
Predominant Role of mTOR Signaling in Skin Diseases with Therapeutic Potential

 , ,
, ,  and
and
Abstract
:1. Introduction
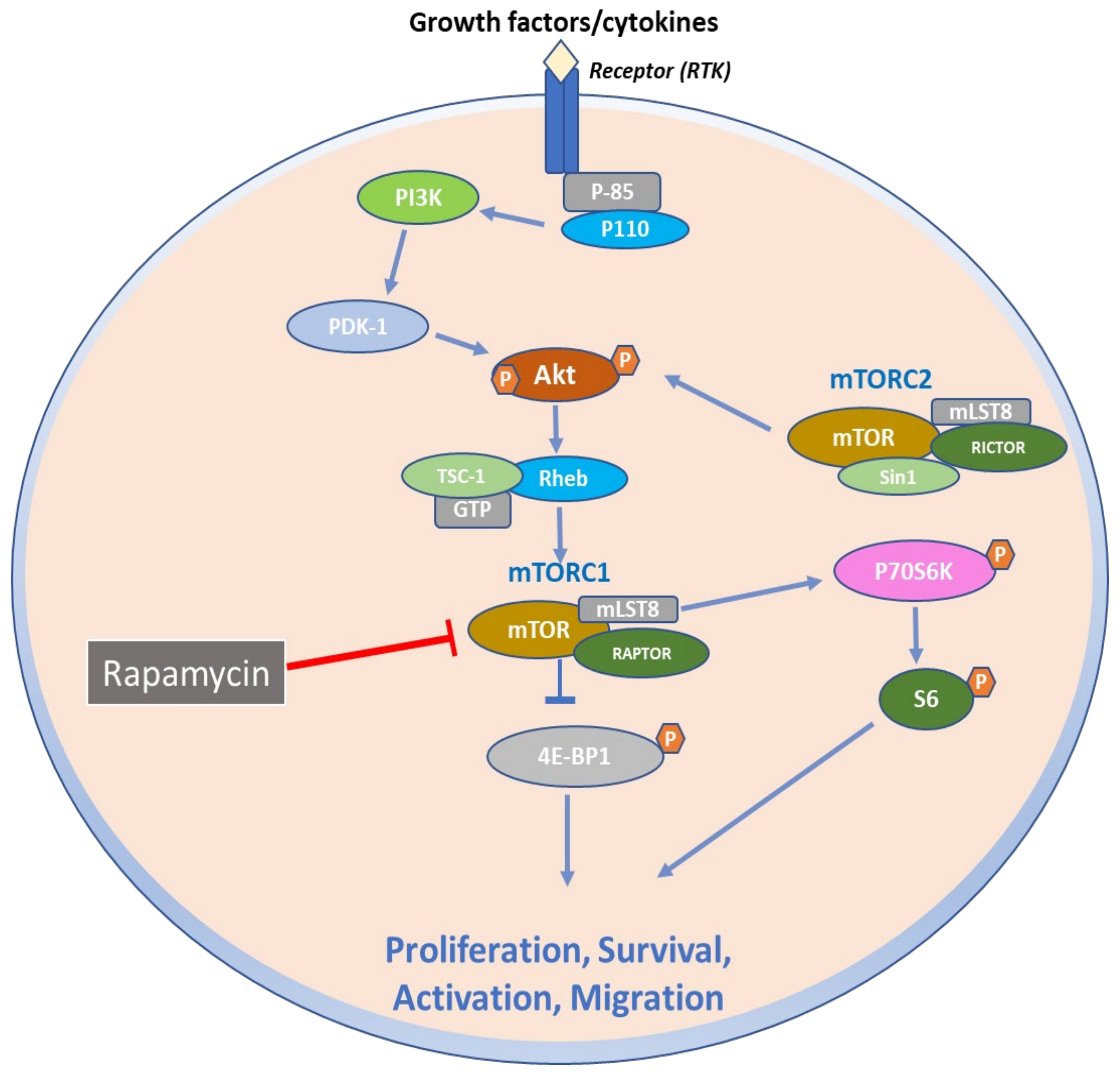
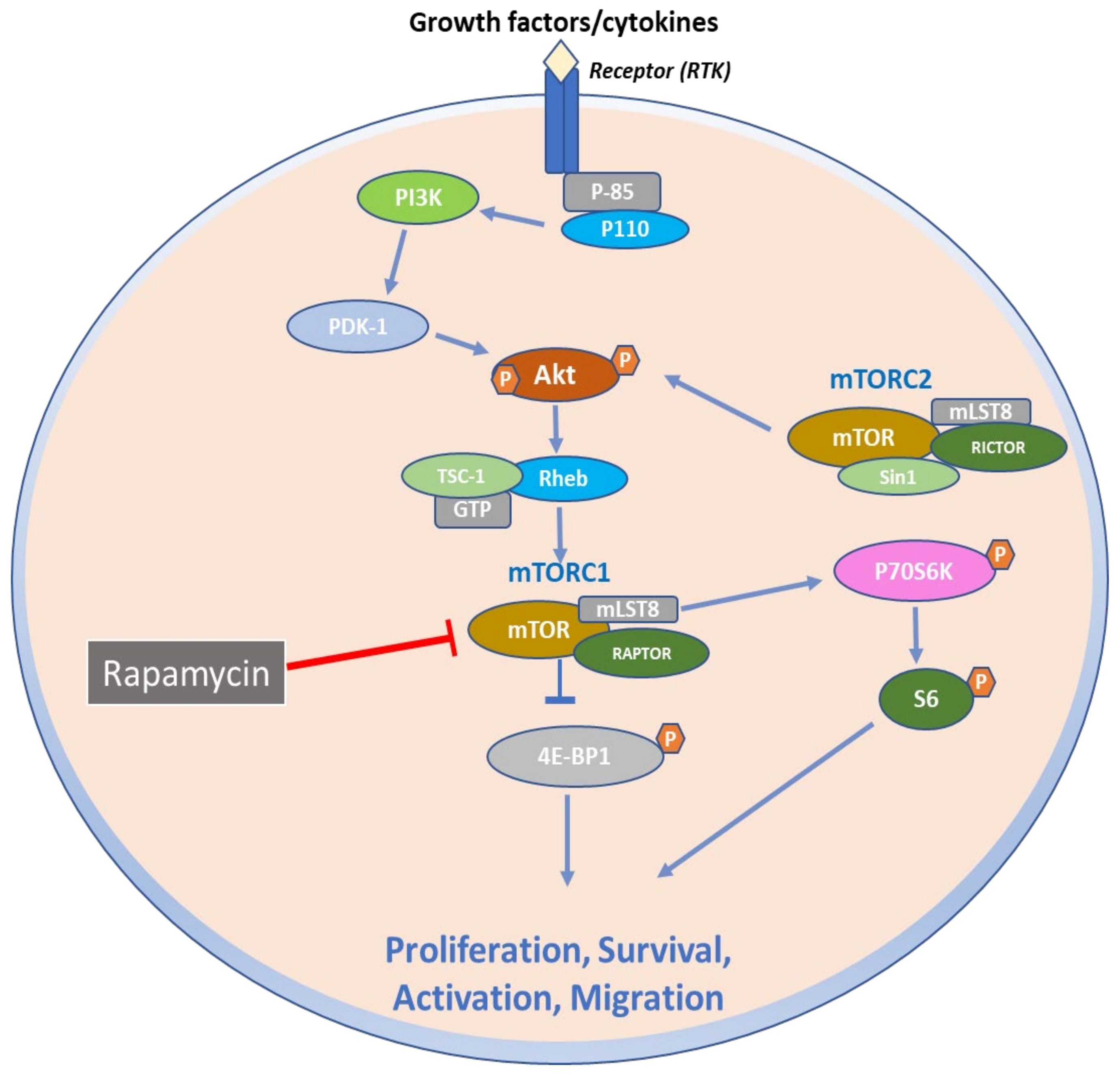
2. Structural and Biochemical Aspects of mTOR Signaling Axis
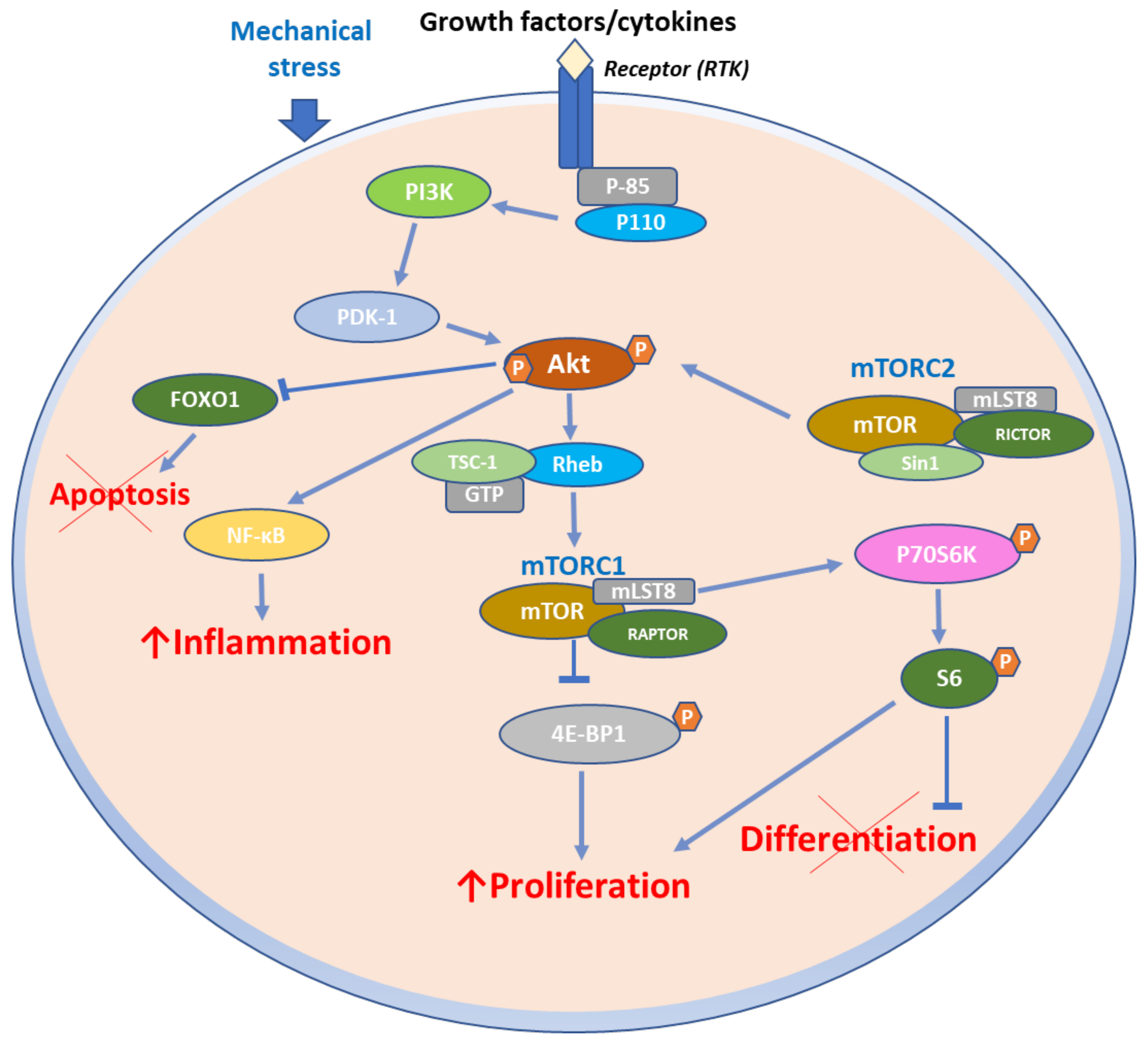
3. Function of mTOR Signaling Pathways
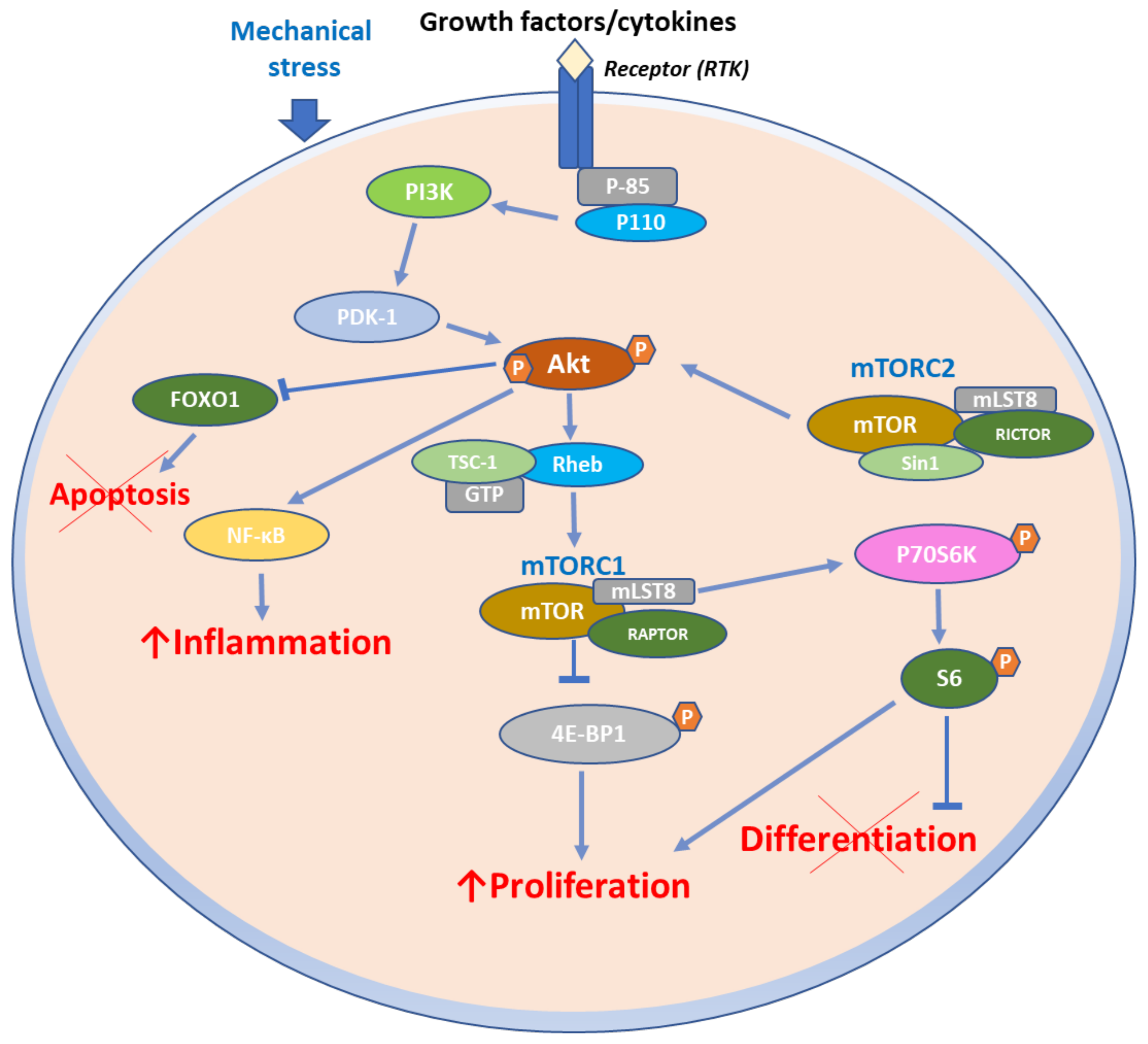
4. Role of mTOR Signaling in Inflammatory Skin Diseases
4.1. mTOR Signaling in Psoriasis
mTOR Pathway in Psoriasis Immunopathogenesis
4.2. mTOR Signaling in Atopic Dermatitis
4.3. mTOR Signaling in Pemphigus
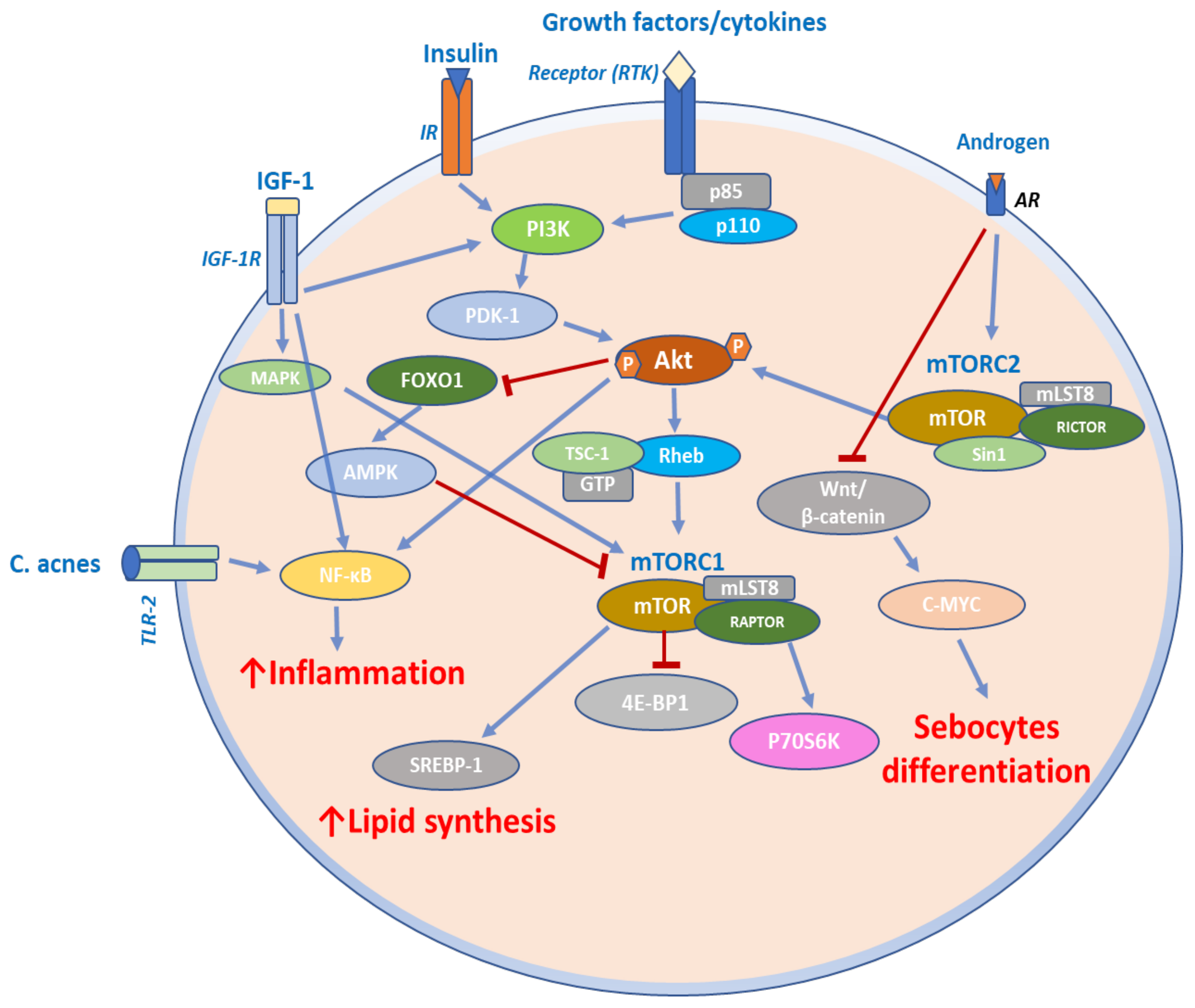
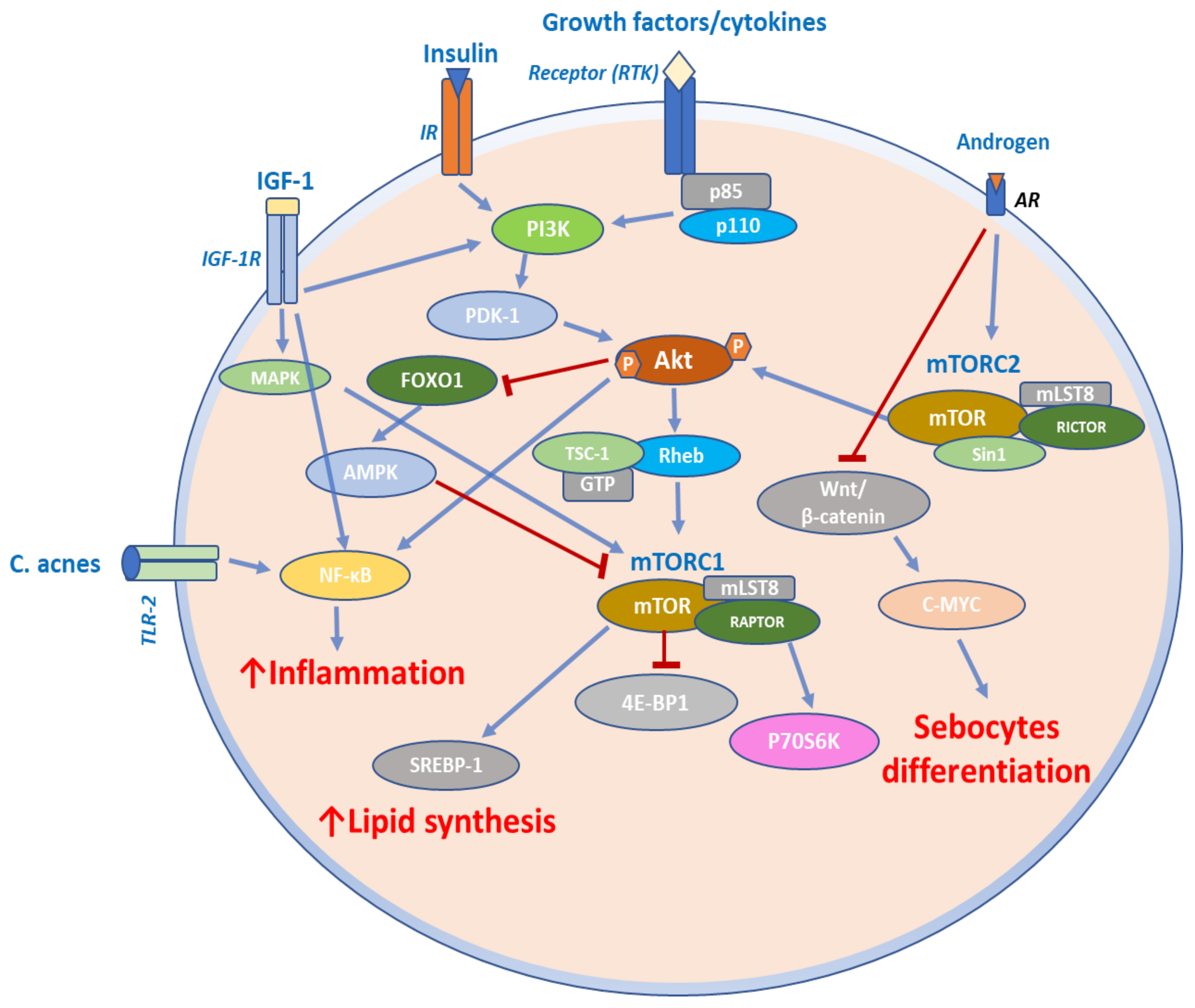
4.4. mTOR Signaling in Acne
5. Role of mTOR Signaling in Skin Cancer
5.1. mTOR Signaling in CTCL
5.2. mTOR Signaling in Melanoma
6. Therapeutic Targeting of mTOR Signaling Axis in Skin Diseases
6.1. Role of mTOR Inhibitors in Psoriasis
6.2. Role of mTOR Inhibitors in CTCL
6.3. Role of mTOR Inhibitors in Melanoma
7. Conclusions—Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| mTOR | Mechanistic Target of Rapamycin |
| PI3K | Phosphatidylinositol 3-kinase |
| Akt | Ak strain transforming |
| mTORC1 | mechanistic target of rapamycin complex 1 |
| mTORC2 | mechanistic target of rapamycin complex 2 |
