
Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC


 ,
,  ,
, 
Abstract
1. Introduction
2. Chemotherapy
2.1. Neo-Adjuvant Setting
2.2. Adjuvant Setting
2.3. Metastatic Setting
3. Main Mechanisms Responsible for Chemoresistance
3.1. CSCs Induction after NACT
3.2. ABC Transporters
3.3. Hypoxia
3.4. Avoidance of Apoptosis
3.5. Receptor Tyrosine Kinases
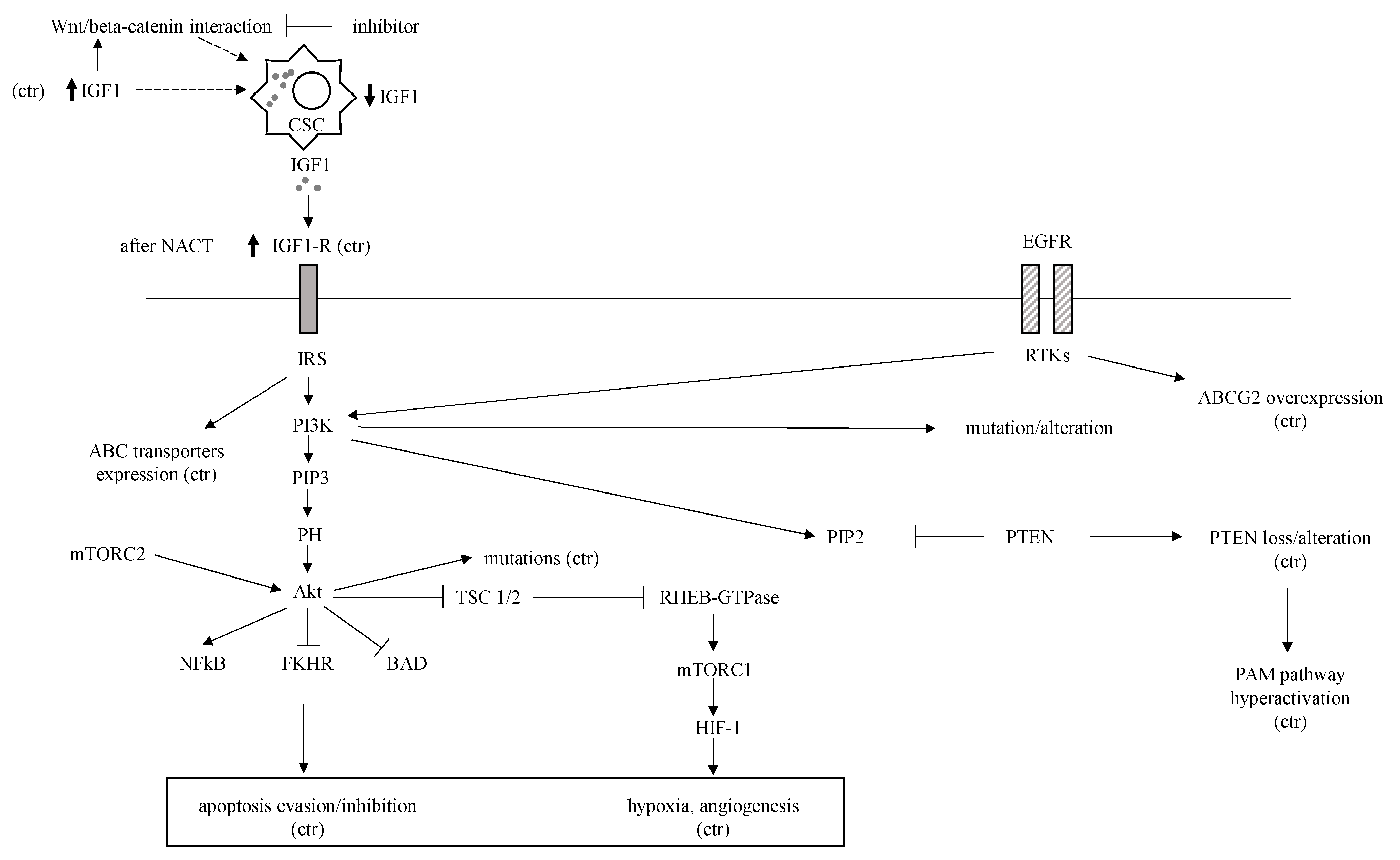
EGFR, IGF-1R
3.6. A Disintegrin and Metalloproteinase 10 (ADAM10)
3.7. Noncoding RNAs, DNA Methylation, and Phosphoproteome including Phosphorylation of Kinases
3.8. Pathological Molecular Pathways
3.8.1. TGF-Beta Pathway
3.8.2. Notch Pathway
3.8.3. Wnt/Beta-Catenin Pathway
3.8.4. Hedgehog (Hh) Pathway
3.8.5. NF-kB Pathway
3.8.6. PTEN and PI3K-AKT-mTOR Pathway
3.8.7. JAK/STAT Pathway
4. Prediction of Resistance to Chemotherapy in TNBC
4.1. Histology and Molecular Subtype
4.2. Tumor Infiltrating Lymphocytes (TILs) and Neoadjuvant Response
TIL Subsets
4.3. Biomarkers Helpful in Predicting Chemoresistance
4.3.1. Biomarkers Predicting Resistance to Platinum-Based Therapy
Lnc DLX6-AS1, miR-105, miR-93-3p and 321 miRNAs including miR-34a
BRCAness and HRD
4.3.2. Biomarkers Predicting Resistance to Taxanes Alone or with Other Agents
BRCAness
IL-6, CXCL8, VEGFA, EGR1, PTGS2, TRIB1 Signature and CXCL8-CXCR1/2 Axis
SYTL4, MITR, SERPINE1, TNFS13 Factors and miR-5195-3p, miR-18a, and miR-1207-5p, MALAT1, CERK, TMPRSS13, PCDH17, JARID2
4.3.3. Biomarkers Predicting Resistance to Anthracyclines
4.3.4. Biomarkers Predicting Resistance to Gemcitabine and CMF
5. Drugs Currently Recommended or Helpful in Chemoresistant TNBC
5.1. Polymerase ADP-Ribose Inhibitors (PARPi) Are Recommended in TNBC BRCA1/2 Germline Mutation Carriers
5.2. Larotrectinib and Entrectinib for NTRK Gene Fusion Carriers
5.3. Anti-Trop2 Antibody Drug Conjugate Therapy
5.4. Other Emerging Targeted Therapies
5.4.1. Targeting Pathological TGF-Beta, Notch, Wnt/Beta-Catenin, Hedgehog, NF-kB, the PI3K-AKT-mTOR, and STAT3/JAK Molecular Pathways
5.4.2. Targeting Apoptosis, miRNAs, EGFR, and AR
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Triple-Negative Breast Cancer. Available online: https://www.cancer.org/cancer/breast-cancer/about/types-of-breast-cancer/triple-negative.html (accessed on 15 January 2022).
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Adamo, B.; Cheang, M.C.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.L.; Cardoso Nunes, N.C.; Izetti, P.; de Mesquita, G.G.; de Melo, A.C. Triple negative breast cancer, A thorough review of biomarkers. Crit. Rev. Oncol. Hematol. 2020, 145, 102855. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast 2015, 24, S36–S40. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, S142–S150. [Google Scholar] [CrossRef]
- NCCN Guidelines 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (accessed on 15 January 2022).
- ESMO Guidelines. Available online: https://www.esmo.org/guidelines/breast-cancer (accessed on 15 January 2022).
