
Different Cell Responses to Hinokitiol Treatment Result in Senescence or Apoptosis in Human Osteosarcoma Cell Lines

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
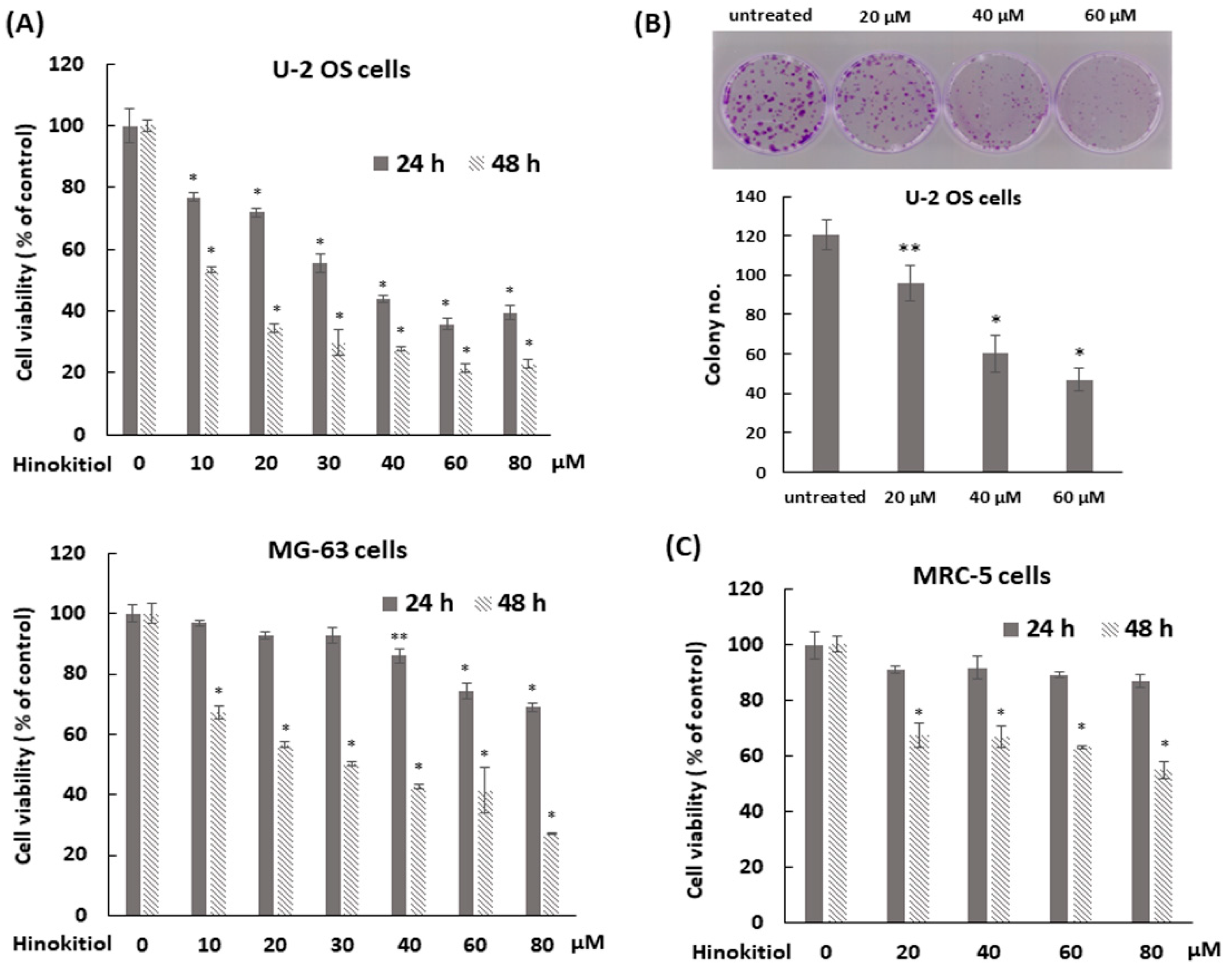
2.1. Hinokitiol Reduces Viability and Proliferation of Osteosarcoma Cell Lines