| mLST8 | mammalian lethal with SEC13 protein 8 |
| RAPTOR | Regulatory-associated protein of mTOR |
| PRAS40 | proline-rich Akt substrate of 40KDa |
| DEPTOR | DEP-domain containing mTOR-interacting protein |
| RICTOR | rapamycin-insensitive companion of mTOR |
| mSIN1 | mammalian stress-activated protein kinase-interacting protein |
| MAPK | Mitogen-activated protein kinase |
| 4E-BPs | eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 |
| PDCD4 | Suppression of Programmed Cell Death 4 |
| elF4E | Eukaryotic translation initiation factor 4E |
| S6K1 | S6 kinase 1 |
| rRNA | ribosomal RNA |
| SREBP1/2 | Sterol regulatory element-binding proteins |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| ATF4 | Activating transcription factor 4 |
| MTHFD2 | Methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2 |
| CAD | Dihydroorotase |
| ULK1 | Unc-51 Like Autophagy Activating Kinase 1 |
| ATG13 | Autophagy-related gene 13 |
| PKCα | Protein kinase Cα |
| PDK1 | Phosphoinositide-dependent protein kinase |
| PKC | Protein kinase C |
| SGK1 | Serum and glucocorticoid-regulated kinase 1 |
| FOXO1/3a | Forkhead transcription factor forkhead box protein O1/3a |
| NAD | Nicotinamide adenine dinucleotide |
| GSK3b | Glycogen synthase kinase 3 beta |
| TSC2 | Tuberous Sclerosis Complex 2 |
