
Intramolecular Charge Transfer of Curcumin and Solvation Dynamics of DMSO Probed by Time-Resolved Raman Spectroscopy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
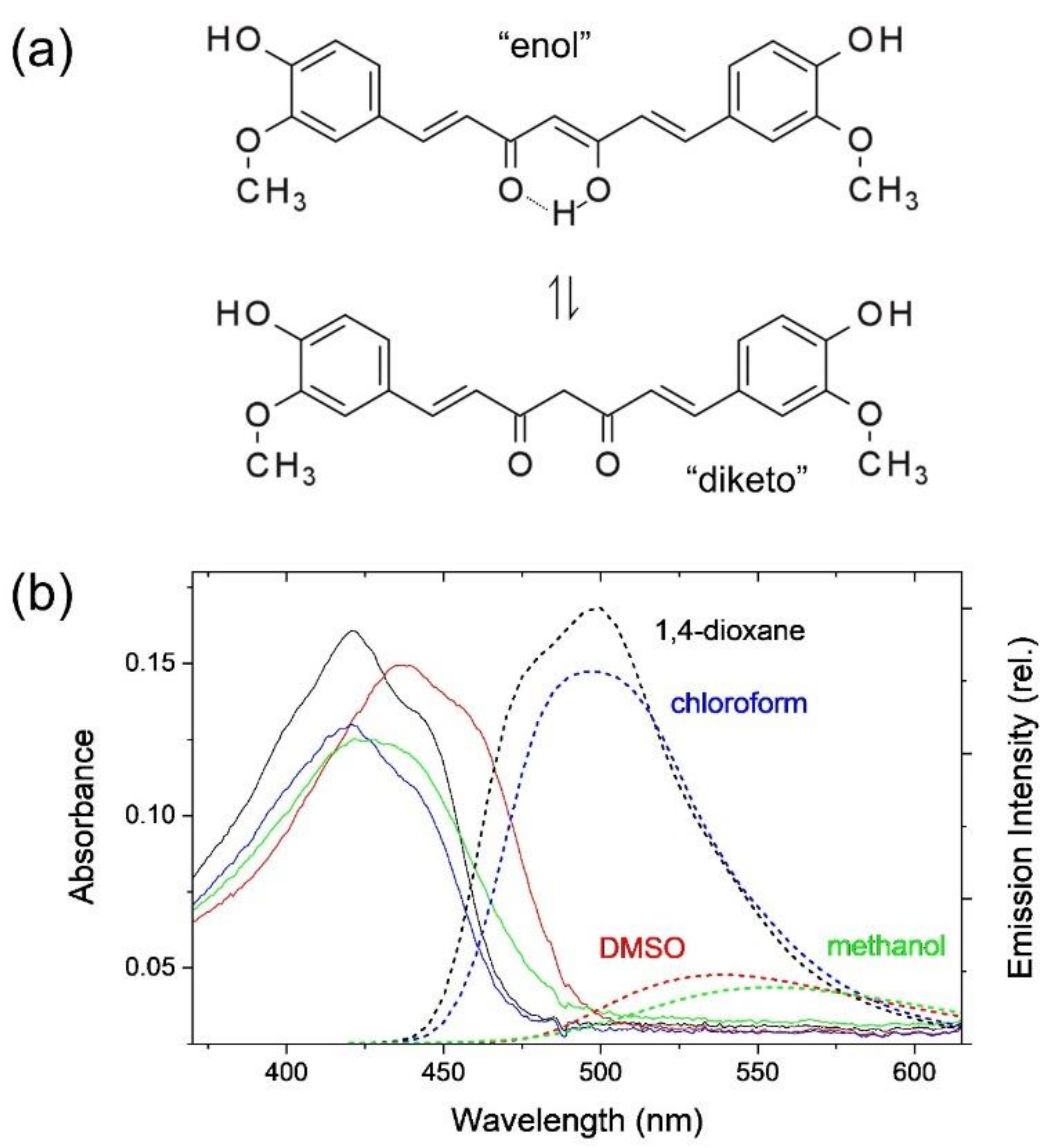
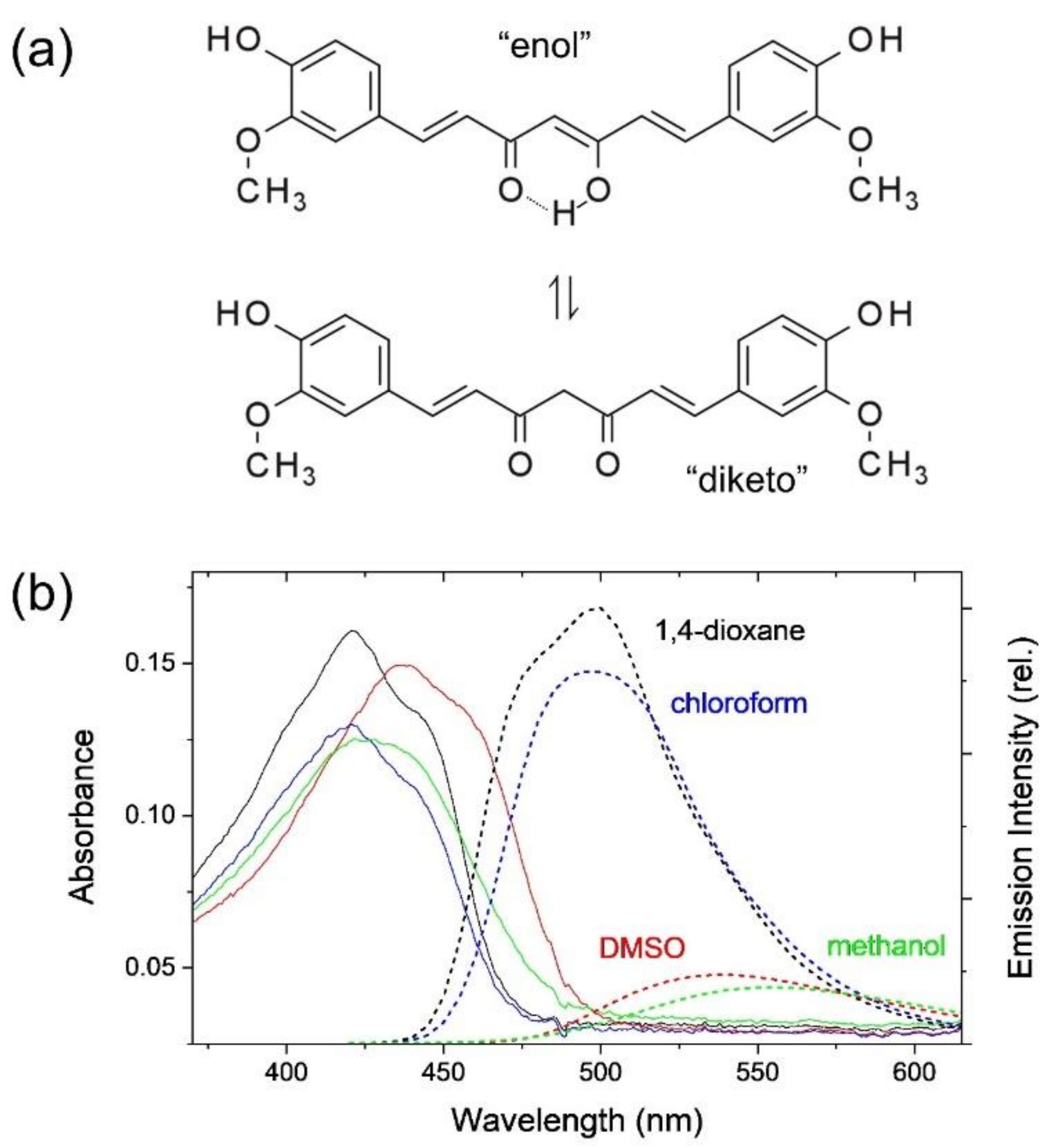
2.1. Intramolecular Charge Transfer of Curcumin
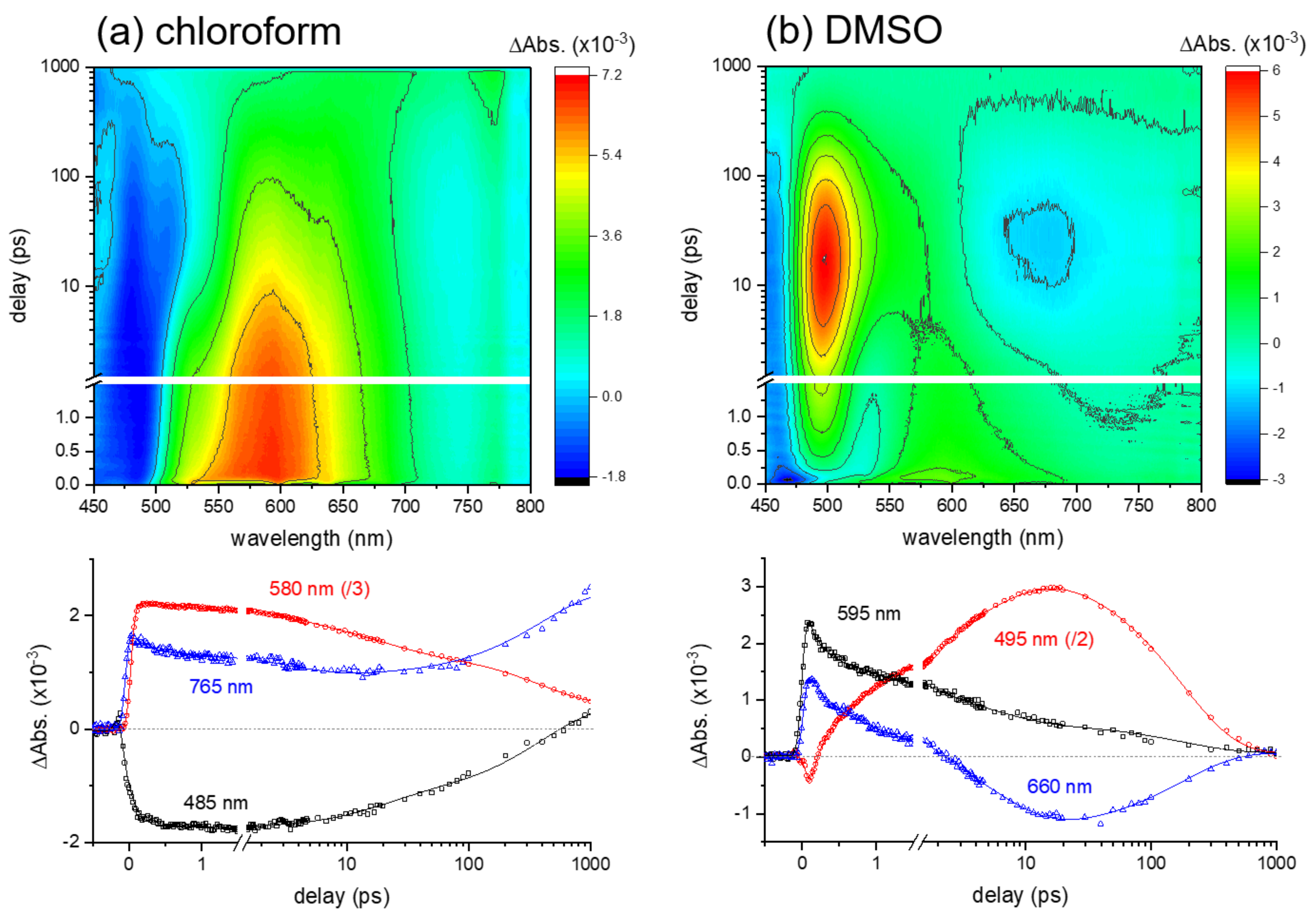
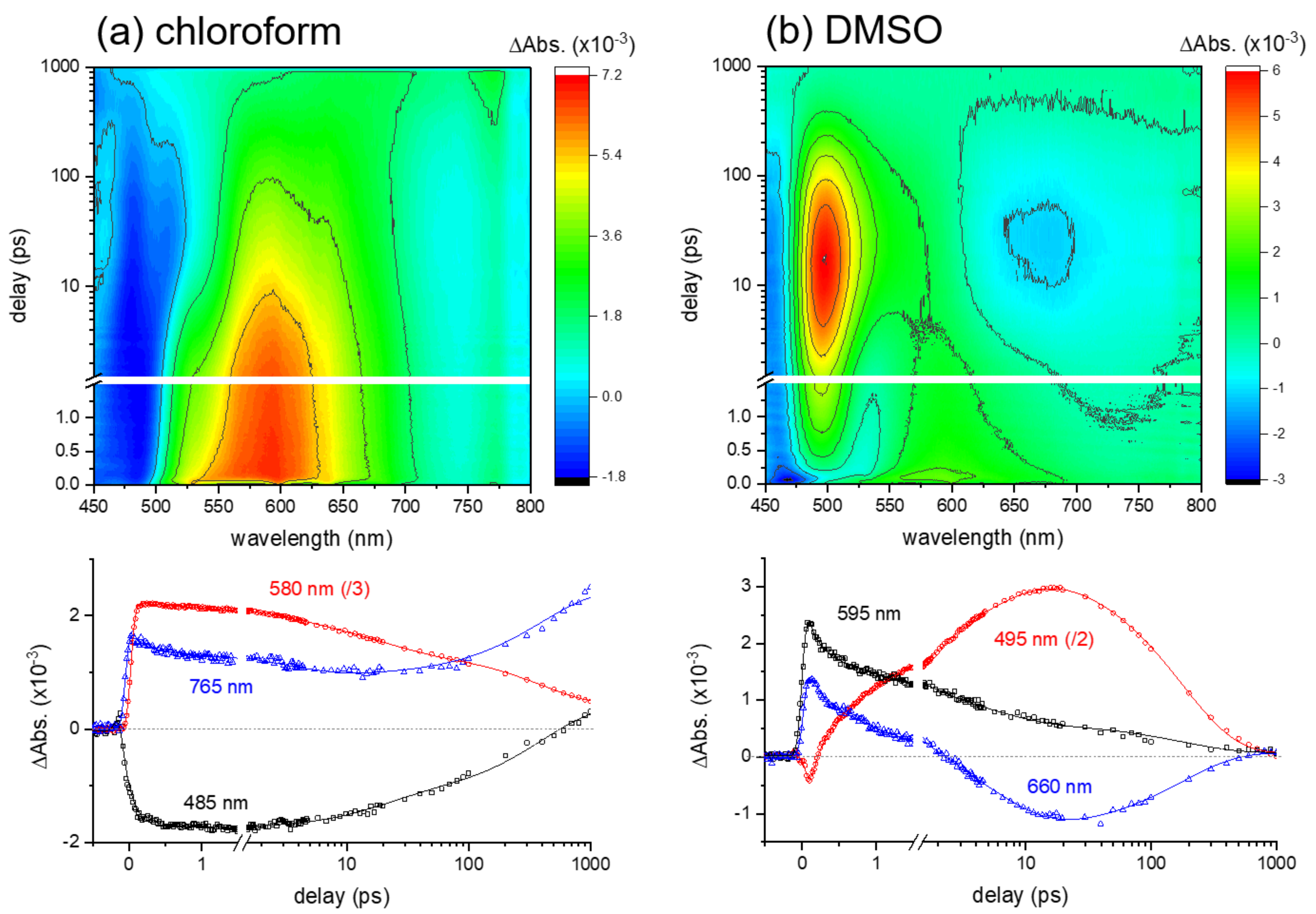
2.2. Excited-State Dynamics of Curcumin by Transient Absorption Spectroscopy
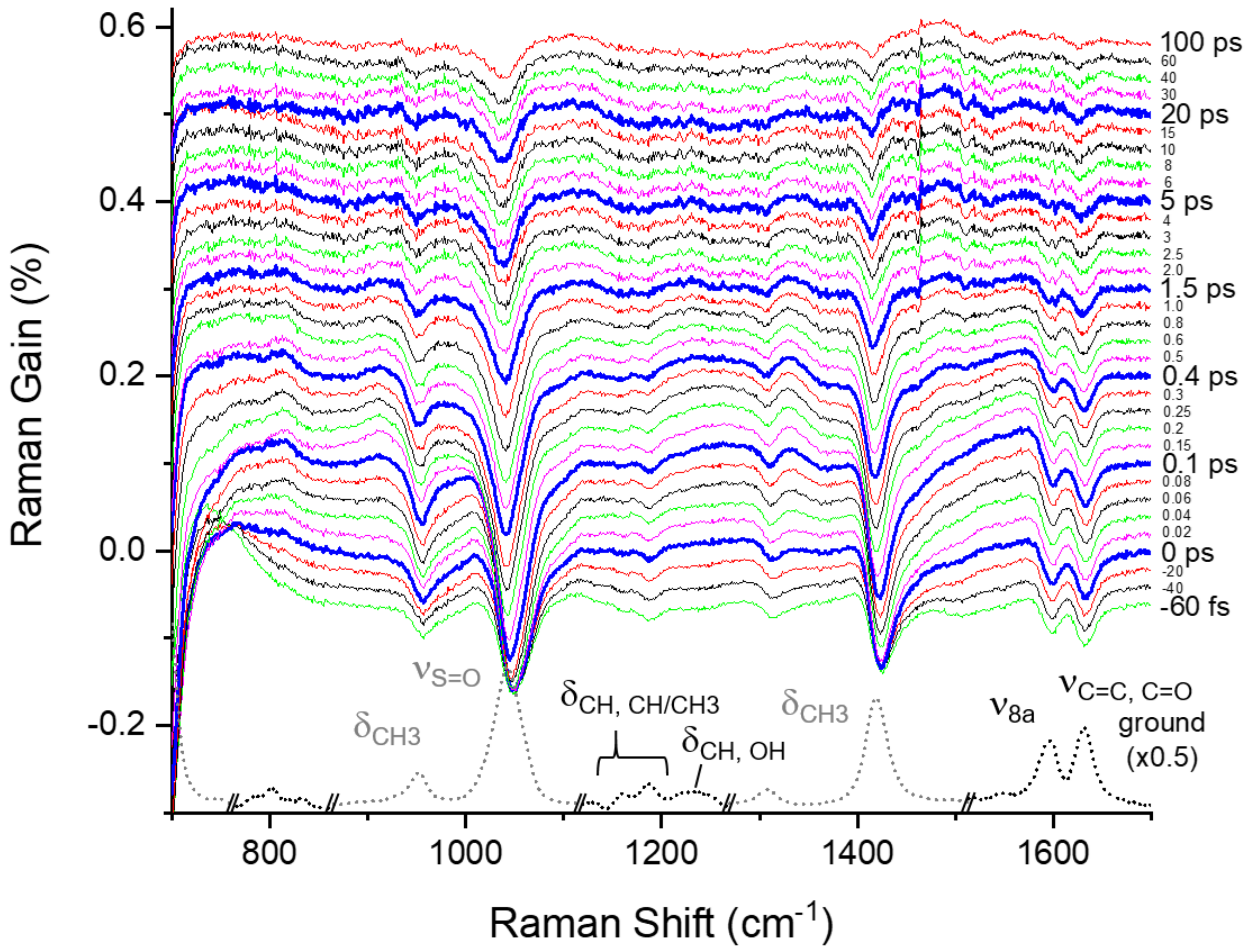
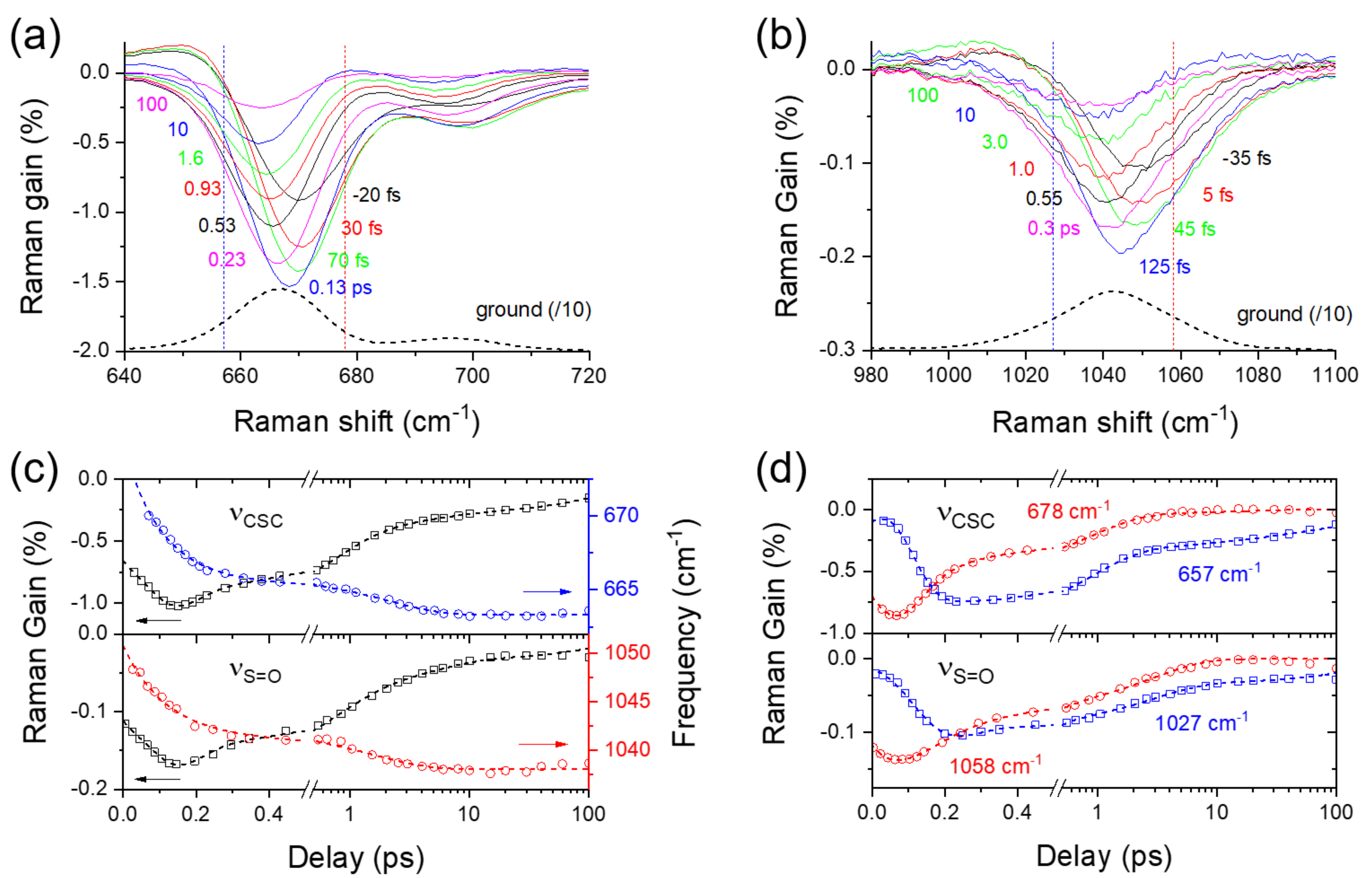
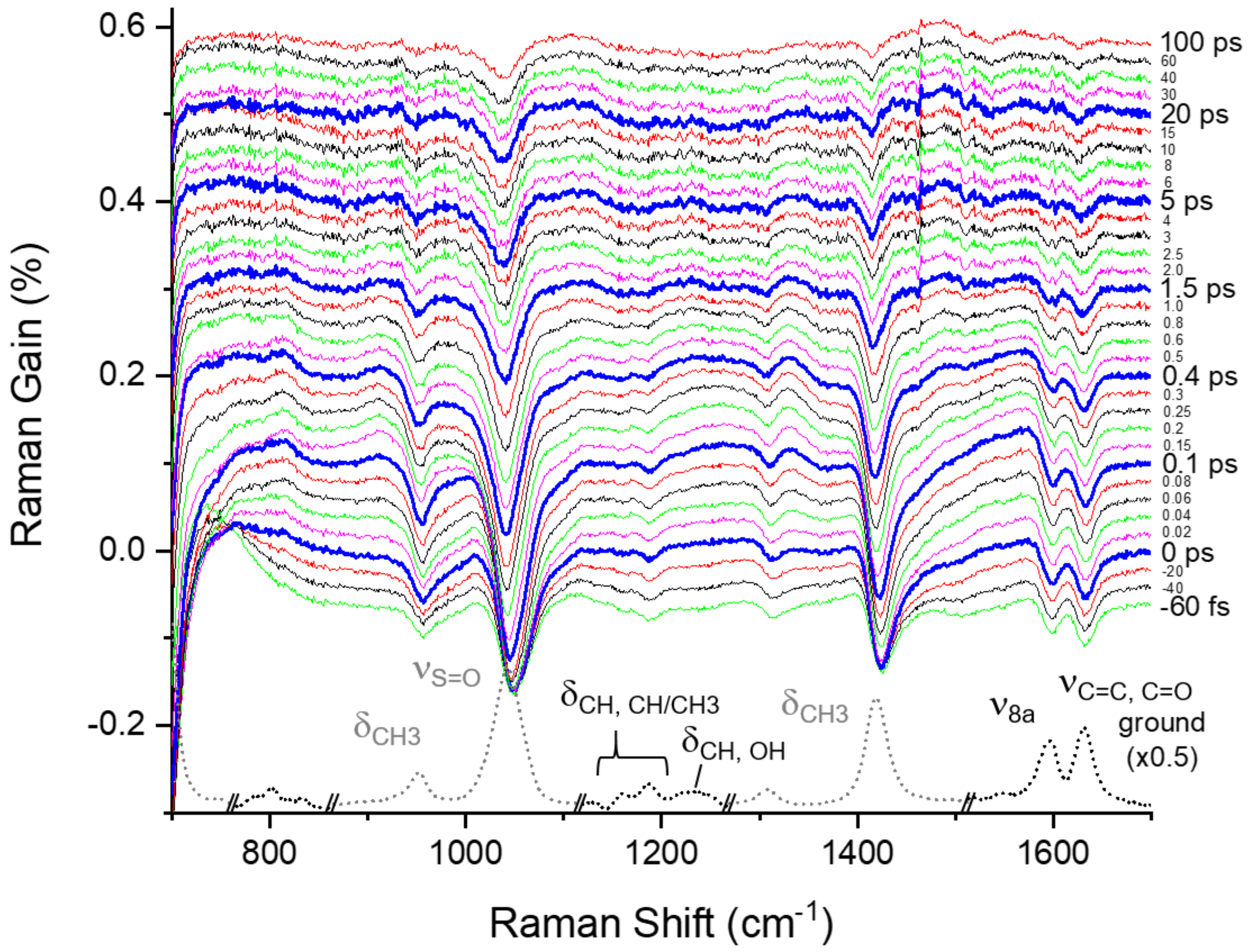
2.3. Excited-State Dynamics of Curcumin by FSRS
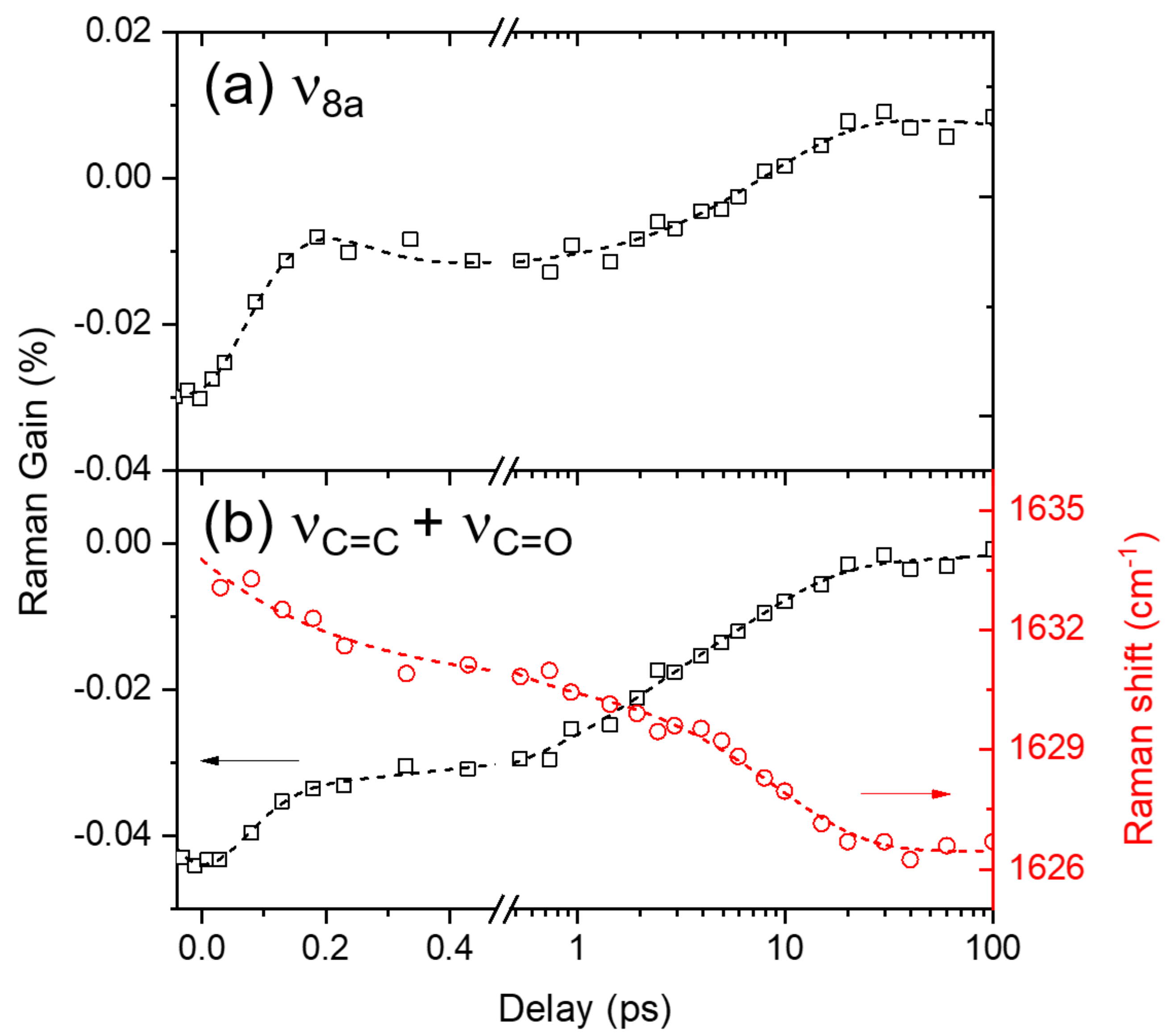
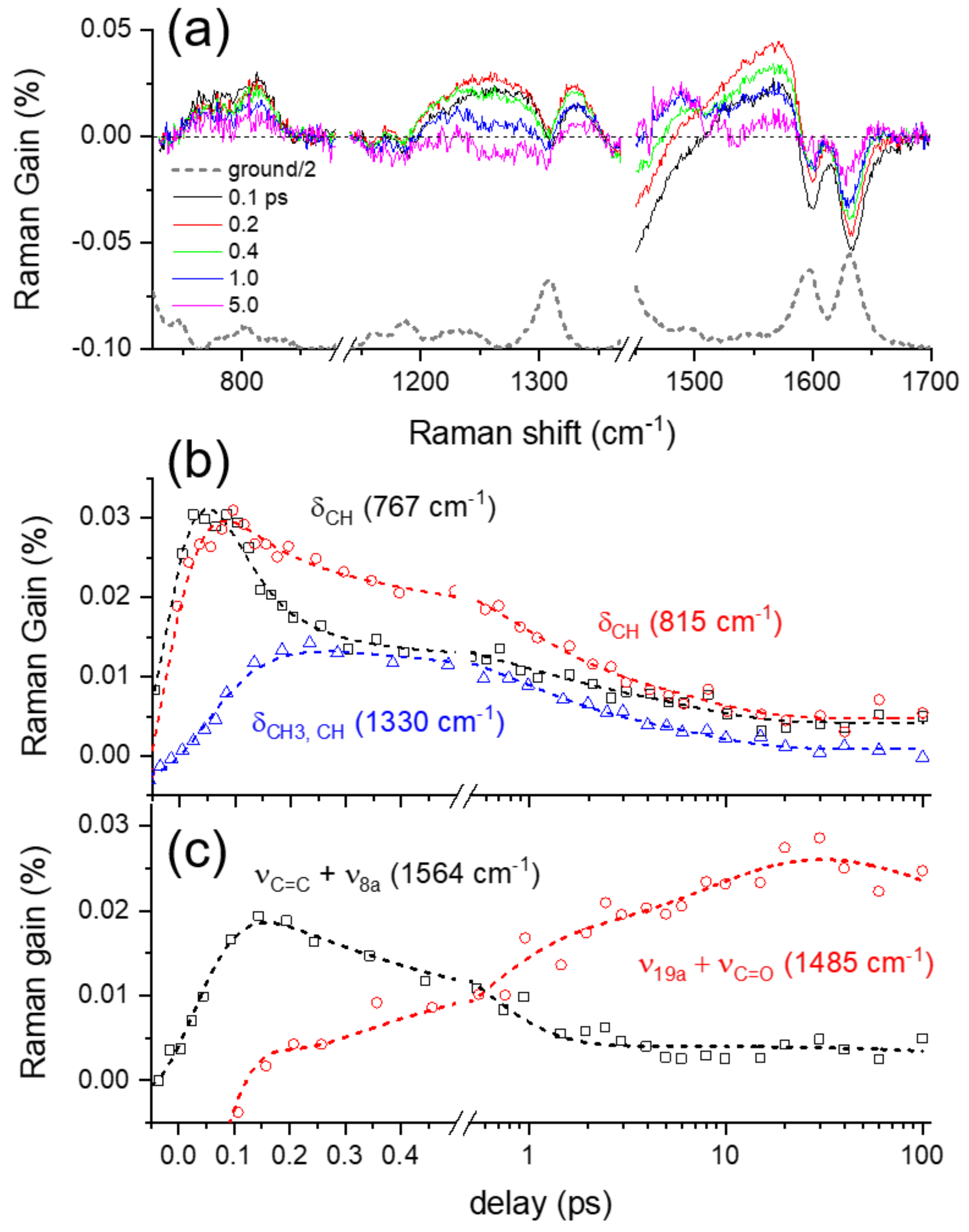
2.4. Ground-State Vibrational Modes of Curcumin
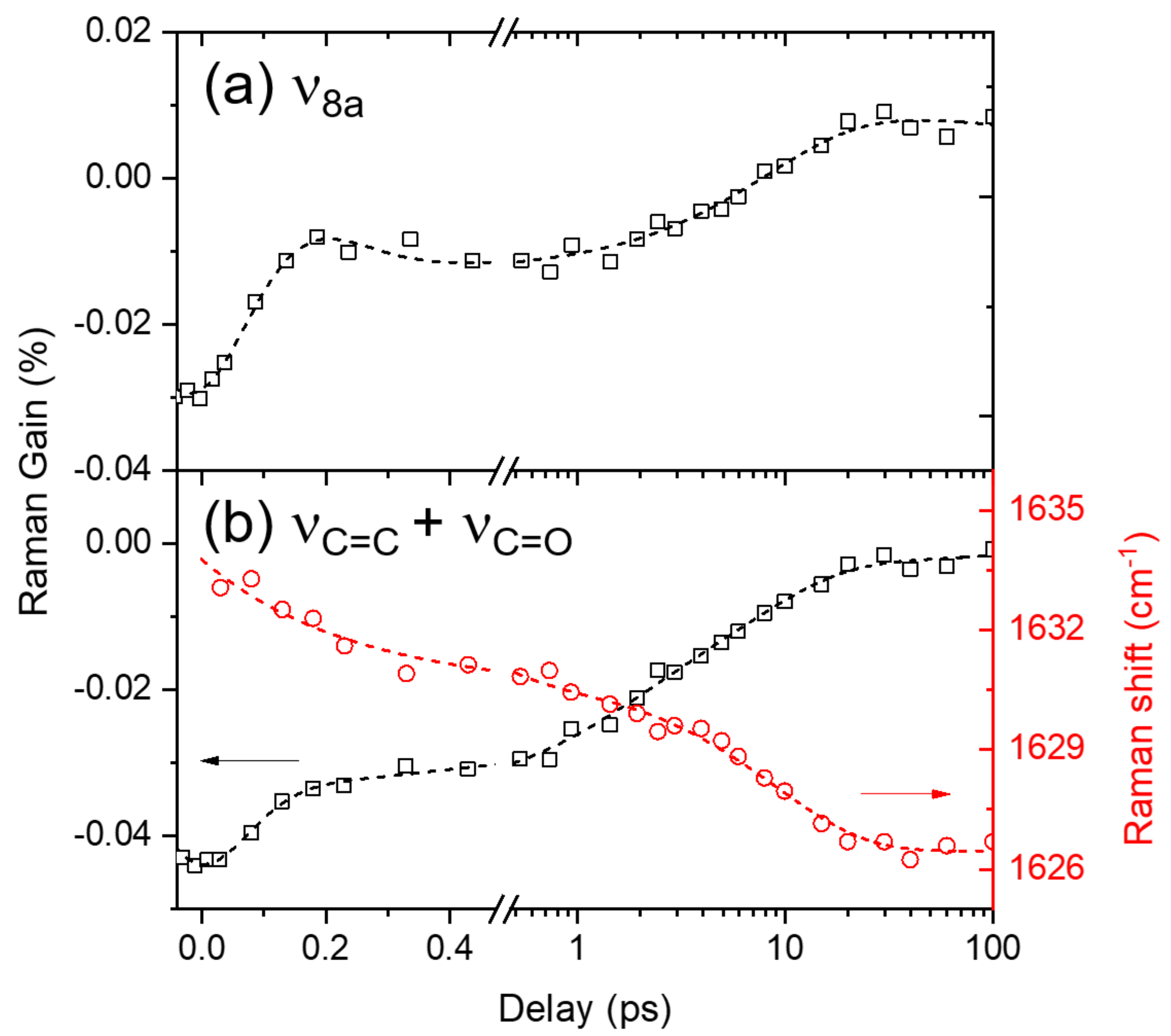
2.5. Solvation Dynamics of DMSO
3. Materials and Methods
3.1. Chemicals
3.2. Femtosecond Transient Absorption Setup
3.3. Femtosecond Stimulated Raman Spectroscopy (FSRS) Setup
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lantz, R.C.; Chen, G.J.; Solyom, A.M.; Jolad, S.D.; Timmermann, B.N. The effect of turmeric extracts on inflammatory mediator production. Phytomedicine 2005, 12, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.P.; Sudheer, A.R. Antioxidant and anti-inflammatory properties of curcumin. Adv. Exp. Med. Biol. 2007, 595, 105–125. [Google Scholar] [PubMed]
- Giordano, A.; Tommonaro, G. Curcumin and Cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Aggarwal, B.B. Turmeric, the Golden Spice. In Herbal Medicine: Biomolecular and Clinical Aspects, 2nd ed.; Benzie, I.F.F., Wachtel-Galoreds, S., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011. [Google Scholar]
- Ghosh, R.; Mondal, J.A.; Palit, D.K. Ultrafast dynamics of the excited states of curcumin in solution. J. Phys. Chem. B 2010, 114, 12129–12143. [Google Scholar] [CrossRef] [PubMed]