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Montemurro, F.; Nuzzolese, I.; Ponzone, R. Neoadjuvant or adjuvant chemotherapy in early breast cancer? Expert Opin. Pharmacother. 2020, 21, 1071–1082. [Google Scholar] [CrossRef]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Hoque, M.O. Targeting Cancer Stem Cells: A Strategy for Effective Eradication of Cancer. Cancers 2019, 11, 732. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Kim, J.H.; Kim, Y.J.; Choi, S.Y.; Kim, S.W.; Kang, E.; Chung, I.Y.; Kim, I.A.; Kim, E.J.; Choi, Y.; et al. An increase in cancer stem cell population after primary systemic therapy is a poor prognostic factor in breast cancer. Br. J. Cancer 2011, 104, 1730–1738. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, H.E.; Li, H.; Shipitsin, M.; Gelman, R.; Polyak, K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin. Cancer Res. 2010, 16, 876–887. [Google Scholar] [CrossRef]
- Ma, F.; Li, H.; Wang, H.; Shi, X.; Fan, Y.; Ding, X.; Lin, C.; Zhan, Q.; Qian, H.; Xu, B. Enriched CD44(+)/CD24(−) population drives the aggressive phenotypes presented in triple-negative breast cancer (TNBC). Cancer Lett. 2014, 353, 153–159. [Google Scholar] [CrossRef]
- Ma, F.; Li, H.; Li, Y.; Ding, X.; Wang, H.; Fan, Y.; Lin, C.; Qian, H.; Xu, B. Aldehyde dehydrogenase 1 (ALDH1) expression is an independent prognostic factor in triple negative breast cancer (TNBC). Medicine 2017, 96, e6561. [Google Scholar] [CrossRef]
- Wang, H.; Wang, L.; Song, Y.; Wang, S.; Huang, X.; Xuan, Q.; Kang, X.; Zhang, Q. CD44+/CD24− phenotype predicts a poor prognosis in triple-negative breast cancer. Oncol. Lett. 2017, 14, 5890–5898. [Google Scholar] [CrossRef]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed]
- Britton, K.M.; Eyre, R.; Harvey, I.J.; Stemke-Hale, K.; Browell, D.; Lennard, T.W.J.; Meeson, A.P. Breast cancer, side population cells and ABCG2 expression. Cancer Lett. 2012, 323, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef]
- Sissung, T.M.; Baum, C.E.; Kirkland, C.T.; Gao, R.; Gardner, E.R.; Figg, W.D. Pharmacogenetics of membrane transporters: An update on current approaches. Mol. Biotechnol. 2010, 44, 152–167. [Google Scholar] [CrossRef]
- Yamada, A.; Ishikawa, T.; Ota, I.; Kimura, M.; Shimizu, D.; Tanabe, M.; Chishima, T.; Sasaki, T.; Ichikawa, Y.; Morita, S.; et al. High expression of ATP-binding cassette transporter ABCC11 in breast tumors is associated with aggressive subtypes and low disease-free survival. Breast Cancer Res. Treat. 2013, 137, 773–782. [Google Scholar] [CrossRef]
- Guestini, F.; Ono, K.; Miyashita, M.; Ishida, T.; Ohuchi, N.; Nakagawa, S.; Hirakawa, H.; Tamaki, K.; Ohi, Y.; Rai, Y.; et al. Impact of Topoisomerase IIα, PTEN, ABCC1/MRP1, and KI67 on triple-negative breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2019, 2, 275–288. [Google Scholar] [CrossRef]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to Smoothened-targeting Hedgehog inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef]
- Arumugam, A.; Subramani, R.; Nandy, S.B.; Terreros, D.; Dwivedi, A.K.; Saltzstein, E.; Lakshmanaswamy, R. Silencing growth hormone receptor inhibits estrogen receptor negative breast cancer through ATP-binding cassette sub-family G member 2. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Oguri, T.; Bessho, Y.; Achiwa, H.; Ozas, H.; Maeno, K.; Maeda, H.; Sato, S.; Ueda, R. MRP8/ABCC11 directly confers resistance to 5-fluorouracil. Mol. Cancer Ther. 2007, 6, 122–127. [Google Scholar] [CrossRef]
- Vaupel, P. Hypoxia and aggressive tumor phenotype: Implications for therapy and prognosis. Oncologist 2008, 13, 21–26. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Vijayappa, S.; Kozin, S. Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol. Cancer Ther. 2006, 5, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Cosse, J.P.; Michiels, C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer. Agents Med. Chem. 2008, 8, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lin, Q.; Glazer, P.M.; Yun, Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Liu, Z.H.; Huan, Q.; Su, P.; Du, G.J.; Wang, Y.; Gao, P.; Zhou, G.Y. Hypoxia-inducible factor-2a is associated with ABCG2 expression, histology-grade and Ki67 expression in breast invasive ductal carcinoma. Diagn. Pathol. 2012, 7, 32. [Google Scholar] [CrossRef]
- Lv, Y.; Zhao, S.; Han, J.; Zheng, L.; Yang, Z.; Zhao, L. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. Onco Targets Ther. 2015, 8, 1941–1948. [Google Scholar] [CrossRef]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef]
- Tan, E.Y.; Yan, M.; Campo, L.; Han, C.; Takano, E.; Turley, H.; Candiloro, I.; Pezzella, F.; Gatter, K.C.; Millar, E.K.; et al. The key hypoxia regulated gene CAIX is upregulated in basal-like breast tumours and is associated with resistance to chemotherapy. Br. J. Cancer 2009, 100, 405–411. [Google Scholar] [CrossRef]
- Simões-Wüst, A.P.; Schürpf, T.; Hall, J.; Stahel, R.A.; Zangemeister-Wittke, U. Bcl-2/bcl-xL bispecific antisense treatment sensitizes breast carcinoma cells to doxorubicin, paclitaxel and cyclophosphamide. Breast Cancer Res. Treat. 2002, 76, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Dhayade, S.; Ferrari, N.; Sims, A.H.; Johnson, E.; Mason, S.M.; Dickson, A.; Ryan, K.M.; Kalna, G.; Edwards, J.; et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 2018, 9, 19. [Google Scholar] [CrossRef]
- Ozretic, P.; Alvir, I.; Sarcevic, B.; Vujaskovic, Z.; Rendic-Miocevic, Z.; Roguljic, A.; Beketic-Oreskovic, L. Apoptosis regulator Bcl-2 is an independent prognostic marker for worse overall survival in triple-negative breast cancer patients. Int. J. Biol. Markers 2018, 33, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef]
- Porcelli, L.; Giovannetti, E.; Assaraf, Y.G.; Jansen, G.; Scheffer, G.L.; Kathman, I.; Azzariti, A.; Paradiso, A.; Peters, G.J. The EGFR pathway regulates BCRP expression in NSCLC cells: Role of erlotinib. Curr. Drug Targets 2014, 15, 1322–1330. [Google Scholar] [CrossRef]
- Zhang, G.N.; Zhang, Y.K.; Wang, Y.J.; Gupta, P.; Ashby, C.R., Jr.; Alqahtani, S.; Deng, T.; Bates, S.E.; Kaddoumi, A.; Wurpel, J.N.D.; et al. Epidermal growth factor receptor (EGFR) inhibitor PD153035 reverses ABCG2-mediated multidrug resistance in non-small cell lung cancer: In vitro and in vivo. Cancer Lett. 2018, 424, 19–29. [Google Scholar] [CrossRef]
- Yuan, J.; Yin, Z.; Tao, K.; Wang, G.; Gao, J. Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy. Oncol. Lett. 2018, 15, 41–47. [Google Scholar] [CrossRef]
- Farabaugh, S.M.; Boone, D.N.; Lee, A.V. Role of IGF1R in Breast Cancer Subtypes, Stemness, and Lineage Differentiation. Front. Endocrinol. 2015, 6, 59. [Google Scholar] [CrossRef]
- Jang, G.B.; Hong, I.S.; Kim, R.J.; Lee, S.Y.; Park, S.J.; Lee, E.S.; Park, J.H.; Yun, C.H.; Chung, J.U.; Lee, K.J.; et al. Wnt/β-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Heskamp, S.; Boerman, O.C.; Molkenboer-Kuenen, J.D.; Wauters, C.A.; Strobbe, L.J.; Mandigers, C.M.; Bult, P.; Oyen, W.J.; van der Graaf, W.T.; van Laarhoven, H.W. Upregulation of IGF-1R expression during neoadjuvant therapy predicts poor outcome in breast cancer patients. PLoS ONE 2015, 10, e0117745. [Google Scholar] [CrossRef]
- Cheng, Y.; Lin, L.; Li, X.; Lu, A.; Hou, C.; Wu, Q.; Hu, X.; Zhou, Z.; Chen, Z.; Tang, F. ADAM10 is involved in the oncogenic process and chemoresistance of triple-negative breast cancer via regulating Notch1 signaling pathway, CD44 and PrPc. Cancer Cell Int. 2021, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, X.; Su, X. Noncoding RNAs in cancer immunity: Functions, regulatory mechanisms, and clinical application. Mol. Cancer 2020, 19, 48. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Zu, X.; Chen, Z.; Wen, G.; Zhong, J. Noncoding RNAs in triple negative breast cancer: Mechanisms for chemoresistance. Cancer Lett. 2021, 523, 100–110. [Google Scholar] [CrossRef]
- Meyer, B.; Clifton, S.; Locke, W.; Luu, P.L.; Du, Q.; Lam, D.; Armstrong, N.J.; Kumar, B.; Deng, N.; Harvey, K.; et al. Identification of DNA methylation biomarkers with potential to predict response to neoadjuvant chemotherapy in triple-negative breast cancer. Clin. Epigenet. 2021, 13, 226. [Google Scholar] [CrossRef]