| IFN | Interferon |
| mDCs | Myeloid dendritic cells |
| Th-1 | Type 1 T helper cells |
| Th-22 | Type 22 T helper cells |
| Th-17 | Type 17 T helper cells |
| TNF-α | Tumour Necrosis Factor alpha |
| IL-17 | Interleukin 17 |
| miRNAs | microRNA |
| p70S6K | 70-kDa ribosomal protein S6 kinase |
| CXCL8 | C-X-C motif ligand 8 |
| VEGF | Vascular endothelial growth factor |
| PBMCs | Peripheral blood mononuclear cells |
| p-mTOR | Phosphorylated mTOR |
| PUVA | Psoralen and ultraviolet light A |
| AD | Atopic dermatitis |
| AMPK | Adenosine monophosphate-activated protein kinase |
| PV | Pemphigus vulgaris |
| Dsg1 | Desmoglein-1 |
| Dsg3 | Desmoglein-3 |
| IgG | Immunoglobin G |
| C.acnes | Cutibacterium acnes |
| ALA-PDT | 5-aminolevulinic acid-photodynamic therapy |
| LS | Lesional skin |
| NLS | Non lesional skin |
| BMI | Body Mass Index |
| PTEN | Phosphatase and TENsin homolog deleted on chromosome 10 |
| CTCL | Cutaneous T-cell Lymphoma |
| MF | Mycosis Fungoides |
| OS | Overall survival |
| SS | Sezary Syndrome |
| HIF1a | Hypoxia-inducible factor-1α |
| GLUT3 | Glucose transporter 3 |
| LDHA | Lactate dehydrogenase A |
| HK2 | hexokinase 2 |
| BRAF | v-raf murine sarcoma viral oncogene homolog B1 |
| MEK | Mitogen-activated protein kinase kinase |