- Nardo, L.; Maspero, A.; Penoni, A.; Palmisano, G.; Ferrari, E.; Saladini, M. Excited state dynamics of bis-dehydroxycurcumin tert-butyl ester, a diketo-shifted derivative of the photosensitizer curcumin. PLoS ONE 2017, 12, e0175225. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, N.K.; Kishor, S.; Katyal, N.; Gogoi, P.; Narang, P.; Deep, S. Effect of pH and temperature on conformational equilibria and aggregation behaviour of curcumin in aqueous binary mixtures of ethanol. RSC Adv. 2016, 6, 103275–103288. [Google Scholar] [CrossRef]
- Shen, L.; Ji, H.-F. Theoretical study on physicochemical properties of curcumin. Spectrochim. Acta A 2007, 67, 619–623. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, H.-Y.; Ji, H.-F. Successful Application of TD-DFT in Transient Absorption Spectra Assignment. Org. Lett. 2005, 7, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Payton, F.; Sandusky, P.; Alworth, W.L. NMR Study of the Solution Structure of Curcumin. J. Nat. Prod. 2007, 70, 143–146. [Google Scholar] [CrossRef]
- Roughley, P.J.; Whiting, D.A. Experiments in the biosynthesis of curcumin. J. Chem. Soc. Perkin Trans. 1973, 2379–2388. [Google Scholar] [CrossRef]
- Khopde, S.M.; Priyadarsini, K.I.; Palit, D.K.; Mukherjee, T. Effect of solvent on the excited-state photophysical properties of curcumin. Photochem. Photobiol. 2000, 72, 625–631. [Google Scholar] [CrossRef]
- Nardo, L.; Paderno, R.; Andreoni, A.; Másson, M.; Haukvik, T.; Tønnesen, H.H. Role of H-bond formation in the photoreactivity of curcumin. Spectroscopy 2008, 22, 187–198. [Google Scholar] [CrossRef]
