancer and neurodegenerative diseases. Nucleic Acids Res. 2021, 49, 7839–7855. [Google Scholar]
- Weiner, A.M. SINEs and LINEs: The art of biting the hand that feeds you. Curr. Opin. Cell Biol. 2002, 14, 343–350. [Google Scholar] [CrossRef]
- Siggers, T.; Gordân, R. Protein-DNA binding: Complexities and multi-protein codes. Nucleic Acids Res. 2014, 42, 2099–2111. [Google Scholar] [CrossRef]
- Balasubramaniyam, T.; Oh, K.I.; Jin, H.S.; Ahn, H.B.; Kim, B.S.; Lee, J.H. Non-Canonical Helical Structure of Nucleic Acids Containing Base-Modified Nucleotides. Int. J. Mol. Sci. 2021, 22, 9552. [Google Scholar] [CrossRef]
- Roy, A.; Raychaudhury, C.; Nandy, A. Novel techniques of graphical representation and analysis of DNA sequences—A review. J. Biosci. 1998, 23, 55–71. [Google Scholar] [CrossRef]
- Cristea, P.D. Conversion of nucleotides sequences into genomic signals. J. Cell. Mol. Med. 2002, 6, 279–303. [Google Scholar] [CrossRef]
- Cristea, P.D. Representation and analysis of DNA sequences. In Genomic Signal Processing and Statistics: EURASIP Book Series in Signal Processing and Communications; Dougherty, E.R., Ed.; Hindawi Pub. Corp.: New York, NY, USA, 2005; Volume 2, pp. 15–66. [Google Scholar]
- Mendizabal-Ruiz, G.; Román-Godínez, I.; Torres-Ramos, S.; Salido-Ruiz, R.A.; Morales, J.A. On DNA numerical representations for genomic similarity computation. PLoS ONE 2017, 12, e0173288. [Google Scholar] [CrossRef]
- Voss, R.F. Evolution of long-range fractal correlations and 1/f noise in DNA base sequences. Phys. Rev. Lett. 1992, 68, 3805–3808. [Google Scholar] [CrossRef]
- Kwan, H.K.; Arniker, S.B. Numerical representation of DNA sequences. In Proceedings of the 2009 IEEE International Conference on Electro/Information Technology, Windsor, ON, Canada, 7–9 June 2009; pp. 307–310. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Brázda, V.; Kolomazník, J.; Lýsek, J.; Hároníková, L.; Coufal, J.; Št’astný, J. Palindrome analyser—A new web-based server for predicting and evaluating inverted repeats in nucleotide sequences. Biochem. Biophys. Res. Commun. 2016, 478, 1739–1745. [Google Scholar] [CrossRef] [PubMed]
- Brazda, V.; Kolomaznik, J.; Mergny, J.L.; Stastny, J. G4Killer web application: A tool to design G-quadruplex mutations. Bioinformatics 2020, 36, 3246–3247. [Google Scholar] [CrossRef]
- Brázda, V.; Kolomazník, J.; Lýsek, J.; Bartas, M.; Fojta, M.; Šťastný, J.; Mergny, J.L. G4Hunter web application: A web server for G-quadruplex prediction. Bioinformatics 2019, 35, 3493–3495. [Google Scholar] [CrossRef] [PubMed]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.I.; Kim, J.; Park, C.J.; Lee, J.H. Dynamics Studies of DNA with Non-canonical Structure Using NMR Spectroscopy. Int. J. Mol. Sci. 2020, 21, 2673. [Google Scholar] [CrossRef]
- Bedrat, A.; Lacroix, L.; Mergny, J.L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016, 44, 1746–1759. [Google Scholar] [CrossRef]
- Macaya, R.F.; Schultze, P.; Smith, F.W.; Roe, J.A.; Feigon, J. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. USA 1993, 90, 3745–3749. [Google Scholar] [CrossRef]
- Demkovičová, E.; Bauer, Ľ.; Krafčíková, P.; Tlučková, K.; Tóthova, P.; Halaganová, A.; Valušová, E.; Víglaský, V. Telomeric G-Quadruplexes: From Human to Tetrahymena Repeats. J. Nucleic Acids 2017, 2017, 9170371. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef]
- Wang, Y.; Patel, D.J. Solution structure of the Tetrahymena telomeric repeat d(T2G4)4 G-tetraplex. Structure 1994, 2, 1141–1156. [Google Scholar] [CrossRef]
- Ambrus, A.; Chen, D.; Dai, J.; Bialis, T.; Jones, R.A.; Yang, D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006, 34, 2723–2735. [Google Scholar] [CrossRef]
- Mathad, R.I.; Hatzakis, E.; Dai, J.; Yang, D. c-MYC promoter G-quadruplex formed at the 5′-end of NHE III1 element: Insights into biological relevance and parallel-stranded G-quadruplex stability. Nucleic Acids Res. 2011, 39, 9023–9033. [Google Scholar] [CrossRef] [PubMed]
- Brcic, J.; Plavec, J. NMR structure of a G-quadruplex formed by four d(G4C2) repeats: Insights into structural polymorphism. Nucleic Acids Res. 2018, 46, 11605–11617. [Google Scholar] [PubMed]