| RTK | Receptor tyrosine kinases |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
References
- Alcalay Joseph Dermatology: A Medical, Surgical and Aesthetic Profession—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/18669131/ (accessed on 14 December 2021).
- Alani, A.; Sadlier, M.; Uddin, A.; Hackett, C.; Ramsay, B.; Ahmad, K. An Analysis of inpatient dermatologic consultations at University Hospital Limerick: Inadequate infrastructure leads to acute skin failure. Iran. J. Med. Sci. 2017, 186, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.J.; Johns, N.E.; Williams, H.C.; Bolliger, I.W.; Dellavalle, R.P.; Margolis, D.J.; Marks, R.; Naldi, L.; Weinstock, M.A.; Wulf, S.K.; et al. The global burden of skin disease in 2010: An analysis of the prevalence and impact of skin conditions. J. Investig. Dermatol. 2014, 134, 1527–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, I.; Rustenbach, S.J.; Zimmer, L.; Augustin, M. Prevalence of skin diseases in a cohort of 48,665 employees in Germany. Dermatology 2008, 217, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Bolognia, J.L.; Schaffer, J.L.; Cerroni, L. Dermatology: 2-Volume Set—9780702062759. Available online: https://www.us.elsevierhealth.com/dermatology-2-volume-set-9780702062759.html (accessed on 14 December 2021).