- Adhikary, R.; Mukherjee, P.; Kee, T.W.; Petrich, J.W. Excited-State Intramolecular Hydrogen Atom Transfer and Solvation Dynamics of the Medicinal Pigment Curcumin. J. Phys. Chem. B 2009, 113, 5255–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikary, R.; Carlson, P.J.; Kee, T.W.; Petrich, J.W. Excited-State Intramolecular Hydrogen Atom Transfer of Curcumin in Surfactant Micelles. J. Phys. Chem. B 2010, 114, 2997–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, R.K.; Das, K. Picosecond Spectral Relaxation of Curcumin Excited State in a Binary Solvent Mixture of Toluene and Methanol. J. Phys. Chem. B 2012, 116, 10357–10363. [Google Scholar] [CrossRef]
- Nardo, L.; Andreoni, A.; Bondani, M.; Másson, M.; Tønnesen, H.H. Studies on curcumin and curcuminoids. XXXIV. Photophysical properties of a symmetrical, non-substituted curcumin analogue. J. Photochem. Photobiol. B Biol. 2009, 97, 77–86. [Google Scholar] [CrossRef]
- Saini, R.K.; Das, K. Picosecond spectral relaxation of curcumin excited state in toluene–alcohol mixtures. J. Lumin. 2013, 144, 169–175. [Google Scholar] [CrossRef]
- Saini, R.K.; Das, K. Photophysics of Curcumin excited state in toluene-polar solvent mixtures: Role of H-bonding properties of the polar solvent. J. Lumin. 2014, 145, 832–837. [Google Scholar] [CrossRef]
- Erez, Y.; Presiado, I.; Gepshtein, R.; Huppert, D. Temperature dependence of the fluorescence properties of curcumin. J. Phys. Chem. A 2011, 115, 10962–10971. [Google Scholar] [CrossRef]
- Patra, D.; Barakat, C. Synchronous fluorescence spectroscopic study of solvatochromic curcumin dye. Spectrochim. Acta A 2011, 79, 1034–1041. [Google Scholar] [CrossRef]
- Toullec, J. Keto–Enol Equilibrium Constants. In The Chemistry of Enols; Rappoport, Z., Ed.; Wiley: Chichester, UK, 1990; pp. 323–398. [Google Scholar]