- Napieralski, R.; Schricker, G.; Auer, G.; Aubele, M.; Perkins, J.; Magdolen, V.; Ulm, K.; Hamann, M.; Walch, A.; Weichert, W.; et al. PITX2 DNA-Methylation: Predictive versus Prognostic Value for Anthracycline-Based Chemotherapy in Triple-Negative Breast Cancer Patients. Breast Care 2021, 16, 523–531. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; Li, S.; Zhang, Z.; Li, X.; Lin, F. Identification of a DNA Methylation-Based Prognostic Signature for Patients with Triple-Negative Breast Cancer. Med. Sci. Monit. 2021, 27, e930025. [Google Scholar] [CrossRef]
- Deng, X.; Kohanfars, M.; Hsu, H.M.; Souda, P.; Capri, J.; Whitelegge, J.P.; Chang, H.R. Combined phosphoproteomics and bioinformatics strategy in deciphering drug resistant related pathways in triple negative breast cancer. Int. J. Proteom. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Cheng, F.; Jia, P.; Wang, Q.; Zhao, Z. Quantitative network mapping of the human kinome interactome reveals new clues for rational kinase inhibitor discovery and individualized cancer therapy. Oncotarget 2014, 5, 3697–3710. [Google Scholar] [CrossRef]
- Tzeng, Y.T.; Liu, P.F.; Li, J.Y.; Liu, L.F.; Kuo, S.Y.; Hsieh, C.W.; Lee, C.H.; Wu, C.H.; Hsiao, M.; Chang, H.T.; et al. Kinome-Wide siRNA Screening Identifies Src-Enhanced Resistance of Chemotherapeutic Drugs in Triple-Negative Breast Cancer Cells. Front. Pharmacol. 2018, 9, 1285. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Robin, T.P.; Ford, H.L. Molecular pathways: Targeting the TGF-β pathway for cancer therapy. Clin. Cancer Res. 2012, 18, 4514–4521. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef]
- Kim, B.; Stephen, S.L.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Richardson, J.; Roundhill, E.A.; Valleley, E.M.; Verghese, E.T.; Williams, B.J.; et al. Chemotherapy induces Notch1-dependent MRP1 upregulation, inhibition of which sensitizes breast cancer cells to chemotherapy. BMC Cancer 2015, 15, 634. [Google Scholar] [CrossRef]
- Li, Z.L.; Chen, C.; Yang, Y.; Wang, C.; Yang, T.; Yang, X.; Liu, S.C. Gamma secretase inhibitor enhances sensitivity to doxorubicin in MDA-MB-231 cells. Int. J. Clin. Exp. Pathol. 2015, 8, 4378–4387. [Google Scholar]
- Qiu, M.; Peng, Q.; Jiang, I.; Carroll, C.; Han, G.; Rymer, I.; Lippincott, J.; Zachwieja, J.; Gajiwala, K.; Kraynov, E.; et al. Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett. 2013, 328, 261–270. [Google Scholar] [CrossRef]
- Ng, L.F.; Kaur, P.; Bunnag, N.; Suresh, J.; Sung, I.C.H.; Tan, Q.H.; Gruber, J.; Tolwinski, N.S. WNT Signaling in Disease. Cells 2019, 8, 826. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Barwick, B.G.; Moreno, C.S.; Ordanic-Kodani, M.; Chen, Z.; Oprea-Ilies, G.; Tang, W.; Catzavelos, C.; Kerstann, K.F.; Sledge, G.W., Jr.; et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [Google Scholar] [CrossRef]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. β-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yan, W.; Yuan, J.; Wang, Z.; Wang, C. Nek2B activates the wnt pathway and promotes triple-negative breast cancer chemotherapy-resistance by stabilizing β-catenin. J. Exp. Clin. Cancer Res. 2019, 38, 243. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Xu, L.; Bonfil, R.D.; Banerjee, S.; Sarkar, F.H.; Sethi, S.; Reddy, K.B. Tumor-initiating cells and FZD8 play a major role in drug resistance in triple-negative breast cancer. Mol. Cancer Ther. 2013, 12, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.G.; Pannell, L.K.; Singh, S.; Samant, R.S.; Shevde, L.A. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene 2012, 31, 3370–3380. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer, A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Sims-Mourtada, J.; Opdenaker, L.M.; Davis, J.; Arnold, K.M.; Flynn, D. Taxane-induced hedgehog signaling is linked to expansion of breast cancer stem-like populations after chemotherapy. Mol. Carcinog. 2015, 54, 1480–1493. [Google Scholar] [CrossRef]
- Arnold, K.M.; Pohlig, R.T.; Sims-Mourtada, J. Co-activation of Hedgehog and Wnt signaling pathways is associated with poor outcomes in triple negative breast cancer. Oncol. Lett. 2017, 14, 5285–5292. [Google Scholar] [CrossRef]
- Fan, Y.; Dutta, J.; Gupta, N.; Fan, G.; Gélinas, C. Regulation of programmed cell death by NF-kappaB and its role in tumorigenesis and therapy. Adv. Exp. Med. Biol. 2008, 615, 223–250. [Google Scholar]
- Ossovskaya, V.; Wang, Y.; Budoff, A.; Xu, Q.; Lituev, A.; Potapova, O.; Vansant, G.; Monforte, J.; Daraselia, N. Exploring molecular pathways of triple-negative breast cancer. Genes Cancer 2011, 2, 870–879. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Rocha, S. Hypoxia Induced NF-κB. Cells 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Ma, J.; Chu, Z.; Wang, X.; Zhao, W.; Zhang, Q. Apatinib-induced NF-κB inactivation sensitizes triple-negative breast cancer cells to doxorubicin. Am. J. Transl. Res. 2020, 12, 3741–3753. [Google Scholar] [PubMed]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ross, A.H. Why is PTEN an important tumor suppressor? J. Cell Biochem. 2007, 102, 1368–1374. [Google Scholar] [CrossRef] [PubMed]
- Ueng, S.H.; Chen, S.C.; Chang, Y.S.; Hsueh, S.; Lin, Y.C.; Chien, H.P.; Lo, Y.F.; Shen, S.C.; Hsueh, C. Phosphorylated mTOR expression correlates with poor outcome in early-stage triple negative breast carcinomas. Int. J. Clin. Exp. Pathol. 2012, 5, 806–813. [Google Scholar]
- Steelman, L.S.; Navolanic, P.M.; Sokolosky, M.L.; Taylor, J.R.; Lehmann, B.D.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Stadelman, K.M.; Terrian, D.M.; et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene 2008, 27, 4086–4095. [Google Scholar] [CrossRef]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Wei, W.; Tweardy, D.J.; Zhang, M.; Zhang, X.; Landua, J.; Petrovic, I.; Bu, W.; Roarty, K.; Hilsenbeck, S.G.; Rosen, J.M.; et al. STAT3 signaling is activated preferentially in tumor-initiating cells in claudin-low models of human breast cancer. Stem Cells 2014, 32, 2571–2582. [Google Scholar] [CrossRef]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Anderson, A.; Harrison, A.; Lange, A.M.; Jin, G.; Watabe, K.; Lo, H.W. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 2018, 37, 2502–2514. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.Y.; Hwu, L.; Wu, C.Y.; Lee, J.S.; Chang, C.W.; Liu, R.S. STAT3/NF-κB-Regulated Lentiviral TK/GCV Suicide Gene Therapy for Cisplatin-Resistant Triple-Negative Breast Cancer. Theranostics 2017, 7, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.P.; da Conceição Braga, L.; Cassali, G.D.; Silva, L.M. STAT3 as a promising chemoresistance biomarker associated with the CD44+/high/CD24-/low/ALDH+ BCSCs-like subset of the triple-negative breast cancer (TNBC) cell line. Exp. Cell Res. 2018, 363, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Soleymani Abyaneh, H.; Gupta, N.; Radziwon-Balicka, A.; Jurasz, P.; Seubert, J.; Lai, R.; Lavasanifar, A. STAT3 but Not HIF-1α Is Important in Mediating Hypoxia-Induced Chemoresistance in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancers 2017, 9, 137. [Google Scholar] [CrossRef]
- Montagna, E.; Maisonneuve, P.; Rotmensz, N.; Cancello, G.; Iorfida, M.; Balduzzi, A.; Galimberti, V.; Veronesi, P.; Luini, A.; Pruneri, G.; et al. Heterogeneity of triple-negative breast cancer: Histologic subtyping to inform the outcome. Clin. Breast Cancer 2013, 13, 31–39. [Google Scholar] [CrossRef]
- Tadros, A.B.; Sevilimedu, V.; Giri, D.D.; Zabor, E.C.; Morrow, M.; Plitas, G. Survival Outcomes for Metaplastic Breast Cancer Differ by Histologic Subtype. Ann. Surg. Oncol. 2021, 28, 4245–4253. [Google Scholar] [CrossRef]
- Corso, G.; Frassoni, S.; Girardi, A.; de Camilli, E.; Montagna, E.; Intra, M.; Bottiglieri, L.; Margherita de Scalzi, A.; Fanianos, D.M.; Magnoni, F.; et al. Metaplastic breast cancer: Prognostic and therapeutic considerations. J. Surg. Oncol. 2021, 123, 61–70. [Google Scholar] [CrossRef]
- Kalaw, E.; Lim, M.; Kutasovic, J.R.; Sokolova, A.; Taege, L.; Johnstone, K.; Bennett, J.; Saunus, J.M.; Niland, C.; Ferguson, K.; et al. Metaplastic breast cancers frequently express immune checkpoint markers FOXP3 and PD-L1. Br. J. Cancer 2020, 123, 1665–1672. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Echavarria, I.; López-Tarruella, S.; Picornell, A.; García-Saenz, J.Á.; Jerez, Y.; Hoadley, K.; Gómez, H.L.; Moreno, F.; Monte-Millan, M.D.; Márquez-Rodas, I.; et al. Pathological Response in a Triple-Negative Breast Cancer Cohort Treated with Neoadjuvant Carboplatin and Docetaxel According to Lehmann’s Refined Classification. Clin. Cancer Res. 2018, 24, 1845–1852. [Google Scholar] [CrossRef]