- Gilliet, M.; Griffiths, C.E.M. The Skin Science Foundation: Promoting skin health through research. J. Investig. Dermatol. 2020, 140, S189–S190. [Google Scholar] [CrossRef]
- Ding, X.; Bloch, W.; Iden, S.; Rüegg, M.A.; Hall, M.N.; Leptin, M.; Partridge, L.; Eming, S.A. MTORC1 and MTORC2 regulate skin morphogenesis and epidermal barrier formation. Nat. Commun. 2016, 7, 13226. [Google Scholar] [CrossRef] [Green Version]
- Madonna, S.; Scarponi, C.; Pallotta, S.; Cavani, A.; Albanesi, C. Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis. 2012, 3, e334. [Google Scholar] [CrossRef] [Green Version]
- Calautti, E.; Li, J.; Saoncella, S.; Brissette, J.L.; Goetinck, P.F. Phosphoinositide 3-kinase signaling to Akt promotes keratinocyte differentiation versus death. J. Biol. Chem. 2005, 280, 32856–32865. [Google Scholar] [CrossRef] [Green Version]
- Keith, C.T.; Schreiber, S.L. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 1995, 270, 50–51. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. MTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.P.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and MTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the MTORC components raptor, rictor, or MLST8 reveals that MTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, G.H.; Christian Crannell, W.; Walensky, L.D. Chemical synthesis of hydrocarbon-stapled peptides for protein interaction research and therapeutic targeting. Curr. Protoc. Chem. Biol. 2011, 3, 99–117. [Google Scholar] [CrossRef] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.I.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (MTOR) partner, raptor, binds the MTOR substrates P70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an MTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the MTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-MTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S. MTOR signaling in metabolism and cancer. Cells 2020, 9, 2278. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. MTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and BetaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef]
- Hannan, K.M.; Brandenburger, Y.; Jenkins, A.; Sharkey, K.; Cavanaugh, A.; Rothblum, L.; Moss, T.; Poortinga, G.; McArthur, G.A.; Pearson, R.B.; et al. MTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell. Biol. 2003, 23, 8862–8877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. MTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Chen, J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through MTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [Green Version]
- Schmeisser, K.; Parker, J.A. Pleiotropic effects of mTOR and autophagy during development and aging. Front. Cell Dev. Biol. 2019, 7, 192. [Google Scholar] [CrossRef] [Green Version]
- Larsson, C. Protein kinase C and the regulation of the actin cytoskeleton. Cell. Signal. 2006, 18, 276–284. [Google Scholar] [CrossRef]
- Schmidt, K.M.; Dietrich, P.; Hackl, C.; Guenzle, J.; Bronsert, P.; Wagner, C.; Fichtner-Feigl, S.; Schlitt, H.J.; Geissler, E.K.; Hellerbrand, C.; et al. Inhibition of MTORC2/RICTOR impairs melanoma hepatic metastasis. Neoplasia 2018, 20, 1198–1208. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stöckli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates MTORC2. Cell Metab. 2013, 17, 1009–1020. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses MTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; Shirsath, N.; Lang, V.; Berard, A.; Diehl, S.; Kaufmann, R.; Boehncke, W.H.; Wolf, P. Inflammation dependent MTORC1 signaling interferes with the switch from keratinocyte proliferation to differentiation. PLoS ONE 2017, 12, e0180853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.L.; Zhang, K.; Lv, S.C.; Xu, G.W.; Zhang, J.F.; Jia, H.Y. LncRNA MEG3 suppresses PI3K/AKT/mTOR signalling pathway to enhance autophagy and inhibit inflammation in TNF-α-treated keratinocytes and psoriatic mice. Cytokine 2021, 148, 155657. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Len, H.; Shi, X.; Ji, J.; Fu, J.; Len, H. MiR-155 promotes cell proliferation and inhibits apoptosis by PTEN signaling pathway in the psoriasis. Biomed. Pharmacother. 2017, 90, 524–530. [Google Scholar] [CrossRef] [PubMed]
- A, R.; Yu, P.; Hao, S.; Li, Y. MiR-876-5p suppresses cell proliferation by targeting Angiopoietin-1 in the psoriasis. Biomed. Pharmacother. 2018, 103, 1163–1169. [Google Scholar] [CrossRef]
- Gargalionis, A.N.; Malakou, L.S.; Adamopoulos, C.; Piperi, C.; Theohari, I.; Nokhbehsaim, M.; Deschner, J.; Kokkalis, G.; Korkolopoulou, P.; Papadavid, E.; et al. Polycystin-1 downregulation induces ERK-dependent MTOR pathway activation in a cellular model of psoriasis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3468–3476. [Google Scholar] [CrossRef]
- Datta Mitra, A.; Raychaudhuri, S.P.; Abria, C.J.; Mitra, A.; Wright, R.; Ray, R.; Kundu-Raychaudhuri, S. 1α,25-Dihydroxyvitamin-D3-3-Bromoacetate regulates AKT/MTOR signaling cascades: A therapeutic agent for psoriasis. J. Investig. Dermatol. 2013, 133, 1556–1564. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Säemann, M.D. The PI3K/Akt/MTOR pathway in innate immune cells: Emerging therapeutic applications. Ann. Rheum. Dis. 2008, 67, iii70–iii74. [Google Scholar] [CrossRef]
- Huang, T.; Lin, X.; Meng, X.; Lin, M. Phosphoinositide-3 kinase/protein kinase-B/mammalian target of rapamycin pathway in psoriasis pathogenesis. A potential therapeutic target? Acta Derm.-Venereol. 2014, 94, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Inoki, K.; Guan, K.L. MTOR pathway as a target in tissue hypertrophy. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 443–467. [Google Scholar] [CrossRef]
- Patel, A.B.; Tsilioni, I.; Weng, Z.; Theoharides, T.C. TNF stimulates IL-6, CXCL8 and VEGF secretion from human keratinocytes via activation of MTOR, inhibited by tetramethoxyluteolin. Exp. Dermatol. 2018, 27, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Varshney, P.; Saini, N. PI3K/AKT/MTOR activation and autophagy inhibition plays a key role in increased cholesterol during IL-17A mediated inflammatory response in psoriasis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Akinduro, O.; Sully, K.; Patel, A.; Robinson, D.J.; Chikh, A.; McPhail, G.; Braun, K.M.; Philpott, M.P.; Harwood, C.A.; Byrne, C.; et al. Constitutive autophagy and nucleophagy during epidermal differentiation. J. Investig. Dermatol. 2016, 136, 1460–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Ouyang, H.; Li, Y.; Guan, K.-L. Signaling by target of rapamycin proteins in cell growth control. Microbiol. Mol. Biol. Rev. 2005, 69, 79–100. [Google Scholar] [CrossRef] [Green Version]