- Kukura, P.; McCamant, D.W.; Yoon, S.; Wandschneider, D.B.; Mathies, R.A. Chemistry: Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science 2005, 310, 1006–1009. [Google Scholar] [CrossRef] [Green Version]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Rhinehart, J.M.; Challa, J.R.; McCamant, D.W. Multimode charge-transfer dynamics of 4-(dimethylamino)benzonitrile probed with ultraviolet femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 10522–10534. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.R.; Conyard, J.; Heisler, I.A.; Jones, G.; Frost, J.; Browne, W.R.; Feringa, B.L.; Meech, S.R. Ultrafast Dynamics in Light-Driven Molecular Rotary Motors Probed by Femtosecond Stimulated Raman Spectroscopy. J. Am. Chem. Soc. 2017, 139, 7408–7414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, Y.; Tang, L.; Oscar, B.G.; Zhu, L.; Fang, C. Panoramic portrait of primary molecular events preceding excited state proton transfer in water. Chem. Sci. 2016, 7, 5484–5494. [Google Scholar] [CrossRef] [Green Version]
- Oscar, B.G.; Liu, W.; Zhao, Y.; Tang, L.; Wang, Y.; Campbell, R.E.; Fang, C. Excited-state structural dynamics of a dual-emission calmodulin-green fluorescent protein sensor for calcium ion imaging. Proc. Natl. Acad. Sci. USA 2014, 111, 10191. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Jen, M.; Pang, Y. Twisted intramolecular charge transfer state of a “push-pull” emitter. Int. J. Mol. Sci. 2020, 21, 7999. [Google Scholar] [CrossRef]
- Hashimoto, H.; Sugisaki, M.; Yoshizawa, M. Ultrafast time-resolved vibrational spectroscopies of carotenoids in photosynthesis. Biochim. Biophys. Acta 2015, 1847, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Weigel, A.; Ernsting, N. Excited stilbene: Intramolecular vibrational redistribution and solvation studied by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2010, 114, 7879–7893. [Google Scholar] [CrossRef]