- Loibl, S.; Müller, B.M.; von Minckwitz, G.; Schwabe, M.; Roller, M.; Darb-Esfahani, S.; Ataseven, B.; du Bois, A.; Fissler-Eckhoff, A.; Gerber, B.; et al. Androgen receptor expression in primary breast cancer and its predictive and prognostic value in patients treated with neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2011, 130, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.S.; Barr, A.R.; Cutts, R.; Beaney, M.; Babina, I.; Sampath, D.; Giltnane, J.; Lacap, J.A.; Crocker, L.; Young, A.; et al. Single-Cell Dynamics Determines Response to CDK4/6 Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Ono, M.; Tsuda, H.; Shimizu, C.; Yamamoto, S.; Shibata, T.; Yamamoto, H.; Hirata, T.; Yonemori, K.; Ando, M.; Tamura, K.; et al. Tumor-infiltrating lymphocytes are correlated with response to neoadjuvant chemotherapy in triple-negative breast cancer. Breast Cancer Res. Treat. 2012, 132, 793–805. [Google Scholar] [CrossRef]
- Oda, N.; Shimazu, K.; Naoi, Y.; Morimoto, K.; Shimomura, A.; Shimoda, M.; Kagara, N.; Maruyama, N.; Kim, S.J.; Noguchi, S. Intratumoral regulatory T cells as an independent predictive factor for pathological complete response to neoadjuvant paclitaxel followed by 5-FU/epirubicin/cyclophosphamide in breast cancer patients. Breast Cancer Res. Treat. 2012, 136, 107–116. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast Cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Cerbelli, B.; Scagnoli, S.; Mezi, S.; de Luca, A.; Pisegna, S.; Amabile, M.I.; Roberto, M.; Fortunato, L.; Costarelli, L.; Pernazza, A.; et al. Tissue Immune Profile: A Tool to Predict Response to Neoadjuvant Therapy in Triple Negative Breast Cancer. Cancers 2020, 12, 2648. [Google Scholar] [CrossRef]
- Brown, J.R.; Wimberly, H.; Lannin, D.R.; Nixon, C.; Rimm, D.L.; Bossuyt, V. Multiplexed quantitative analysis of CD3, CD8, and CD20 predicts response to neoadjuvant chemotherapy in breast cancer. Clin. Cancer Res. 2014, 20, 5995–6005. [Google Scholar] [CrossRef]
- Seo, A.N.; Lee, H.J.; Kim, E.J.; Kim, H.J.; Jang, M.H.; Lee, H.E.; Kim, Y.J.; Kim, J.H.; Park, S.Y. Tumour-infiltrating CD8+ lymphocytes as an independent predictive factor for pathological complete response to primary systemic therapy in breast cancer. Br. J. Cancer 2013, 109, 2705–2713. [Google Scholar] [CrossRef]
- Verma, C.; Kaewkangsadan, V.; Eremin, J.M.; Cowley, G.P.; Ilyas, M.; El-Sheemy, M.A.; Eremin, O. Natural killer (NK) cell profiles in blood and tumour in women with large and locally advanced breast cancer (LLABC) and their contribution to a pathological complete response (PCR) in the tumour following neoadjuvant chemotherapy (NAC): Differential restoration of blood profiles by NAC and surgery. J. Transl. Med. 2015, 13, 180. [Google Scholar] [PubMed]
- Kim, R.; Kawai, A.; Wakisaka, M.; Funaoka, Y.; Yasuda, N.; Hidaka, M.; Morita, Y.; Ohtani, S.; Ito, M.; Arihiro, K. A potential role for peripheral natural killer cell activity induced by preoperative chemotherapy in breast cancer patients. Cancer Immunol. Immunother. 2019, 68, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, M.L.; Idowu, M.O.; Zhao, Y.; Khalak, H.; Payne, K.K.; Wang, X.Y.; Dumur, C.I.; Bedognetti, D.; Tomei, S.; Ascierto, P.A.; et al. Molecular signatures mostly associated with NK cells are predictive of relapse free survival in breast cancer patients. J. Transl. Med. 2013, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Chagollan, M.; Carranza-Torres, I.E.; Carranza-Rosales, P.; Guzmán-Delgado, N.E.; Ramírez-Montoya, H.; Martínez-Silva, M.G.; Mariscal-Ramirez, I.; Barrón-Gallardo, C.A.; Pereira-Suárez, A.L.; Aguilar-Lemarroy, A.; et al. Expression of NK Cell Surface Receptors in Breast Cancer Tissue as Predictors of Resistance to Antineoplastic Treatment. Technol. Cancer Res. Treat. 2018, 17, 1533033818764499. [Google Scholar] [CrossRef]
- Antonio, N.; Bønnelykke-Behrndtz, M.L.; Ward, L.C.; Collin, J.; Christensen, I.J.; Steiniche, T.; Schmidt, H.; Feng, Y.; Martin, P. The wound inflammatory response exacerbates growth of pre-neoplastic cells and progression to cancer. EMBO J. 2015, 34, 2219–2236. [Google Scholar] [CrossRef]
- Kaewkangsadan, V.; Verma, C.; Eremin, J.M.; Cowley, G.; Ilyas, M.; Satthaporn, S.; Eremin, O. The Differential Contribution of the Innate Immune System to a Good Pathological Response in the Breast and Axillary Lymph Nodes Induced by Neoadjuvant Chemotherapy in Women with Large and Locally Advanced Breast Cancers. J. Immunol. Res. 2017, 2017, 1049023. [Google Scholar] [CrossRef]
- Fitzpatrick, A.; Tutt, A. Controversial issues in the neoadjuvant treatment of triple-negative breast cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919882581. [Google Scholar] [CrossRef]
- Du, C.; Wang, Y.; Zhang, Y.; Zhang, J.; Zhang, L.; Li, J. LncRNA DLX6-AS1 Contributes to Epithelial-Mesenchymal Transition and Cisplatin Resistance in Triple-negative Breast Cancer via Modulating Mir-199b-5p/Paxillin Axis. Cell Transplant. 2020, 29, 963689720929983. [Google Scholar] [CrossRef]
- Li, H.Y.; Liang, J.L.; Kuo, Y.L.; Lee, H.H.; Calkins, M.J.; Chang, H.T.; Lin, F.C.; Chen, Y.C.; Hsu, T.I.; Hsiao, M.; et al. miR-105/93–3p promotes chemoresistance and circulating miR-105/93–3p acts as a diagnostic biomarker for triple negative breast cancer. Breast Cancer Res. 2017, 19, 133. [Google Scholar] [CrossRef]
- Kahraman, M.; Röske, A.; Laufer, T.; Fehlmann, T.; Backes, C.; Kern, F.; Kohlhaas, J.; Schrörs, H.; Saiz, A.; Zabler, C.; et al. MicroRNA in diagnosis and therapy monitoring of early-stage triple-negative breast cancer. Sci. Rep. 2018, 8, 11584. [Google Scholar] [CrossRef]
- Sukumar, J.; Gast, K.; Quiroga, D.; Lustberg, M.; Williams, N. Triple-negative breast cancer: Promising prognostic biomarkers currently in development. Expert Rev. Anticancer. Ther. 2021, 21, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, Y.; Yamamoto, Y.; Tanino, H.; Nishimiya, H.; Yamamoto-Ibusuki, M.; Hirota, Y.; Iwase, H.; Nakamura, S.; Akashi-Tanaka, S. BRCAness as an Important Prognostic Marker in Patients with Triple-Negative Breast Cancer Treated with Neoadjuvant Chemotherapy: A Multicenter Retrospective Study. Diagnostics 2020, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Chen, M.; Tsai, L.W.; Lo, C.; Yen, T.C.; Huang, T.Y.; Chen, C.K.; Fan, S.C.; Kuo, S.H.; Huang, C.S. Using next-generation sequencing to redefine BRCAness in triple-negative breast cancer. Cancer Sci. 2020, 111, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Telli, M.L.; Hellyer, J.; Audeh, W.; Jensen, K.C.; Bose, S.; Timms, K.M.; Gutin, A.; Abkevich, V.; Peterson, R.N.; Neff, C.; et al. Homologous recombination deficiency (HRD) status predicts response to standard neoadjuvant chemotherapy in patients with triple-negative or BRCA1/2 mutation-associated breast cancer. Breast Cancer Res. Treat. 2018, 168, 625–630. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, W.; Ji, W.; Yang, F.; Guan, X. Predictive biomarkers for triple negative breast cancer treated with platinum-based chemotherapy. Cancer Biol. Ther. 2017, 18, 369–378. [Google Scholar] [CrossRef][Green Version]
- Loibl, S.; Weber, K.E.; Timms, K.M.; Elkin, E.P.; Hahnen, E.; Fasching, P.A.; Lederer, B.; Denkert, C.; Schneeweiss, A.; Braun, S.; et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response-final results from GeparSixto. Ann. Oncol. 2018, 29, 2341–2347. [Google Scholar] [CrossRef]
- Kaklamani, V.G.; Jeruss, J.S.; Hughes, E.; Siziopikou, K.; Timms, K.M.; Gutin, A.; Abkevich, V.; Sangale, Z.; Solimeno, C.; Brown, K.L.; et al. Phase II neoadjuvant clinical trial of carboplatin and eribulin in women with triple negative early-stage breast cancer (NCT01372579). Breast Cancer Res. Treat. 2015, 151, 629–638. [Google Scholar] [CrossRef]
- Teraoka, S.; Muguruma, M.; Takano, N.; Miyahara, K.; Kawate, T.; Kaise, H.; Yamada, K.; Miyazawa, K.; Ishikawa, T. Association of BRCA Mutations and BRCAness Status with Anticancer Drug Sensitivities in Triple-Negative Breast Cancer Cell Lines. J. Surg. Res. 2020, 250, 200–208. [Google Scholar] [CrossRef]
- Liu, L.; Matsunaga, Y.; Tsurutani, J.; Akashi-Tanaka, S.; Masuda, H.; Ide, Y.; Hashimoto, R.; Inuzuka, M.; Watanabe, C.; Taruno, K.; et al. BRCAness as a prognostic indicator in patients with early breast cancer. Sci. Rep. 2020, 10, 21173. [Google Scholar] [CrossRef] [PubMed]