- Tlučková, K.; Marušič, M.; Tóthová, P.; Bauer, L.; Šket, P.; Plavec, J.; Viglasky, V. Human papillomavirus G-quadruplexes. Biochemistry 2013, 52, 7207–7216. [Google Scholar] [CrossRef]
- Kocman, V.; Plavec, J. A tetrahelical DNA fold adopted by tandem repeats of alternating GGG and GCG tracts. Nat. Commun. 2014, 5, 5831. [Google Scholar] [CrossRef][Green Version]
- Trachman, R.J., 3rd; Autour, A.; Jeng, S.C.Y.; Abdolahzadeh, A.; Andreoni, A.; Cojocaru, R.; Garipov, R.; Dolgosheina, E.V.; Knutson, J.R.; Ryckelynck, M.; et al. Structure and functional reselection of the Mango-III fluorogenic RNA aptamer. Nat. Chem. Biol. 2019, 15, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Sjekloća, L.; Ferré-D’Amaré, A.R. Binding between G Quadruplexes at the Homodimer Interface of the Corn RNA Aptamer Strongly Activates Thioflavin T Fluorescence. Cell Chem. Biol. 2019, 26, 1159–1168.e4. [Google Scholar] [CrossRef]
- Anastassiou, D. Frequency-domain analysis of biomolecular sequences. Bioinformatics 2000, 16, 1073–1081. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | xz-Projection Area, r = 3 | xy-Projection Area, r = 3 | G4hunter Score | xz-Projection Area, r = 3, ψ = 15° # | xz-Projection Area, r = 3, ψ = 30° # | G4 Formation |
|---|---|---|---|---|---|---|
| HTR | 1.56 | −0.39 | 1.57 | 1.66 | 1.75 | yes |
| c-myc | 2.22 | 0 | 2.11 | 2.22 | 2.22 | yes |
| TBA | 1.59 | −1.2 | 2.20 | 1.90 | 2.19 | yes |
| G4C2 | 1.13 | 0 | 2.08 | 1.13 | 1.13 | yes |
| G4T2 | 2.00 | −0.87 | 2.67 | 2.23 | 2.43 | yes |
| HPV25-2 | 1.16 | +0.34 | 1.68 | 1.07 | 0.99 | no |
| VK | 1.1 | +0.60 | 1.4 | 0.94 | 0.80 | no |
| palindrom * | 0 | 0 | 0 | 0 | 0 | no |
| cs-Mango III | 1.35 | −0.41 | 0.85 | 1.46 | 1.55 | yes |
| cs-Corn | 1.41 | +0.18 | 0.94 | 1.36 | 1.32 | yes |
| Ala/A | GCU {i, −i, −1} | GCC {i, −i, −i} | GCA {i, −i, 1} | GCG {i, −i, i} | ||
| Arg/R | CGU {−i, i, −1} | CGC {i, i, −i} | CGA {−i, i, 1} | CGG {−i, i, i} | AGA {1, i, 1} | AGG {1, i, i} |
| Asn/N | AAU {1, 1, −1} | AAC {1, 1, −i} | ||||
| Asp/D | GAU {i, 1, −1} | GAC {i, 1, −i} | ||||
| Cys/C | UGU {−1, i, −1} | UGC {−1, i, −i} | ||||
| Gln/Q | CAA {−i, 1, 1} | CAG {−i, 1, i} | ||||
| Glu/E | GAA {i, 1, 1} | GAG {i, 1, i} | ||||
| Gly/G | GGU {i, i, −1} | GGC {i, i, −i} | GGA {i, i, 1} | GGG {i, i, i} | ||
| His/H | CAU {−i, 1, −1} | CAC {−i, 1, −i} | ||||
| Ile/I | AUU {1, −1, −1} | AUC {1, −1, -i} | AUA {1, −1, 1} | |||
| Leu/L | UUA {−1, −1, 1} | UUG {−1, −1, i} | CUU {−i, −1, −1} | CUC {−i, −1, −i} | CUA {−i, −1, 1} | CUG {−i, −1, i} |
| Lys/K | AAA {1, 1, 1} | AAG {1, 1, i} | ||||
| Met/M | AUG {1, −1, i} | |||||
| Phe/F | UUU {−1, −1, −1} | UUC {−1, −1, −i} | ||||
| Pro/P | CCU {−i, −i, −1} | CCC {−i, −i, −i} | CCA {−i, −i, 1} | CCG {−i, −i, i} | ||
| Ser/S | UCU {−1, −i, −1} | UCC {−1, −i, −i} | UCA {−1, −i, 1} | UCG {−1, −i, i} | AGU {1, i, −1} | AGC {1, i, −i} |
| Thr/T | ACU {1, −i, −1} | ACC {1, −i, −i} | ACA {1, −i, 1} | ACG {1, −i, i} | ||
| Trp/W | UGG {−1, i, i} | |||||
| Tyr/Y | UAU {−1, 1, −1} | UAC {−1, 1, −i} | ||||
| Val/V | GUU {i, −1, −1} | GUC {i, −1, −i} | GUA {i, −1, 1} | GUG {i, −1, i} | ||
| STOP | UAG {−1, 1, i} | UGA {−1, i, 1} | UAA {−1, 1, 1} |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Víglaský, V. Hidden Information Revealed Using the Orthogonal System of Nucleic Acids. Int. J. Mol. Sci. 2022, 23, 1804. https://doi.org/10.3390/ijms23031804
Víglaský V. Hidden Information Revealed Using the Orthogonal System of Nucleic Acids. International Journal of Molecular Sciences. 2022; 23(3):1804. https://doi.org/10.3390/ijms23031804
Chicago/Turabian StyleVíglaský, Viktor. 2022. "Hidden Information Revealed Using the Orthogonal System of Nucleic Acids" International Journal of Molecular Sciences 23, no. 3: 1804. https://doi.org/10.3390/ijms23031804
APA StyleVíglaský, V. (2022). Hidden Information Revealed Using the Orthogonal System of Nucleic Acids. International Journal of Molecular Sciences, 23(3), 1804. https://doi.org/10.3390/ijms23031804