- Batignani, G.; Pontecorvo, E.; Ferrante, C.; Aschi, M.; Elles, C.G.; Scopigno, T. Visualizing Excited-State Dynamics of a Diaryl Thiophene: Femtosecond Stimulated Raman Scattering as a Probe of Conjugated Molecules. J. Phys. Chem. Lett. 2016, 7, 2981–2988. [Google Scholar] [CrossRef]
- Jen, M.; Jeon, K.; Lee, S.; Hwang, S.; Chung, W.J.; Pang, Y. Ultrafast intramolecular proton transfer reactions and solvation dynamics of DMSO. Struct. Dyn. 2019, 6, 064901. [Google Scholar] [CrossRef] [Green Version]
- Jen, M.; Lee, S.; Jeon, K.; Hussain, S.; Pang, Y. Ultrafast Intramolecular Proton Transfer of Alizarin Investigated by Femtosecond Stimulated Raman Spectroscopy. J. Phys. Chem. B 2017, 121, 4129–4136. [Google Scholar] [CrossRef] [PubMed]
- Kamlet, M.J.; Abboud, J.L.; Taft, R.W. The solvatochromic comparison method. 6. The.pi.* scale of solvent polarities. J. Am. Chem. Soc. 1977, 99, 6027–6038. [Google Scholar] [CrossRef]
- Malik, P.K.; Tripathy, M.; Kajjam, A.B.; Patel, S. Preferential solvation of p-nitroaniline in a binary mixture of chloroform and hydrogen bond acceptor solvents: The role of specific solute–solvent hydrogen bonding. Phys. Chem. Chem. Phys. 2020, 22, 3545–3562. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Song, J.; Lee, D.; Pang, Y. Metal-enhanced fluorescence and excited state dynamics of carotenoids in thin polymer films. Sci. Rep. 2019, 9, 3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horng, M.L.; Gardecki, J.A.; Papazyan, A.; Maroncelli, M. Subpicosecond Measurements of Polar Solvation Dynamics: Coumarin 153 Revisited. J. Phys. Chem. 1995, 99, 17311–17337. [Google Scholar] [CrossRef]
- Prasad, R.; Gupta, K.M.; Poornachary, S.K.; Dalvi, S.V. Elucidating the Polymorphic Behavior of Curcumin during Antisolvent Crystallization: Insights from Raman Spectroscopy and Molecular Modeling. Cryst. Growth Des. 2020, 20, 6008–6023. [Google Scholar] [CrossRef]