- Jurj, A.; Pop, L.A.; Zanoaga, O.; Ciocan-Cârtiţă, C.A.; Cojocneanu, R.; Moldovan, C.; Raduly, L.; Pop-Bica, C.; Trif, M.; Irimie, A.; et al. New Insights in Gene Expression Alteration as Effect of Paclitaxel Drug Resistance in Triple Negative Breast Cancer Cells. Cell Physiol. Biochem. 2020, 54, 648–664. [Google Scholar]
- Wang, R.X.; Ji, P.; Gong, Y.; Shao, Z.M.; Chen, S. Value of CXCL8-CXCR1/2 axis in neoadjuvant chemotherapy for triple-negative breast cancer patients: A retrospective pilot study. Breast Cancer Res. Treat. 2020, 181, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Jiang, W.; Ma, D.; Ge, L.P.; Yang, Y.S.; Gou, Z.C.; Xu, X.E.; Shao, Z.M.; Jiang, Y.Z. SYTL4 downregulates microtubule stability and confers paclitaxel resistance in triple-negative breast cancer. Theranostics 2020, 10, 10940–10956. [Google Scholar] [CrossRef] [PubMed]
- Lian, B.; Pei, Y.C.; Jiang, Y.Z.; Xue, M.Z.; Li, D.Q.; Li, X.G.; Zheng, Y.Z.; Liu, X.Y.; Qiao, F.; Sun, W.L.; et al. Truncated HDAC9 identified by integrated genome-wide screen as the key modulator for paclitaxel resistance in triple-negative breast cancer. Theranostics 2020, 10, 11092–11109. [Google Scholar] [CrossRef]
- Zhang, Q.; Lei, L.; Jing, D. Knockdown of SERPINE1 reverses resistance of triple-negative breast cancer to paclitaxel via suppression of VEGFA. Oncol. Rep. 2020, 44, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Kuei, C.H.; Lee, H.H.; Lin, C.H.; Chen, Y.L.; Chen, C.L.; Lin, Y.F. TNFSF13 upregulation confers chemotherapeutic resistance via triggering autophagy initiation in triple-negative breast cancer. J. Mol. Med. 2020, 98, 1255–1267. [Google Scholar] [CrossRef]
- Liu, M.; Gong, C.; Xu, R.; Chen, Y.; Wang, X. MicroRNA-5195–3p enhances the chemosensitivity of triple-negative breast cancer to paclitaxel by downregulating EIF4A2. Cell Mol. Biol. Lett. 2019, 24, 47. [Google Scholar] [CrossRef]
- Sha, L.Y.; Zhang, Y.; Wang, W.; Sui, X.; Liu, S.K.; Wang, T.; Zhang, H. MiR-18a upregulation decreases Dicer expression and confers paclitaxel resistance in triple negative breast cancer. Eur. Rev. Med. Pharmacol Sci. 2016, 20, 2201–2208. [Google Scholar]
- Hou, X.; Niu, Z.; Liu, L.; Guo, Q.; Li, H.; Yang, X.; Zhang, X. miR-1207–5p regulates the sensitivity of triple-negative breast cancer cells to Taxol treatment via the suppression of LZTS1 expression. Oncol. Lett. 2019, 17, 990–998. [Google Scholar] [CrossRef]
- Shaath, H.; Vishnubalaji, R.; Elango, R.; Khattak, S.; Alajez, N.M. Single-cell long noncoding RNA (lncRNA) transcriptome implicates MALAT1 in triple-negative breast cancer (TNBC) resistance to neoadjuvant chemotherapy. Cell Death Discov. 2021, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Bornancin, F. Ceramide kinase: The first decade. Cell Signal. 2011, 23, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Munoz, A. The role of ceramide 1-phosphate in tumor cell survival and dissemination. Adv. Cancer Res. 2018, 140, 217–234. [Google Scholar] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Tumor suppressive functions of ceramide: Evidence and mechanisms. Apoptosis 2015, 20, 689–711. [Google Scholar] [CrossRef]
- Che, J.; Huang, Y.; Xu, C.; Zhang, P. Increased ceramide production sensitizes breast cancer cell response to chemotherapy. Cancer Chemother. Pharmacol. 2017, 79, 933–941. [Google Scholar] [CrossRef]
- Zhu, S.; Xu, Y.; Wang, L.; Liao, S.; Wang, Y.; Shi, M.; Tu, Y.; Zhou, Y.; Wei, W. Ceramide kinase mediates intrinsic resistance and inferior response to chemotherapy in triple-negative breast cancer by upregulating Ras/ERK and PI3K/Akt pathways. Cancer Cell Int. 2021, 21, 42. [Google Scholar] [CrossRef]
- Murray, A.S.; Hyland, T.E.; Sala-Hamrick, K.E.; Mackinder, J.R.; Martin, C.E.; Tanabe, L.M.; Varela, F.A.; List, K. The cell-surface anchored serine protease TMPRSS13 promotes breast cancer progression and resistance to chemotherapy. Oncogene 2020, 39, 6421–6436. [Google Scholar] [CrossRef]
- Kong, D.D.; Fu, R.Z.; Li, L.; Wang, W.; Wang, S.B. Association between the methylation status of PCDH17 and the efficacy of neoadjuvant chemotherapy in triple-negative breast cancer. Oncol. Lett. 2020, 20, 1649–1656. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.; Yang, Q.; Wang, Y.; Li, X.; Liu, Y.; Shan, B. Tumor mutation burden and JARID2 gene alteration are associated with short disease-free survival in locally advanced triple-negative breast cancer. Ann. Transl. Med. 2020, 8, 1052. [Google Scholar] [CrossRef]
- Dou, D.; Ren, X.; Han, M.; Xu, X.; Ge, X.; Gu, Y.; Wang, X.; Zhao, S. CircUBE2D2 (hsa_circ_0005728) promotes cell proliferation, metastasis and chemoresistance in triple-negative breast cancer by regulating miR-512–3p/CDCA3 axis. Cancer Cell Int. 2020, 20, 454. [Google Scholar] [CrossRef]
- Tormo, E.; Ballester, S.; Adam-Artigues, A.; Burgués, O.; Alonso, E.; Bermejo, B.; Menéndez, S.; Zazo, S.; Madoz-Gúrpide, J.; Rovira, A.; et al. The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Sci. Rep. 2019, 9, 5316. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, Y.; Sang, Y.; Song, X.; Zhang, H.; Liu, Y.; Jiang, L.; Yang, Q. MiR-770 suppresses the chemoresistance and metastasis of triple negative breast cancer via direct targeting of STMN1. Cell Death Dis. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, D.; Braun, M.; Kordek, R.; Sadej, R.; Romanska, H. MicroRNAs in regulation of triple-negative breast cancer progression. J. Cancer Res. Clin. Oncol. 2018, 144, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Ciocan-Cartita, C.A.; Jurj, A.; Zanoaga, O.; Cojocneanu, R.; Pop, L.A.; Moldovan, A.; Moldovan, C.; Zimta, A.A.; Raduly, L.; Pop-Bica, C.; et al. New insights in gene expression alteration as effect of doxorubicin drug resistance in triple negative breast cancer cells. J. Exp. Clin. Cancer Res. 2020, 39, 241. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, W.; Xu, L.; Chen, Y.; Xu, Y.; Yuan, L. Long Non-Coding RNA PVT1 Regulates the Resistance of the Breast Cancer Cell Line MDA-MB-231 to Doxorubicin via Nrf2. Technol. Cancer Res. Treat. 2020, 19, 1533033820980763. [Google Scholar] [CrossRef]
- Przanowski, P.; Lou, S.; Tihagam, R.D.; Mondal, T.; Conlan, C.; Shivange, G.; Saltani, I.; Singh, C.; Xing, K.; Morris, B.B.; et al. Oncogenic TRIM37 Links Chemoresistance and Metastatic Fate in Triple-Negative Breast Cancer. Cancer Res. 2020, 80, 4791–4804. [Google Scholar] [CrossRef]
- Zheng, Q.; Yao, D.; Cai, Y.; Zhou, T. NLRP3 augmented resistance to gemcitabine in triple-negative breast cancer cells via EMT/IL-1β/Wnt/β-catenin signaling pathway. Biosci. Rep. 2020, 40, BSR20200730. [Google Scholar] [CrossRef]
- Wu, C.; Zhao, A.; Tan, T.; Wang, Y.; Shen, Z. Overexpression of microRNA-620 facilitates the resistance of triple negative breast cancer cells to gemcitabine treatment by targeting DCTD. Exp. Ther. Med. 2019, 18, 550–558. [Google Scholar] [CrossRef]
- Elsharawy, K.A.; Althobiti, M.; Mohammed, O.J.; Aljohani, A.I.; Toss, M.S.; Green, A.R.; Rakha, E.A. Nucleolar protein 10 (NOP10) predicts poor prognosis in invasive breast cancer. Breast Cancer Res. Treat. 2021, 185, 615–627. [Google Scholar] [CrossRef]
- Damaskos, C.; Garmpis, N.; Garmpi, A.; Nikolettos, K.; Sarantis, P.; Georgakopoulou, V.E.; Nonni, A.; Schizas, D.; Antoniou, E.A.; Karamouzis, M.V.; et al. Investigational Drug Treatments for Triple-Negative Breast Cancer. J. Pers. Med. 2021, 11, 652. [Google Scholar] [CrossRef]
- Copson, E.R.; Maishman, T.C.; Tapper, W.J.; Cutress, R.I.; Greville-Heygate, S.; Altman, D.G.; Eccles, B.; Gerty, S.; Durcan, L.T.; Jones, L.; et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): A prospective cohort study. Lancet Oncol. 2018, 19, 169–180. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef]
- Fasching, P.A.; Link, T.; Hauke, J.; Seither, F.; Jackisch, C.; Klare, P.; Schmatloch, S.; Hanusch, C.; Huober, J.; Stefek, A.; et al. Neoadjuvant paclitaxel/olaparib in comparison to paclitaxel/carboplatinum in patients with HER2-negative breast cancer and homologous recombination deficiency (GeparOLA study). Ann. Oncol. 2021, 32, 49–57. [Google Scholar] [CrossRef]
- Litton, J.K.; Scoggins, M.E.; Hess, K.R.; Adrada, B.E.; Murthy, R.K.; Damodaran, S.; DeSnyder, S.M.; Brewster, A.M.; Barcenas, C.H.; Valero, V.; et al. Neoadjuvant Talazoparib for Patients with Operable Breast Cancer with a Germline BRCA Pathogenic Variant. J. Clin. Oncol. 2020, 38, 388–394. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Ross, J.S.; Chung, J.H.; Elvin, J.A.; Vergilio, J.; Ramkissoon, S.H.; Suh, J.H.; Severson, E.; Daniel, S.; Frampton, G.M.; Fabrizio, D.A.; et al. NTRK fusions in breast cancer: Clinical, pathologic and genomic findings. Cancer Res. 2018, 78, P2-09-15. [Google Scholar] [CrossRef]