- Buerger, C. Epidermal MTORC1 signaling contributes to the pathogenesis of psoriasis and could serve as a therapeutic target. Front. Immunol. 2018, 9, 2786. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Raychaudhuri, S.K.; Raychaudhuri, S.P. IL-22 induced cell proliferation is regulated by PI3K/Akt/MTOR signaling cascade. Cytokine 2012, 60, 38–42. [Google Scholar] [CrossRef]
- Pike, M.C.; Lee, C.S.; Elder, J.T.; Voorhees, J.J.; Fisher, G.J. Increased phosphatidylinositol kinase activity in psoriatic epidermis. J. Investig. Dermatol. 1989, 92, 791–797. [Google Scholar] [CrossRef] [Green Version]
- Ochaion, A.; Bar-Yehuda, S.; Cohen, S.; Barer, F.; Patoka, R.; Amital, H.; Reitblat, T.; Reitblat, A.; Ophir, J.; Konfino, I.; et al. The anti-inflammatory target A(3) adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn’s disease. Cell. Immunol. 2009, 258, 115–122. [Google Scholar] [CrossRef]
- Ainali, C.; Valeyev, N.; Perera, G.; Williams, A.; Gudjonsson, J.E.; Ouzounis, C.A.; Nestle, F.O.; Tsoka, S. Transcriptome classification reveals molecular subtypes in psoriasis. BMC Genom. 2012, 13, 472. [Google Scholar] [CrossRef] [Green Version]
- Buerger, C.; Malisiewicz, B.; Eiser, A.; Hardt, K.; Boehncke, W.H. Mammalian target of rapamycin and its downstream signalling components are activated in psoriatic skin. Br. J. Dermatol. 2013, 169, 156–159. [Google Scholar] [CrossRef]
- Shirsath, N.; Mayer, G.; Singh, T.P.; Wolf, P. 8-Methoxypsoralen plus UVA (PUVA) therapy normalizes signalling of phosphorylated component of MTOR pathway in psoriatic skin of K5. HTGFβ1 transgenic mice. Exp. Dermatol. 2015, 24, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Cibrian, D.; de la Fuente, H.; Sánchez-Madrid, F. Metabolic pathways that control skin homeostasis and inflammation. Trends Mol. Med. 2020, 26, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.N.; Zeng, Y.P. Rapamycin blocks the IL-13-induced deficiency of epidermal barrier related proteins via upregulation of MiR-143 in HaCaT keratinocytes. Int. J. Med. Sci. 2020, 17, 2087–2094. [Google Scholar] [CrossRef]
- Hou, T.; Sun, X.; Zhu, J.; Hon, K.L.; Jiang, P.; Chu, I.M.T.; Tsang, M.S.M.; Lam, C.W.K.; Zeng, H.; Wong, C.K. IL-37 ameliorating allergic inflammation in atopic dermatitis through regulating microbiota and AMPK-MTOR signaling pathway-modulated autophagy mechanism. Front. Immunol. 2020, 11, 752. [Google Scholar] [CrossRef] [PubMed]
- Cork, M.J.; Robinson, D.A.; Vasilopoulos, Y.; Ferguson, A.; Moustafa, M.; MacGowan, A.; Duff, G.W.; Ward, S.J.; Tazi-Ahnini, R. New perspectives on epidermal barrier dysfunction in atopic dermatitis: Gene-environment interactions. J. Allergy Clin. Immunol. 2006, 118, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.S.; Tommasi, C.; Cole, C.; Brown, S.J.; Zhu, Y.; Way, B.; Willis Owen, S.A.G.; Moffatt, M.; Cookson, W.O.; Harper, J.I.; et al. A mechanistic target of rapamycin complex 1/2 (MTORC1)/V-Akt murine thymoma viral oncogene homolog 1 (AKT1)/cathepsin H axis controls filaggrin expression and processing in skin, a novel mechanism for skin barrier disruption in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2017, 139, 1228–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodo, D.; Sarig, O.; Sprecher, E. The genetics of pemphigus vulgaris. Front. Med. 2018, 5, 226. [Google Scholar] [CrossRef] [Green Version]
- Di Lullo, G.; Calabresi, V.; Mariotti, F.; Zambruno, G.; Lanzavecchia, A.; Di Zenzo, G. Identification of a novel non-desmoglein autoantigen in pemphigus vulgaris. Front. Immunol. 2019, 10, 1391. [Google Scholar] [CrossRef]
- Schmidt, E.; Gutberlet, J.; Siegmund, D.; Berg, D.; Wajant, H.; Waschke, J. Apoptosis is not required for acantholysis in pemphigus vulgaris. Am. J. Physiol. Cell Physiol. 2009, 296, 162–172. [Google Scholar] [CrossRef]
- Grando, S.A.; Bystryn, J.C.; Chernyavsky, A.I.; Frušić-zlotkin, M.; Gniadecki, R.; Lotti, R.; Milner, Y.; Pittelkow, M.R.; Pincelli, C. Apoptolysis: A novel mechanism of skin blistering in pemphigus vulgaris linking the apoptotic pathways to basal cell shrinkage and suprabasal acantholysis. Exp. Dermatol. 2009, 18, 764–770. [Google Scholar] [CrossRef]
- Lai, K.; Zhang, W.; Li, S.; Zhang, Z.; Xie, S.; Xu, M.; Li, C.; Zeng, K. MTOR pathway regulates the differentiation of peripheral blood Th2/Treg cell subsets in patients with pemphigus vulgaris. Acta Biochim. Biophys. Sin. 2021, 53, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Fabbrocini, G.; Annunziata, M.C.; D’Arco, V.; de Vita, V.; Lodi, G.; Mauriello, M.C.; Pastore, F.; Monfrecola, G. Acne scars: Pathogenesis, classification and treatment. Dermatol. Res. Pract. 2010, 2010, 893080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuo, J.; Wang, Q.; Liu, Y.; Ma, Y.; Ma, L.; Ying, J.; Zhang, C.; Xiang, L. ALA-PDT suppressing the cell growth and reducing the lipogenesis in human SZ95 sebocytes by MTOR signaling pathway in vitro. Photodiagn. Photodyn. Ther. 2017, 18, 295–301. [Google Scholar] [CrossRef]
- Monfrecola, G.; Lembo, S.; Caiazzo, G.; de Vita, V.; di Caprio, R.; Balato, A.; Fabbrocini, G. Mechanistic target of rapamycin (MTOR) expression is increased in acne patients’ skin. Exp. Dermatol. 2016, 25, 153–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agamia, N.F.; Abdallah, D.M.; Sorour, O.; Mourad, B.; Younan, D.N. Skin expression of mammalian target of rapamycin and forkhead box transcription factor O1, and serum insulin-like growth factor-1 in patients with acne vulgaris and their relationship with diet. Br. J. Dermatol. 2016, 174, 1299–1307. [Google Scholar] [CrossRef]
- Melnik, B.C. Is nuclear deficiency of FoxO1 due to increased growth factor/PI3K/Akt-signalling in acne vulgaris reversed by isotretinoin treatment? Br. J. Dermatol. 2010, 162, 1398–1400. [Google Scholar] [CrossRef]
- Melnik, B.C. Isotretinoin and FoxO1: A scientific hypothesis. Dermato-Endocrinology 2011, 3, 141–165. [Google Scholar] [CrossRef] [Green Version]
- Melnik, B.C.; Schmitz, G. Are Therapeutic effects of antiacne agents mediated by activation of FoxO1 and inhibition of MTORC1? Exp. Dermatol. 2013, 22, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Melnik, B.C. P53: Key conductor of all anti-acne therapies. J. Transl. Med. 2017, 15, 195. [Google Scholar] [CrossRef] [Green Version]
- Pink, A.; Anzengruber, F.; Navarini, A.A. Acne and hidradenitis suppurativa. Br. J. Dermatol. 2018, 178, 619–631. [Google Scholar] [CrossRef]