- Van Nong, H.; Hung, L.X.; Thang, P.N.; Chinh, V.D.; Vu, L.V.; Dung, P.T.; Van Trung, T.; Nga, P.T. Fabrication and vibration characterization of curcumin extracted from turmeric (Curcuma longa) rhizomes of the northern Vietnam. SpringerPlus 2016, 5, 1147. [Google Scholar] [CrossRef] [Green Version]
- Mohan, P.R.K.; Sreelakshmi, G.; Muraleedharan, C.V.; Joseph, R. Water soluble complexes of curcumin with cyclodextrins: Characterization by FT-Raman spectroscopy. Vib. Spectrosc. 2012, 62, 77–84. [Google Scholar] [CrossRef]
- Kolev, T.M.; Velcheva, E.A.; Stamboliyska, B.A.; Spiteller, M. DFT and experimental studies of the structure and vibrational spectra of curcumin. Int. J. Quantum Chem. 2005, 102, 1069–1079. [Google Scholar] [CrossRef]
- Batignani, G.; Fumero, G.; Pontecorvo, E.; Ferrante, C.; Mukamel, S.; Scopigno, T. Genuine Dynamics vs Cross Phase Modulation Artifacts in Femtosecond Stimulated Raman Spectroscopy. ACS Photonics 2019, 6, 492–500. [Google Scholar] [CrossRef]
- Oh, K.-I.; Rajesh, K.; Stanton, J.F.; Baiz, C.R. Quantifying Hydrogen-Bond Populations in Dimethyl Sulfoxide/Water Mixtures. Angew. Chem. Int. Ed. 2017, 56, 11375–11379. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-I.; Baiz, C.R. Crowding Stabilizes DMSO–Water Hydrogen-Bonding Interactions. J. Phys. Chem. B 2018, 122, 5984–5990. [Google Scholar] [CrossRef] [PubMed]
- Luzar, A.; Chandler, D. Structure and hydrogen bond dynamics of water–dimethyl sulfoxide mixtures by computer simulations. J. Chem. Phys. 1993, 98, 8160–8173. [Google Scholar] [CrossRef] [Green Version]
- Chudoba, C.; Nibbering, E.T.J.; Elsaesser, T. Ultrafast Structural Response of Hydrogen Bonded Complexes to Electronic Excitation in the Liquid Phase. J. Phys. Chem. A 1999, 103, 5625–5628. [Google Scholar] [CrossRef]