- Jacob, J.D.; Hodge, C.; Franko, J.; Pezzi, C.M.; Goldman, C.D.; Klimberg, V.S. Rare breast cancer: 246 invasive secretory carcinomas from the National Cancer Data Base. J. Surg. Oncol. 2016, 113, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef]
- Krings, G.; Joseph, N.M.; Bean, G.R.; Solomon, D.; Onodera, C.; Talevich, E.; Yeh, I.; Grenert, J.P.; Hosfield, E.; Crawford, E.D.; et al. Genomic profiling of breast secretory carcinomas reveals distinct genetics from other breast cancers and similarity to mammary analog secretory carcinomas. Mod. Pathol. 2017, 30, 1086–1099. [Google Scholar] [CrossRef] [PubMed]
- Laé, M.; Fréneaux, P.; Sastre-Garau, X.; Chouchane, O.; Sigal-Zafrani, B.; Vincent-Salomon, A. Secretory breast carcinomas with ETV6-NTRK3 fusion gene belong to the basal-like carcinoma spectrum. Mod. Pathol. 2009, 22, 291–298. [Google Scholar] [CrossRef]
- Hoda, R.S.; Brogi, E.; Pareja, F.; Nanjangud, G.; Murray, M.P.; Weigelt, B.; Reis-Filho, J.S.; Wen, H.Y. Secretory carcinoma of the breast: Clinicopathologic profile of 14 cases emphasising distant metastatic potential. Histopathology 2019, 75, 213–224. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Shukla, N.; Peled, N.; Landman, Y.; Onitilo, A.; Montez, S.; Ku, N.C.; Hyman, D.M.; Drilon, A.; Hong, D.S. Activity of larotrectinib, a highly selective inhibitor of tropomyosin receptor kinase, in TRK fusion breast cancers. Cancer Res. 2019, 79, P6-20-02. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Stein, R.; Sharkey, R.M. The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget 2018, 9, 28989–29006. [Google Scholar] [CrossRef]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; Vahdat, L.T.; Isakoff, S.J.; Guarino, M.; Messersmith, W.A.; Picozzi, V.J.; Mayer, I.A.; Wegener, W.A.; et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer 2017, 123, 3843–3854. [Google Scholar] [CrossRef]
- Rugo, H.S.; Bardia, A.; Tolaney, S.M.; Arteaga, C.; Cortes, J.; Sohn, J.; Marmé, F.; Hong, Q.; Delaney, R.J.; Hafeez, A.; et al. TROPiCS-02: A Phase III study investigating sacituzumab govitecan in the treatment of HR+/HER2− metastatic breast cancer. Future Oncol. 2020, 16, 705–715. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients with Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Broner, E.C.; Alpert, G.; Gluschnaider, U.; Mondshine, A.; Solomon, O.; Sloma, I.; Rauch, R.; Izumchenko, E.; Aster, J.C.; Davis, M.; et al. AL101 mediated tumor inhibition in notch-altered TNBC PDX models. J. Clin. Oncol. 2019, 37, 1064. [Google Scholar] [CrossRef]
- Locatelli, M.A.; Aftimos, P.; Dees, E.C.; LoRusso, P.M.; Pegram, M.D.; Awada, A.; Huang, B.; Cesari, R.; Jiang, Y.; Shaik, M.N.; et al. Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triple-negative breast cancer. Oncotarget 2017, 8, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Gangrade, A.; Pathak, V.; Augelli-Szafran, C.E.; Wei, H.X.; Oliver, P.; Suto, M.; Buchsbaum, D.J. Preferential Inhibition of Wnt/β-Catenin Signaling by Novel Benzimidazole Compounds in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1524. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, H.; Li, X.; Han, L.; Xu, N.; Shi, A. Signaling pathway inhibitors target breast cancer stem cells in triple-negative breast cancer. Oncol Rep. 2019, 41, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Koval, A.; Xu, J.; Bodmer, A.; Katanaev, V.L. Towards the first targeted therapy for triple-negative breast cancer: Repositioning of clofazimine as a chemotherapy-compatible selective Wnt pathway inhibitor. Cancer Lett. 2019, 449, 45–55. [Google Scholar] [CrossRef]
- Xie, W.; Zhang, Y.; He, Y.; Zhang, K.; Wan, G.; Huang, Y.; Zhou, Z.; Huang, G.; Wang, J. A novel recombinant human Frizzled-7 protein exhibits antitumor activity against triple negative breast cancer via abating Wnt/β-catenin pathway. Int. J. Biochem. Cell Biol. 2018, 103, 45–55. [Google Scholar] [CrossRef]
- Bhateja, P.; Cherian, M.; Majumder, S.; Ramaswamy, B. The Hedgehog Signaling Pathway: A Viable Target in Breast Cancer? Cancers 2019, 11, 1126. [Google Scholar] [CrossRef]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef]
- Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T.; Kurebayashi, J. Anti-cell growth and anticancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer 2017, 24, 683–693. [Google Scholar] [CrossRef]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Messeha, S.S.; Zarmouh, N.O.; Mendonca, P.; Alwagdani, H.; Kolta, M.G.; Soliman, K.F.A. The inhibitory effects of plumbagin on the NF-қB pathway and CCL2 release in racially different triple-negative breast cancer cells. PLoS ONE 2018, 13, e0201116. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Zhou, W.; He, W.; Liu, X.; Ding, Q.; Ling, L.; Zha, X.; Wang, S. Genistein inhibits MDA-MB-231 triple-negative breast cancer cell growth by inhibiting NF-κB activity via the Notch-1 pathway. Int. J. Mol. Med. 2012, 30, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Labbozzetta, M.; Poma, P.; Vivona, N.; Gulino, A.; D’Alessandro, N.; Notarbartolo, M. Epigenetic changes and nuclear factor-κB activation, but not microRNA-224, downregulate Raf-1 kinase inhibitor protein in triple-negative breast cancer SUM 159 cells. Oncol. Lett. 2015, 10, 3807–3815. [Google Scholar] [CrossRef]
- Dent, R.; Oliveira, M.; Isakoff, S.J.; Im, S.A.; Espié, M.; Blau, S.; Tan, A.R.; Saura, C.; Wongchenko, M.J.; Xu, N.; et al. Final results of the double-blind placebo-controlled randomized phase 2 LOTUS trial of first-line ipatasertib plus paclitaxel for inoperable locally advanced/metastatic triple-negative breast cancer. Breast Cancer Res. Treat. 2021, 189, 377–386. [Google Scholar]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel ss First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin. Oncol. 2020, 38, 423–433. [Google Scholar] [CrossRef]
- Qin, J.J.; Yan, L.; Zhang, J.; Zhang, W.D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 195. [Google Scholar] [CrossRef]
- Koff, J.L.; Ramachandiran, S.; Bernal-Mizrachi, L. A time to kill: Targeting apoptosis in cancer. Int. J. Mol. Sci. 2015, 16, 2942–2955. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Varley, K.E.; Abramson, V.G.; Li, Y.; Vaklavas, C.; Lin, N.U.; Liu, M.C.; Rugo, H.S.; Nanda, R.; Storniolo, A.M.; et al. TBCRC 019: A Phase II Trial of Nanoparticle Albumin-Bound Paclitaxel with or without the Anti-Death Receptor 5 Monoclonal Antibody Tigatuzumab in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 21, 2722–2729. [Google Scholar] [CrossRef]
- Greer, Y.E.; Gilbert, S.F.; Gril, B.; Narwal, R.; Peacock Brooks, D.L.; Tice, D.A.; Steeg, P.S.; Lipkowitz, S. MEDI3039, a novel highly potent tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 2 agonist, causes regression of orthotopic tumors and inhibits outgrowth of metastatic triple-negative breast cancer. Breast Cancer Res. 2019, 21, 27. [Google Scholar] [CrossRef]
- Wang, S.; Bai, L.; Lu, J.; Liu, L.; Yang, C.Y.; Sun, H. Targeting inhibitors of apoptosis proteins (IAPs) for new breast cancer therapeutics. J. Mammary Gland Biol. Neoplasia 2012, 17, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Parton, M.; Kümmel, S.; Estévez, L.G.; Huang, C.S.; Cortés, J.; Ruiz-Borrego, M.; Telli, M.L.; Martin-Martorell, P.; López, R.; et al. Paclitaxel with Inhibitor of Apoptosis Antagonist, LCL161, for Localized Triple-Negative Breast Cancer, Prospectively Stratified by Gene Signature in a Biomarker-Driven Neoadjuvant Trial. J. Clin. Oncol. 2018, 36, 3126–3133. [Google Scholar] [CrossRef]
- Petrovic, N.; Ergun, S. miRNAs as Potential Treatment Targets and Treatment Options in Cancer. Mol. Diagn. Ther. 2018, 22, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Devulapally, R.; Sekar, N.M.; Sekar, T.V.; Foygel, K.; Massoud, T.F.; Willmann, J.K.; Paulmurugan, R. Polymer nanoparticles mediated codelivery of antimiR-10b and antimiR-21 for achieving triple negative breast cancer therapy. ACS Nano 2015, 9, 2290–2302. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, Z.S.; Moghadam, M.F.; Farokhimanesh, S.; Rajabibazl, M.; Sadroddiny, E. Inhibition of breast cancer metastasis by co-transfection of miR-31/193b-mimics. Iran. J. Basic Med. Sci. 2018, 21, 427–433. [Google Scholar] [PubMed]