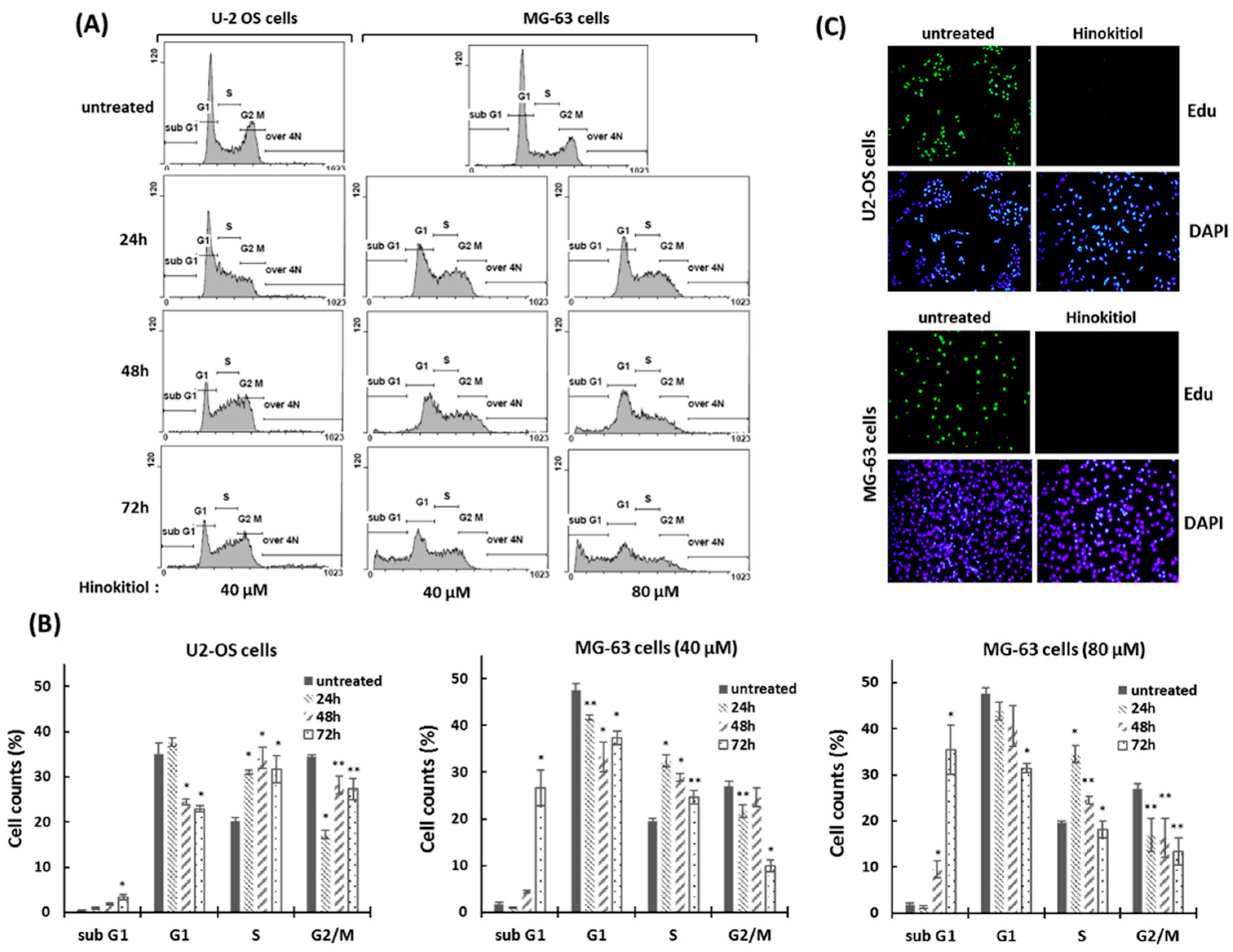
2.2. Hinokitiol Induces S-Phase Arrest of Cell Cycle and DNA Damage Response