- Pavlidis, A.; Piperi, C.; Papadavid, E. Novel therapeutic approaches for cutaneous T cell lymphomas. Exp. Rev. Clin. Immunol. 2021, 17, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Kremer, M.; Sliva, K.; Klemke, C.D.; Schnierle, B.S. Cutaneous T-cell lymphoma cells are sensitive to rapamycin. Exp. Dermatol. 2010, 19, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Krejsgaard, T.; Vetter-Kauczok, C.S.; Woetmann, A.; Lovato, P.; Labuda, T.; Eriksen, K.W.; Zhang, Q.; Becker, J.C.; Ødum, N. Jak3- and JNK-dependent vascular endothelial growth factor expression in cutaneous T-cell lymphoma. Leukemia 2006, 20, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Kittipongdaja, W.; Wu, X.; Garner, J.; Liu, X.; Komas, S.M.; Hwang, S.T.; Schieke, S.M. Rapamycin suppresses tumor growth and alters the metabolic phenotype in T-cell lymphoma. J. Investig. Dermatol. 2015, 135, 2301–2308. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Huang, H.; Wang, S.; Chen, Y.; Yin, X.; Zhang, X.; Zhang, Y. Molecular profiling of TOX-deficient neoplastic cells in cutaneous T cell lymphoma. Arch. Dermatol. Res. 2020, 312, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Fecher, L.A.; Cummings, S.D.; Keefe, M.J.; Alani, R.M. Toward a molecular classification of melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 1606–1620. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, W.; Zhang, G.; Kwong, L.; Lu, H.; Tan, J.; Sadek, N.; Xiao, M.; Zhang, J.; Labrie, M.; et al. Targeting MTOR signaling overcomes acquired resistance to combined BRAF and MEK inhibition in BRAF-mutant melanoma. Oncogene 2021, 40, 5590–5599. [Google Scholar] [CrossRef]
- Liu, X.; Wu, J.; Qin, H.; Xu, J. The role of autophagy in the resistance to BRAF inhibition in BRAF-mutated melanoma. Target. Oncol. 2018, 13, 437–446. [Google Scholar] [CrossRef]
- Shao, Z.; Bao, Q.; Jiang, F.; Qian, H.; Fang, Q.; Hu, X. VS-5584, a novel PI3K-MTOR dual inhibitor, inhibits melanoma cell growth in vitro and in vivo. PLoS ONE 2015, 10, e0132655. [Google Scholar] [CrossRef]
- Foster, R.S.; Bint, L.J.; Halbert, A.R. Topical 0.1% rapamycin for angiofibromas in paediatric patients with tuberous sclerosis: A pilot study of four patients. Australas. J. Dermatol. 2012, 53, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, A.D.; Shah, S.A.A.; Copeland, P.; Omar, G.; Winfield, A. Treatment of psoriasis with topical sirolimus: Preclinical development and a randomized, double-blind trial. Br. J. Dermatol. 2005, 152, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Frigerio, E.; Colombo, M.D.; Franchi, C.; Altomare, A.; Garutti, C.; Altomare, G.F. Severe psoriasis treated with a new macrolide: Everolimus. Br. J. Dermatol. 2007, 156, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Reitamo, S.; Spuls, P.; Sassolas, B.; Lahfa, M.; Claudy, A.; Griffiths, C.E.M. Efficacy of sirolimus (rapamycin) administered concomitantly with a subtherapeutic dose of cyclosporin in the treatment of severe psoriasis: A randomized controlled trial. Br. J. Dermatol. 2001, 145, 438–445. [Google Scholar] [CrossRef]
- Wei, K.C.; Lai, P.C. Combination of everolimus and tacrolimus: A potentially effective regimen for recalcitrant psoriasis. Dermatol. Ther. 2015, 28, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Chamcheu, J.C.; Chaves-Rodriquez, M.I.; Adhami, V.M.; Siddiqui, I.A.; Wood, G.S.; Jack Longley, B.; Mukhtar, H. Upregulation of PI3K/AKT/MTOR, FABP5 and PPARβ/δ in human psoriasis and imiquimod-induced murine psoriasiform dermatitis model. Acta Derm.-Venereol. 2016, 96, 854–856. [Google Scholar] [CrossRef] [Green Version]
- Buerger, C.; Shirsath, N.; Lang, V.; Diehl, S.; Kaufmann, R.; Weigert, A.; Han, Y.Y.; Ringel, C.; Wolf, P. Blocking MTOR signalling with rapamycin ameliorates imiquimod-induced psoriasis in mice. Acta Derm.-Venereol. 2017, 97, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Si, X. Rapamycin ameliorates psoriasis by regulating the expression and methylation levels of tropomyosin via ERK1/2 and MTOR pathways in vitro and in vivo. Exp. Dermatol. 2018, 27, 1112–1119. [Google Scholar] [CrossRef]
- Zhou, H.-Y.; Huang, S.-L. Current development of the second generation of MTOR inhibitors as anticancer agents. Chin. J. Cancer 2012, 31, 8–18. [Google Scholar] [CrossRef]
- Fasolo, A.; Sessa, C. Targeting MTOR pathways in human malignancies. Curr. Pharm. Des. 2012, 18, 2766–2777. [Google Scholar] [CrossRef]
- Lee, J.H.S.; Vo, T.T.; Fruman, D.A. Targeting MTOR for the treatment of B cell malignancies. Br. J. Clin. Pharmacol. 2016, 82, 1213–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresin, A.; Cristofoletti, C.; Caprini, E.; Cantonetti, M.; Monopoli, A.; Russo, G.; Narducci, M.G. Preclinical evidence for targeting PI3K/MTOR signaling with dual-inhibitors as a therapeutic strategy against cutaneous T-cell lymphoma. J. Investig. Dermatol. 2020, 140, 1045–1053.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molhoek, K.R.; Brautigan, D.L.; Slingluff, C.L. Synergistic inhibition of human melanoma proliferation by combination treatment with B-Raf inhibitor BAY43-9006 and MTOR inhibitor rapamycin. J. Transl. Med. 2005, 3, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leducq, S.; Giraudeau, B.; Tavernier, E.; Maruani, A. Topical use of mammalian target of rapamycin inhibitors in dermatology: A systematic review with meta-analysis. J. Am. Acad. Dermatol. 2019, 80, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Greger, J.G.; Eastman, S.D.; Zhang, V.; Bleam, M.R.; Hughes, A.M.; Smitheman, K.N.; Dickerson, S.H.; Laquerre, S.G.; Liu, L.; Gilmer, T.M. Combinations of BRAF, MEK, and PI3K/MTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 2012, 11, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; White, E.P.; Mehnert, J.M. Coordinate autophagy and MTOR pathway inhibition enhances cell death in melanoma. PLoS ONE 2013, 8, e55096. [Google Scholar] [CrossRef]
- Calero, R.; Morchon, E.; Martinez-Argudo, I.; Serrano, R. Synergistic anti-tumor effect of 17AAG with the PI3K/MTOR inhibitor NVP-BEZ235 on human melanoma. Cancer Lett. 2017, 406, 1–11. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Disease | Study | Type of Study/Sample Type/Main Findings | Reference |
|---|---|---|---|
| Psoriasis | Pike et al., 1989 | Skin biopsies: PI3K activity was increased by 6.7-fold in the epidermis of psoriatic plaques compared to normal skin. | [49] |
| Calautti et al., 2005 | In vitro: Kinase activities of PI3K and Akt are induced, Akt involved in suppression of cell apoptosis. | [9] | |
| Ochaion et al., 2009 | PBMCs: PI3K and Akt were elevated in PBMCs of patients with psoriasis compared to healthy subjects. | [50] | |
| Ainali et al., 2012 | A large-scale study of gene expression in different samples of psoriatic skin detected an overexpression of the PI3K/Akt pathway in plaque psoriatic skin. | [51] | |
| Mitra et al., 2012 | In vitro: IL-22 promotes growth of keratinocytes via the Akt/mTOR pathway. | [48] | |