- Nakajima, H.; Ishikawa, Y.; Furuya, M.; Sano, T.; Ohno, Y.; Horiguchi, J.; Oyama, T. Protein expression, gene amplification, and mutational analysis of EGFR in triple-negative breast cancer. Breast Cancer 2014, 21, 66–74. [Google Scholar] [CrossRef]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pêgo, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef]
- Matsuda, N.; Wang, X.; Lim, B.; Krishnamurthy, S.; Alvarez, R.H.; Willey, J.S.; Parker, C.A.; Song, J.; Shen, Y.; Hu, J.; et al. Safety and Efficacy of Panitumumab Plus Neoadjuvant Chemotherapy in Patients with Primary HER2-Negative Inflammatory Breast Cancer. JAMA Oncol. 2018, 4, 1207–1213. [Google Scholar] [CrossRef]
- Cowherd, S.; Miller, L.D.; Melin, S.A.; Akman, S.; Isom, S.; Cole, J.; Pullikuth, A.; Lawrence, J.A. A phase II clinical trial of weekly paclitaxel and carboplatin in combination with panitumumab in metastatic triple negative breast cancer. Cancer Biol. Ther. 2015, 16, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, S.; Zhang, Y.; Wang, C.; Hu, X.; Kong, D.; Liang, G.; Yuan, X.; Li, Y.; Wang, X. Apatinib enhances the antitumor effect of paclitaxel via the PI3K/p65/Bcl-xl pathway in triple-negative breast cancer. Ann. Transl. Med. 2021, 9, 1001. [Google Scholar] [CrossRef]
- Gao, Z.; Shi, M.; Wang, Y.; Chen, J.; Ou, Y. Apatinib enhanced antitumor activity of cisplatin on triple-negative breast cancer through inhibition of VEGFR-2. Pathol. Res. Pract. 2019, 215, 152422. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Zhou, Y.; Wang, Y.W.; Tong, L.; Jiang, R.X.; Xiao, L.; Zhang, G.J.; Xing, S.S.; Qian, F.; Feng, J.Q.; et al. Comparison of apatinib and capecitabine (Xeloda) with capecitabine (Xeloda) in advanced triple-negative breast cancer as third-line therapy: A retrospective study. Medicine 2018, 97, e12222. [Google Scholar] [CrossRef] [PubMed]
- El Guerrab, A.; Bamdad, M.; Kwiatkowski, F.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Anti-EGFR monoclonal antibodies and EGFR tyrosine kinase inhibitors as combination therapy for triple-negative breast cancer. Oncotarget 2016, 7, 73618–73637. [Google Scholar] [CrossRef] [PubMed]
- Nabholtz, J.M.; Abrial, C.; Mouret-Reynier, M.A.; Dauplat, M.M.; Weber, B.; Gligorov, J.; Forest, A.M.; Tredan, O.; Vanlemmens, L.; Petit, T.; et al. Multicentric neoadjuvant phase II study of panitumumab combined with an anthracycline/taxane-based chemotherapy in operable triple-negative breast cancer: Identification of biologically defined signatures predicting treatment impact. Ann. Oncol. 2014, 25, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells Through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 4490. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef]
- Bonnefoi, H.; Grellety, T.; Tredan, O.; Saghatchian, M.; Dalenc, F.; Mailliez, A.; L’Haridon, T.; Cottu, P.; Abadie-Lacourtoisie, S.; You, B.; et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12–1). Ann. Oncol. 2016, 27, 812–818. [Google Scholar] [CrossRef]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef]
- Speers, C.; Zhao, S.G.; Chandler, B.; Liu, M.; Wilder-Romans, K.; Olsen, E.; Nyati, S.; Ritter, C.; Alluri, P.G.; Kothari, V.; et al. Androgen receptor as a mediator and biomarker of radioresistance in triple-negative breast cancer. NPJ Breast Cancer 2017, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Fallahpour, S.; Navaneelan, T.; De, P.; Borgo, A. Breast cancer survival by molecular subtype: A population-based analysis of cancer registry data. CMAJ Open 2017, 5, E734–E739. [Google Scholar] [CrossRef]
- Malorni, L.; Shetty, P.B.; de Angelis, C.; Hilsenbeck, S.; Rimawi, M.F.; Elledge, R.; Osborne, C.K.; De Placido, S.; Arpino, G. Clinical and biologic features of triple-negative breast cancers in a large cohort of patients with long-term follow-up. Breast Cancer Res. Treat. 2012, 136, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Olivier, T.; Prasad, V. Sacituzumab govitecan in metastatic triple negative breast cancer (TNBC): Four design features in the ASCENT trial potentially favored the experimental arm. Transl Oncol. 2021, 15, 101248. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Han, M.; Liu, A.; Shi, B. Economic Evaluation of Sacituzumab Govitecan for the Treatment of Metastatic Triple-Negative Breast Cancer in China and the US. Front. Oncol. 2021, 11, 734594. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.T.R.; Emens, L.A. Emerging combination immunotherapy strategies for breast cancer: Dual immune checkpoint modulation, antibody-drug conjugates and bispecific antibodies. Breast Cancer Res. Treat. 2021, 191, 291–302. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Biological Condition/ Component | Status | Mechanism | References |
|---|---|---|---|
| Cancer stem cells | Intrinsically enriched, increased after NACT through HIFs and ABC B1 upregulation | Quiescence, ABCG2 transporter overexpression, tumor-initiating cells enrichment | [17,18,19,20,21,22,23,24,25,26,27] |
| ABC Transporters | ABCC1/MRP1, ABCG2/BCRP, ABCC11/MRP8 intrinsic increase or after NACT or Hh pathway | Transporter-mediated efflux through ATP | [29,30,31,32,33] |
| Hypoxia | Morphological features characteristics of hypoxia (expression of CAIX) | Insufficient drug penetration and multiple other mechanisms due to the promoted TME changes (see text) | [34,35,36,37,38,39,40,41,42,43] |
| Apoptosis | Malfunction (BCL-2 and Mcl-1 protein expression) | Evasion of apoptosis | [44,45,46,47,48] |
| Factor | |||
| EGFR | Increased expression (from 13% to 76%) | ABCG2-mediated, apoptosis inhibition, angiogenesis, and cell proliferation involvement | [6,49,50,51] |
| IGF-1R | Expressed in 46% and increased after NACT | ABCG2-mediated, apoptosis inhibition, angiogenesis and cell proliferation involvement, Wnt-beta-catenin interaction, CSCs self-renewal decrease | [52,53,54,55] |
| ADAM10 | Highly expressed in cell lines | Notch signaling downregulation; proliferation, migration, invasion increase | [56] |
| NcRNAs | Aberrant expression | Promotion of apoptosis resistance, EMT, ABC transporters upregulation; cell cycle arrest, CSCs, DNA repair and autophagy inhibition | [57,58] |
| DNA methylation | Strong hypomethylation and low gains of methylation | Significantly differentially methylated regions | [59,60,61] |
| Phosphoproteome, phosphorylation of kinases | Activation of protein kinases and phosphatases through phosphorylation | Changes in phosphorylated proteins, phosphorylation and signal transduction involvement, aberrant expression or activation of protein kinases | [62] |
| Pathologic Molecular Pathway | |||
| TGF-beta | Signaling increase after NACT | CSCs upregulation, EMT increase | [24,66,67,68,69] |
| Notch | Signaling increase after NACT | CSCs maintenance, ABCC1 overexpression | [70,71,72,73] |
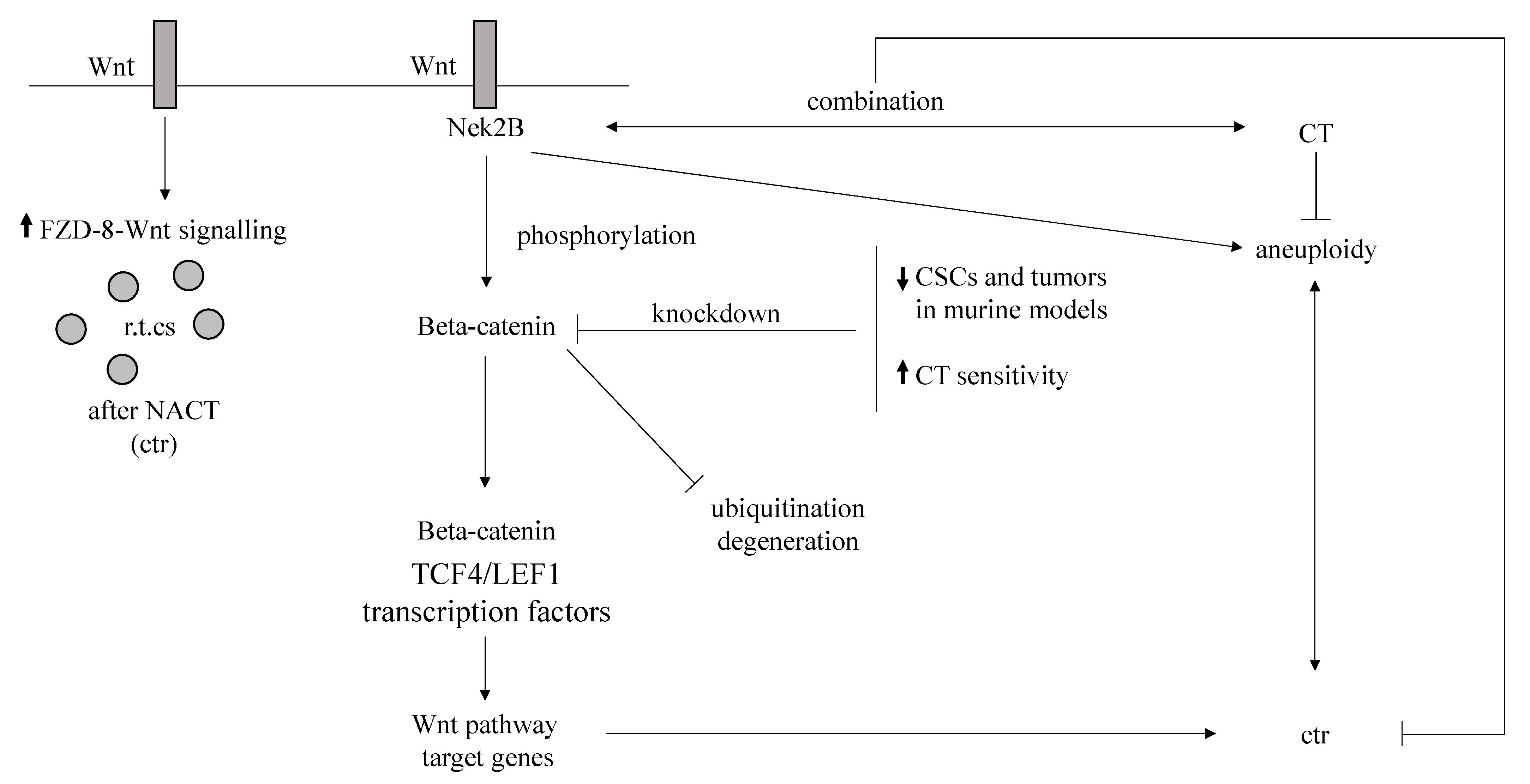
| Wnt/beta-catenin | Signaling deregulation | CSCs increase, beta-catenin synergistic effect with NeK2B FLD8-mediated signaling increase | [74,75,76,77,78] |
| Hedgehog (Hh) | Signaling activation by cytotoxic drugs | CSCs expansion through GLI1/2 activation, promotion of expression of ABC transporters | [31,79,80,81,82] |
| NF-kB | Overexpression | Apoptosis inhibition | [83,84,85,86] |
| PTEN and PI3K-AKT-mTOR (PAM) | Hyperactivation due to PTEN loss | PTEN loss, HIF-1 induction by Akt | [87,88,89,90] |
| JAK/STAT | STAT3 hyperexpression downstream of IL-6/8 extracellular ligands | STAT3-NFkB interaction, STAT3 HIF-1 and ABC transporters expression upregulation | [91,92,93,94,95,96,97] |
| A. | |||||
|---|---|---|---|---|---|
| Predictive Modality | Setting | Objective | References | ||
| CS, ES | Kind | Outcome | Drug | ||
| Histologic/Molecular subtype | |||||
| Metaplastic | CS | Neoadjuvant | Low pCR | Anthracycline/taxane-based NACT with or without carboplatin | [100] |
| LAR and MES | Carboplatin plus docetaxel | [104] | |||
| BL1 and BL2 | ES | NA | High proliferation | Cisplatin | [102] |
| TILs | |||||
| Whole TILs | CS | Neoadjuvant | High pCR (positive correlation) | Anthracycline/taxane-based NACT | [107,108,109,110] |
| TILs, PD-L1, CD73 (TNP) | [111] | ||||
| CD3+ cells | [112] | ||||
| CD4+, CD8+, FOXP3+ cells | [113] | ||||
| CD20+ cells | [105,107] | ||||
| NK cells | [114,115] | ||||
| Blood PMN neutrophils | [119] | ||||
| Blood DCs | [119] | ||||
| Biomarkers | |||||
| HE LncDLX6-AS1 | ES | NA | R | Cisplatin | [121] |