- Nibbering, E.T.J.; Tschirschwitz, F.; Chudoba, C.; Elsaesser, T. Femtochemistry of Hydrogen Bonded Complexes after Electronic Excitation in the Liquid Phase: The Case of Coumarin 102. J. Phys. Chem. A 2000, 104, 4236–4246. [Google Scholar] [CrossRef]
- Martens, W.N.; Frost, R.L.; Kristof, J.; Theo Kloprogge, J. Raman spectroscopy of dimethyl sulphoxide and deuterated dimethyl sulphoxide at 298 and 77 K. J. Raman Spectrosc. 2002, 33, 84–91. [Google Scholar] [CrossRef]
- Singh, S.; Krueger, P.J. Raman spectral studies of aqueous solutions of non-electrolytes: Dimethylsulfoxide, acetone and acetonitrile. J. Raman Spectrosc. 1982, 13, 178–188. [Google Scholar] [CrossRef]
- Shikata, T.; Sugimoto, N. Dimeric Molecular Association of Dimethyl Sulfoxide in Solutions of Nonpolar Liquids. J. Phys. Chem. A 2012, 116, 990–999. [Google Scholar] [CrossRef]
- Wang, Z.; Pang, Y.; Dlott, D.D. The vibrational stokes shift of water (HOD in D2O). J. Chem. Phys. 2004, 120, 8345–8348. [Google Scholar] [CrossRef]
- Wang, Z.; Pakoulev, A.; Pang, Y.; Dlott, D.D. Vibrational Substructure in the OH Stretching Transition of Water and HOD. J. Phys. Chem. A 2004, 108, 9054–9063. [Google Scholar] [CrossRef]
- Jumper, C.C.; Arpin, P.C.; Turner, D.B.; McClure, S.D.; Rafiq, S.; Dean, J.C.; Cina, J.A.; Kovac, P.A.; Mirkovic, T.; Scholes, G.D. Broad-Band Pump–Probe Spectroscopy Quantifies Ultrafast Solvation Dynamics of Proteins and Molecules. J. Phys. Chem. Lett. 2016, 7, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.; Jen, M.; Lee, S.; Jang, T.; Pang, Y. Intramolecular Charge Transfer of 1-Aminoanthraquinone and Ultrafast Solvation Dynamics of Dimethylsulfoxide. Int. J. Mol. Sci. 2021, 22, 11926. [Google Scholar] [CrossRef] [PubMed]