| Buerger et al., 2017 | In vitro and punch biopsies: Akt activation detected in basal Ki-67+ proliferating cells as well as in all epidermal layers affected in psoriatic lesions. | [34] | |
| Madonna et al., 2012 | In vitro and skin biopsies: Akt was strongly active in all epidermal layers of psoriatic lesions; phosphorylated Akt was elevated in lesional psoriatic skin in vivo as well as in activated psoriatic keratinocytes in vitro. | [8] | |
| Buerger et al., 2013 | Clinical: higher p-mTOR levels were detected in the basal layer along with increased S6K1 in suprabasal layers in punch biopsies of patients with plaque psoriasis, suggesting the important role of mTORC1 in disease pathogenesis. | [52] | |
| Xu et al., 2017 | In vitro: miR-155 knockdown led to a significant decrease in cell proliferation; the expression of several apoptosis-related factors was dramatically changed, such as PTEN, PIP3, AKT, p-AKT, Bax and Bcl-2. Clinical: miR-155 mRNA expression was up-regulated in psoriasis tissues compared with adjacent noncancerous tissues. | [36] | |
| Rongna et al., 2018 | In vitro: miR-876-5p restrains proliferation, cell cycle, cell invasion and adhesion in psoriatic cells. Clinical: low-level of miR-876-5p in psoriatic tissues and blood compared to the respective normal samples. | [37] | |
| Gargalionis et al., 2018 | In vitro: PC1 knockdown in HaCaT cells led to an elevated mRNA expression of psoriasis-related biomarkers Ki-67, IL-6, TNF-α, VEGF and Bcl-2; PC1 functional inhibition was accompanied by increased cell proliferation and migration of HaCaT cells. | [38] | |
| Atopic Dermatitis | Jia et al., 2020 | In vitro: IL-13 increased the expression levels of p-mTOR, p-S6K1, and p-Akt. | [55] |
| Pemphigus vulgaris | Grando et al., 2009 | In vivo: p-mTOR detected at the basal cells of PV IgG injected mice compared to a scattered localization observed in control mice injected with normal human serum. | [62] |
| Lai et al., 2021 | Clinical: PV patients showed elevated serum IL-4 when compared with HCs, and serum IL-4 level was positively correlated with the titer of anti-Dsg1/3 antibody and disease severity; elevated mRNA levels of PI3K, AKT, mTOR and protein levels of PI3K (P85), AKT, p-AKT (Ser473), mTOR, p-mTOR (Ser2448), p-p70S6K (Thr389), GATA3; reduced protein of forkhead box protein 3. | [63] | |
| CTCL | Kremer et al., 2010 | In vitro: Constitutive activation of mTOR kinase in MyLa, HUT78, SeAx and MK-1; rapamycin induced cell cycle arrest in G1 phase and delayed cell growth of CTCL cell lines and primary CD4+ cells isolated from Sézary patients; rapamycin treatment inhibits mTOR, which regulates HIF-1α and consequently decreases VEGF expression in CTCL cell lines. In vivo: Rapamycin treatment delays tumor growth in MyLa xenotransplant model. | [74] |
| Krejsgaard et al., 2006 | In vitro: VEGF expression in MyLA and SeAx cell lines regulated by mTOR signaling. In vivo: VEGF expression in dermal lesions of different stages of CTCL patients through mTOR regulation. | [75] | |
| Kittipongdaja et al., 2015 | In vitro: Rapamycin suppressed tumor growth and mTOR activity in MBL2, HH and Hu78 cell lines. Additionally, rapamycin-treated MBL2, HH, and Hu78 cell lines exhibited reduce aerobic glycolysis and decreased glucose utilization. In vivo: Rapamycin treatment demonstrated suppression of tumor growth and reduce tumor mass in CTCL xenotransplant model. | [76] | |
| Shi et al., 2011 | In vitro: mTOR, via HIF-1α dependent transcriptional program, mediated glycolytic activity and contributed to the lineage selection between Th17 and Tregs. | [77] | |
| Xu et al., 2020 | In vitro: Pathway analysis revealed mTORC1 activation in CTCL cell lines; rapamycin inhibited mTORC1 signaling and restrain the growth of CTCL cells. | [78] | |
| Melanoma | Wang et al., 2021 | In vitro: BRAF/MEK inhibitors combination restored mTORC1 activity, in resistance-associated mTORC1 signaling melanoma cells. | [80] |
| Shao et al., 2015 | In vitro: BRAF/MAPK and PI3K/mTORC1 regulated cooperatively the activation of 4E-PB1 p70S6K, ribosomal protein S6 and, mTORC1 downstream targets.. | [82] | |
| In vivo: The transition of primary benign and malignant melanomas progression to invasive stage was associated with Akt/mTOR activation. | [82] |
| Disease | Drug Name/Approach | Type of Study/Effects | Reference |
|---|---|---|---|
| Psoriasis | Everolimus combined with cyclosporin | Case report (psoriasis patient)
| [85] |
| Sirolimus combined with cyclosporin | Phase 2 randomized controlled trial (N = 150)
| [86] | |
| Everolimus combined with tacrolimus | Case Report (renal transplant patient with psoriasis)
| [87] | |
| Sirolimus | In vitro
| [84] | |
| Rapamycin | In vivo (murine imiquimod-induced psoriasis model)
| [89] | |
| Rapamycin | In vitro
| [90] | |
| Atopic Dermatitis | Rapamycin | In vitro
| [55] |
| Pemphigus vulgaris | Rapamycin | In vivo
| [62] |
| Rapamycin | In vitro
| [63] | |
| CTCL | PF-502 | In vitro
| [94] |
| PF-502 | Xenograft mouse model
| [94] | |
| Melanoma | Rapamycin combined with NVP-BEZ235 | In vitro
| [95] |
| Everolimus | In vitro
| [95] | |
| Temsirolimus | In vitro
| [98] | |
| Rapamycin combined with BAY43-9006 | In vitro
| [95] | |
| GSK2118436 combined with GSK1120212 | In vitro
| [97] | |
| GSK2118436 combined with GSK1120212 and GSK2126458 | In vitro
| [97] | |
| Combination of the lysosomotropic agent and autophagy inhibitor hydroxychloroquine (HCQ) with temsirolimus | In vitro
| [98] | |
| HSP90 inhibitor 17AAG with the PI3K/mTOR inhibitor NVP-BEZ235 | In vitro
| [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karagianni, F.; Pavlidis, A.; Malakou, L.S.; Piperi, C.; Papadavid, E. Predominant Role of mTOR Signaling in Skin Diseases with Therapeutic Potential. Int. J. Mol. Sci. 2022, 23, 1693. https://doi.org/10.3390/ijms23031693
Karagianni F, Pavlidis A, Malakou LS, Piperi C, Papadavid E. Predominant Role of mTOR Signaling in Skin Diseases with Therapeutic Potential. International Journal of Molecular Sciences. 2022; 23(3):1693. https://doi.org/10.3390/ijms23031693
Chicago/Turabian StyleKaragianni, Fani, Antreas Pavlidis, Lina S. Malakou, Christina Piperi, and Evangelia Papadavid. 2022. "Predominant Role of mTOR Signaling in Skin Diseases with Therapeutic Potential" International Journal of Molecular Sciences 23, no. 3: 1693. https://doi.org/10.3390/ijms23031693
APA StyleKaragianni, F., Pavlidis, A., Malakou, L. S., Piperi, C., & Papadavid, E. (2022). Predominant Role of mTOR Signaling in Skin Diseases with Therapeutic Potential. International Journal of Molecular Sciences, 23(3), 1693. https://doi.org/10.3390/ijms23031693