| HE miR-105 and miR-93-3p | [122] | ||||
| 321 miRNAs (including miR-34a) expression change | CS | Neoadjuvant | High pCR | Carboplatin/paclitaxel | [125] |
| High HRD score | Anthracycline and/or taxane-based NACT | [129] | |||
| HRD | Platinum-containing NACT | [131,132] | |||
| Low BRCA1-like score | ES | NA | R | Cisplatin, docetaxel | [133] |
| BRCAness | CS | Neoadjuvant | Low pCR | Taxane-based NACT | [134] |
| IL-6, CXCL8, VEGFA, EGR1, PTGS2, TRIB1 signature | ES | NA | R | Paclitaxel | [135] |
| LE CXCL8-CXCR1/2 axis | CS | Neoadjuvant | High pCR | Carboplatin plus paclitaxel | [136] |
| B. | |||||
| Predictive Modality | Setting | Objective | References | ||
| CS, ES | Kind | Outcome | Drug | ||
| Biomarkers | |||||
| HE SYTL4 | CS/ES | Neoadjuvant/NA | R | Paclitaxel | [137] |
| HE MITR | ES | NA | [138] | ||
| HE SERPINE1 | [139] | ||||
| HE TNFS13 | Paclitaxel, anthracycline | [140] | |||
| LE miR-5195-3p | Paclitaxel | [141] | |||
| HE miR-18a | CS | Neoadjuvant | Paclitaxel-containing NACT | [142] | |
| HE miR-1207-5p | ES | NA | Paclitaxel | [143] | |
| HE Long nc RNA MALAT-1 | CS | Neoadjuvant | Paclitaxel/doxorubicin | [144] | |
| HE CERK | CS/ES | Metastatic/NA | [149] | ||
| HE TMPRSS13 | ES | NA | Paclitaxel/carboplatin | [150] | |
| High PCDH17 methylation | CS | Neoadjuvant | Taxane/anthracycline-based NACT | [151] | |
| JARID2 mutation | Short DFS in patients without pCR | [152] | |||
| circUBE2D2/miR-512-3p/CDCA3 axis | ES | NA | R | Doxorubicin | [153] |
| HE miRNA-449 family | CS | Neoadjuvant | S | [154] | |
| HE miR-770 | ES | NA | [155] | ||
| LE miR221/222 and miR200 family | CS | ND | R | [156] | |
| A cluster of 15 overexpressed genes | ES | NA | [157] | ||
| HE PVT1 | [158] | ||||
| HE TRIM37 network | [159] | ||||
| HE NLRP3 | Gemcitabine | [160] | |||
| HE mir-620 | [161] | ||||
| NOP10 | CS | Adjuvant | Short OS | CMF-treated | [162] |
| A. | ||||
|---|---|---|---|---|
| Drug | Target/Mechanism of Action | CS/ES | Outcome | Reference/NCT Number |
| Currently recommended | ||||
| Olaparib | PARP inhibitor | Metastatic, in HER2 negative BC pts with a germline BRCA mutation (CS) | Higher objective RR and PFS | [165] |
| Talazoparib | Advanced, in BC pts with germline BRCA mutation (CS) | [166] | ||
| Larotrectinib | Inhibitor of tropomyosin receptor kinase (TRK) | Advanced, in NTRK gene fusion-positive solid tumours (CS) | ORR 71% | [170] |
| Entrectinib | ORR 57%; Median duration of response 10 months | [172] | ||
| Sacituzumab govitecan | Anti-Trop2 antibody drug conjugate | Metastatic, in heavily pretreated pts (CS) | RR 33.3%; median duration of response 7.7 months; clinical benefit rate 45.4%; median PFS 5.5 months; OS 13.0 months | [184] |
| Under investigation | ||||
| Galunisertib | TGF beta type I receptor inhibitor | Metastatic, in combination with CT (CS) | NA | NCT02672475 (phase I) |
| PF-03084014 | Gamma secretase inhibitor | Advanced, in combination with docetaxel (CS) | Median PFS 4.1 months | [186] |
| AL101 | Patient-derived xenografts with abnormal Notch signaling (ES) | Inhibition of tumor growth | [185] | |
| SRI33576, SRI35889 | wnt/beta-catenin inhibitors | Cell lines (ES) | Pro-apoptotic effects by downregulating LRP6 | [187] |
| Salinomycin | Breast CSCs (ES) | inhibition of proliferation, invasion, and self-renewal while inducing apoptosis | [185,186] | |
| CWP232228 | Xenograft models (ES) | Inhibition of tumor growth | [54] | |
| Clofazimine | Cells and xenograft models (ES) | inhibition of proliferation; | [189] | |
| Frizzled-7 protein antagonist (rhFzd7) | Cells and xenografts (ES) | Inhibition of proliferation, invasion, and angiogenesis while sensitizing cells to docetaxel | [190] | |
| LGK974 | Advanced, in pts with wnt-ligand dependent malignancies, including TNBC (CS) | NA | NCT01351103 (phase I) | |
| PTK7-ADC | Metastatic, in combination with gedatolisib (dual PI3K-mTORC1/2 inhibitor) (CS) | NCT03243331 (phase I) | ||
| B. | ||||
| Drug | Target/Mechanism of Action | CS/ES | Outcome | Reference/NCT Number |
| Under investigation | ||||
| GANT61 | Hh/direct GLI inhibitor | Cell lines (ES) | promoted apoptosis, reduced proliferation, and decreased CSC population | [192,193] |
| Plumbagin | Non-specific NF-kB inhibitor | Decreased cell viability and promoted apoptosis | [195] | |
| Genistein | NF-kB inhibitor | Anti-growth and pro-apoptotic effects | [196] | |
| DHMEQ | Nuclear translocation of NF-B inhibitor | Decreased growth and induction of apoptosis | [197] | |
| Everolimus | mTOR inhibitor | Advanced, in combination with carboplatin (CS) | NA | NCT02531932 (phase II) |
| Advanced, in combination with cisplatin (CS) | NCT01931163 (phase II) | |||
| BKM120 | PI3K inhibitor | Metastatic (CS) | NCT01629615 (phase II) | |
| Alpelisib | Neoadjuvant, in combination with nab-paclitaxel in anthracycline refractory pts with PIK3CA or PTEN alterations (CS) | NCT04216472 (phase I) | ||
| Ipatasertib | AKT inhibitor | Locally advanced/metastatic, first line (phase II), in combination with paclitaxel (CS) | Prolonged PFS and OS | [198] |
| Ipatasertib | Advanced, in PIK3CA/AKT1/PTEN-altered pts, in combination with paclitaxel versus placebo + paclitaxel (CS) | NA | NCT03337724 (phase III) | |
| Uprosertib | Metastatic, in combination with trametinib (CS) | NCT01964924 (phase II) | ||
| AZD5363 | Metastatic, in combination with CT (CS) | Prolonged OS | [201] | |
| Ruxolitinib | JAK1/2 inhibitor | Neoadjuvant, in combination with CT (CS) | NA | NCT02876302 (phase II) |
| C. | ||||
| Drug | Target/Mechanism of Action | CS/ES | Outcome | Reference/NCT Number |
| Under investigation | ||||
| AZD9150 | Antisense nucleotide inhibitor of STAT3 | Metastatic, in combination with durvalumab and paclitaxel (CS) | NA | NCT03742102 (phase I/II) |
| MEDI3039 | Apoptosis/DR agonist | In-vitro and in-murine models (ES) | Tumor growth inhibition | [205] |
| Debio 1143 | IAP antagonist | Advanced, solid tumors including TNBC (CS) | NA | NCT01078649, NCT01930292 (phase I) |
| LCL161 | SMAC analog | Neoadjuvant, in combination with paclitaxel (CS) | Doubled pCR rate in a group preselected for the tumor necrosis factor (TNF) gene expression profile | [205] |
| antisense-miRNA-21 and antisense-miRNA-10b co-delivery | Inhibition of oncogenic miRNAs | Murine models (ES) | reduced tumor growth | [207] |
| miR-mimic recombinant vectors | Restoration of tumor suppressor miRNAs | Cell line (ES) | Reduced migration and invasion | [209] |
| Panitumumab | anti-EGFR mAb | Neoadjuvant, in combination with CT | NA | NCT02876107 (phase II) NCT01036087 (phase II) |
| Apatinib | Anti-EGFR TKI | Advanced, alone or in combination with CT (CS) | NCT05019690 (phase I/II) NCT03932526 (phase II) NCT03254654 (phase II) | |
| Icotinib | Metastatic, pre-treated (CS) | Under evaluation | NCT02362230 (phase II) | |
| Bicalutamide | AR antagonist | Metastatic, AR-positive (CS) | six-month CBR 19%, median PFS 12 weeks | [221] |
| Abiraterone acetate | Advanced or metastatic, AR-positive pts, in combination with prednisone (CS) | six-month CBR 20.0%, ORR 6.7%, median PFS 2.8 months | [222] | |
| Enzalutamide | Locally advanced or metastatic AR-positive pts (CS) | 16 weeks CBR 33%, median PFS 3.3 months, median OS 17.6 months | [223] | |
| Darolutamide | Locally recurrent or metastatic, in AR-positive pts (CS) | NA | NCT03383679 (phase II) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, P.; Scatena, C.; Ghilli, M.; Bargagna, I.; Lorenzini, G.; Nicolini, A. Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC. Int. J. Mol. Sci. 2022, 23, 1665. https://doi.org/10.3390/ijms23031665
Ferrari P, Scatena C, Ghilli M, Bargagna I, Lorenzini G, Nicolini A. Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC. International Journal of Molecular Sciences. 2022; 23(3):1665. https://doi.org/10.3390/ijms23031665
Chicago/Turabian StyleFerrari, Paola, Cristian Scatena, Matteo Ghilli, Irene Bargagna, Giulia Lorenzini, and Andrea Nicolini. 2022. "Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC" International Journal of Molecular Sciences 23, no. 3: 1665. https://doi.org/10.3390/ijms23031665
APA StyleFerrari, P., Scatena, C., Ghilli, M., Bargagna, I., Lorenzini, G., & Nicolini, A. (2022). Molecular Mechanisms, Biomarkers and Emerging Therapies for Chemotherapy Resistant TNBC. International Journal of Molecular Sciences, 23(3), 1665. https://doi.org/10.3390/ijms23031665