- Worth, G.A.; Cederbaum, L.S. Beyond Born-oppenheimer: Molecular Dynamics Through a Conical Intersection. Annu. Rev. Phys. Chem. 2004, 55, 127–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollitrault, P.J.; Mazzola, G.; Tavernelli, I. Nonadiabatic Molecular Quantum Dynamics with Quantum Computers. Phys. Rev. Lett. 2020, 125, 260511. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Jen, M.; Pang, Y. Metal-enhanced fluorescence: Wavelength-dependent ultrafast energy transfer. J. Phys. Chem. C 2015, 119, 23285–23291. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.; Pang, Y. Excited state intramolecular proton transfer of 1,2-dihydroxyanthraquinone by femtosecond transient absorption spectroscopy. Curr. Appl. Phys. 2015, 15, 1492–1499. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jen, M.; Lee, S.; Lee, G.; Lee, D.; Pang, Y. Intramolecular Charge Transfer of Curcumin and Solvation Dynamics of DMSO Probed by Time-Resolved Raman Spectroscopy. Int. J. Mol. Sci. 2022, 23, 1727. https://doi.org/10.3390/ijms23031727
Jen M, Lee S, Lee G, Lee D, Pang Y. Intramolecular Charge Transfer of Curcumin and Solvation Dynamics of DMSO Probed by Time-Resolved Raman Spectroscopy. International Journal of Molecular Sciences. 2022; 23(3):1727. https://doi.org/10.3390/ijms23031727
Chicago/Turabian StyleJen, Myungsam, Sebok Lee, Gisang Lee, Daedu Lee, and Yoonsoo Pang. 2022. "Intramolecular Charge Transfer of Curcumin and Solvation Dynamics of DMSO Probed by Time-Resolved Raman Spectroscopy" International Journal of Molecular Sciences 23, no. 3: 1727. https://doi.org/10.3390/ijms23031727
APA StyleJen, M., Lee, S., Lee, G., Lee, D., & Pang, Y. (2022). Intramolecular Charge Transfer of Curcumin and Solvation Dynamics of DMSO Probed by Time-Resolved Raman Spectroscopy. International Journal of Molecular Sciences, 23(3), 1727. https://doi.org/10.3390/ijms23031727