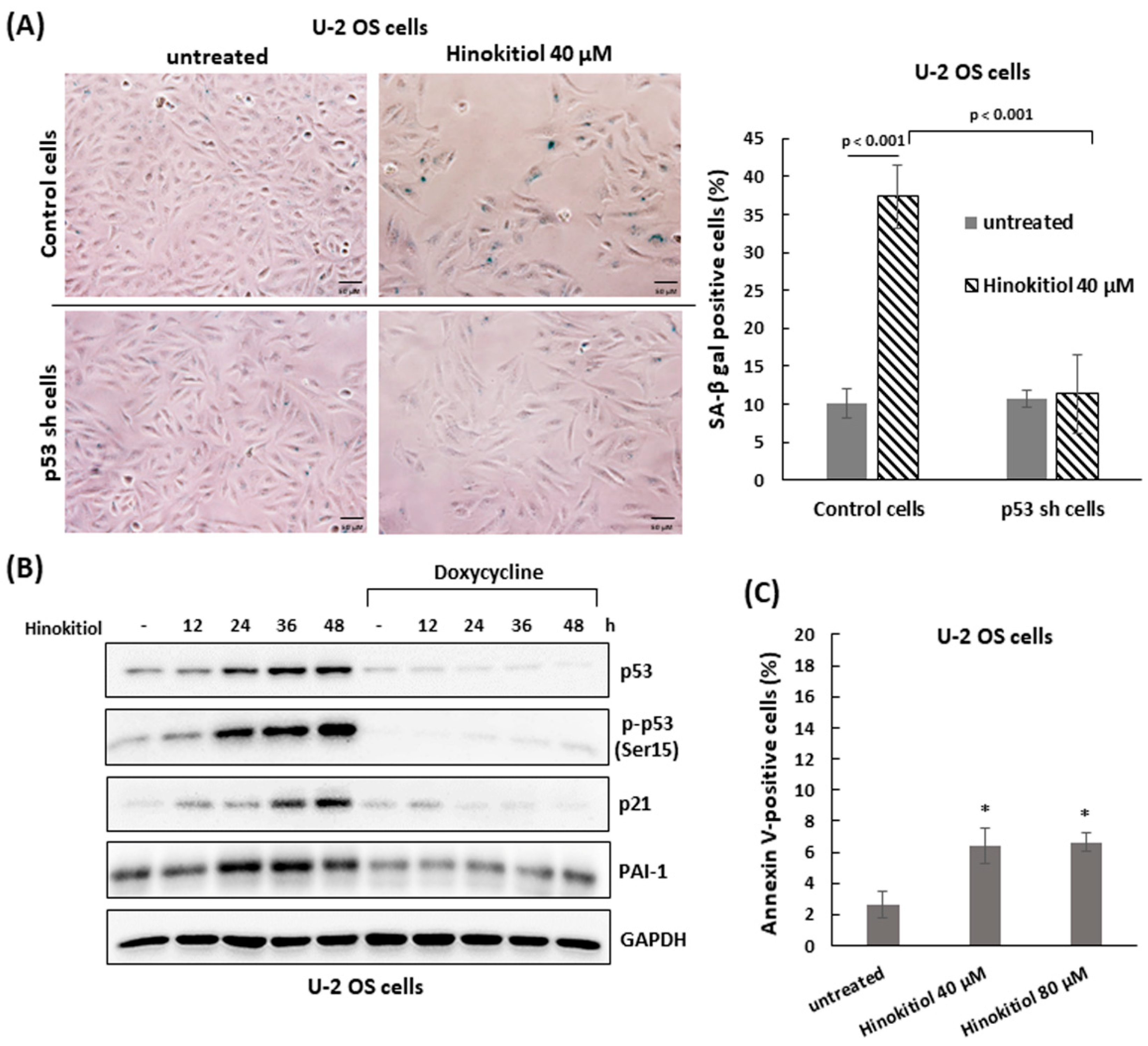
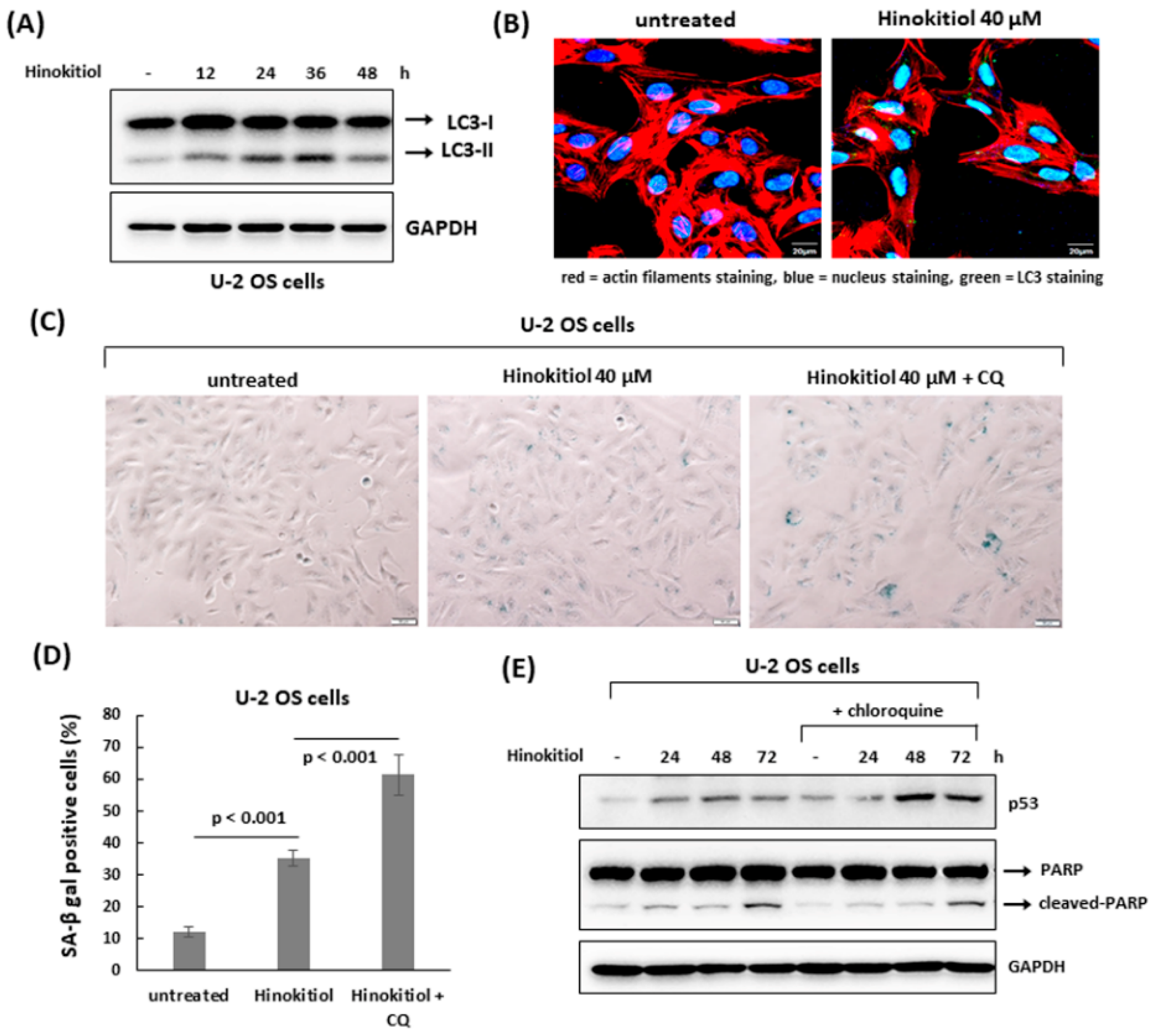
2.3. Hinokitiol Induces Senescence in U-2 OS Cells
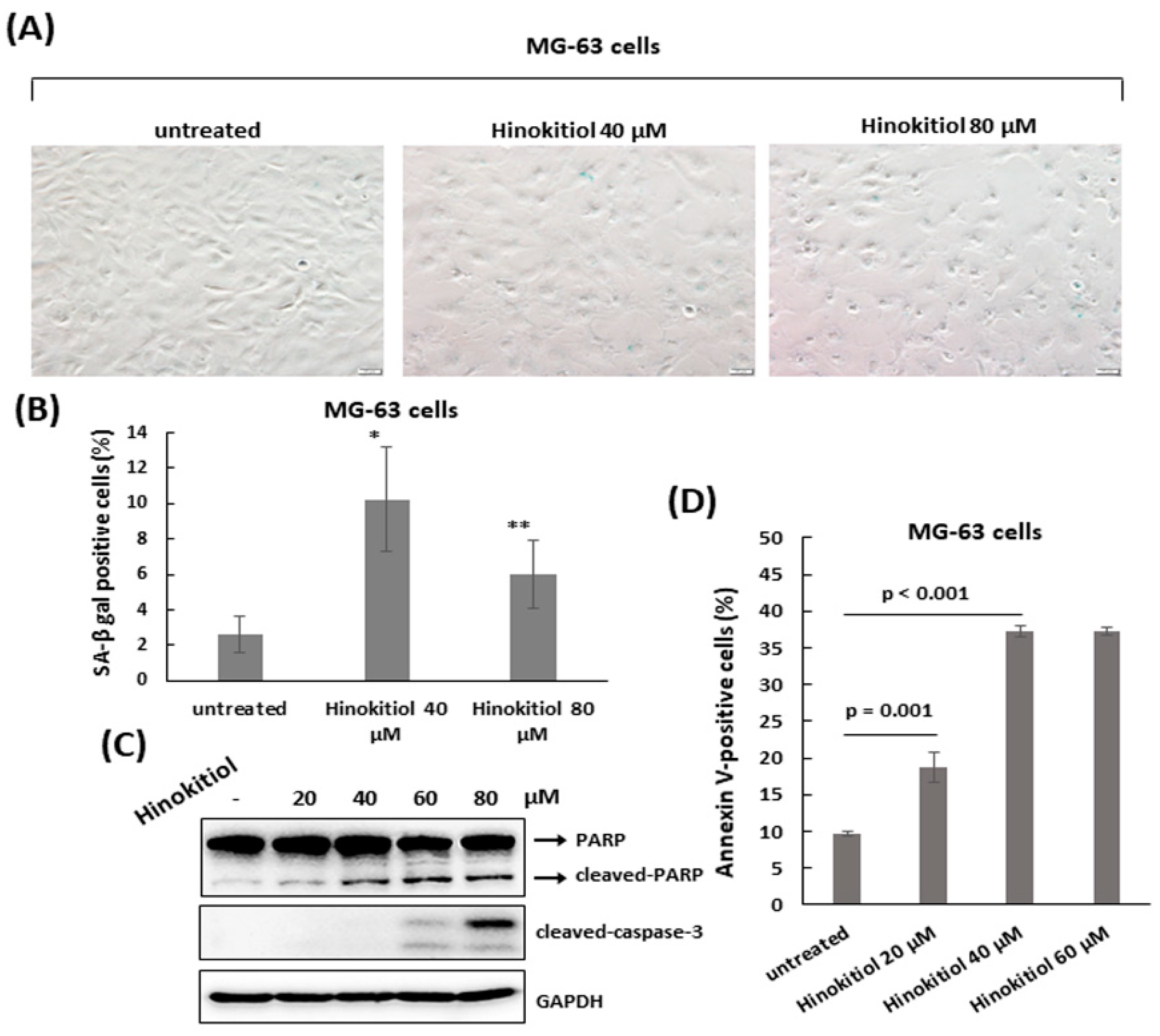
2.4. Hinokitiol Triggers Apoptosis in MG-63 Cells
2.5. The Suppression of Autophagy Enhances Hinokitiol-Induced Response
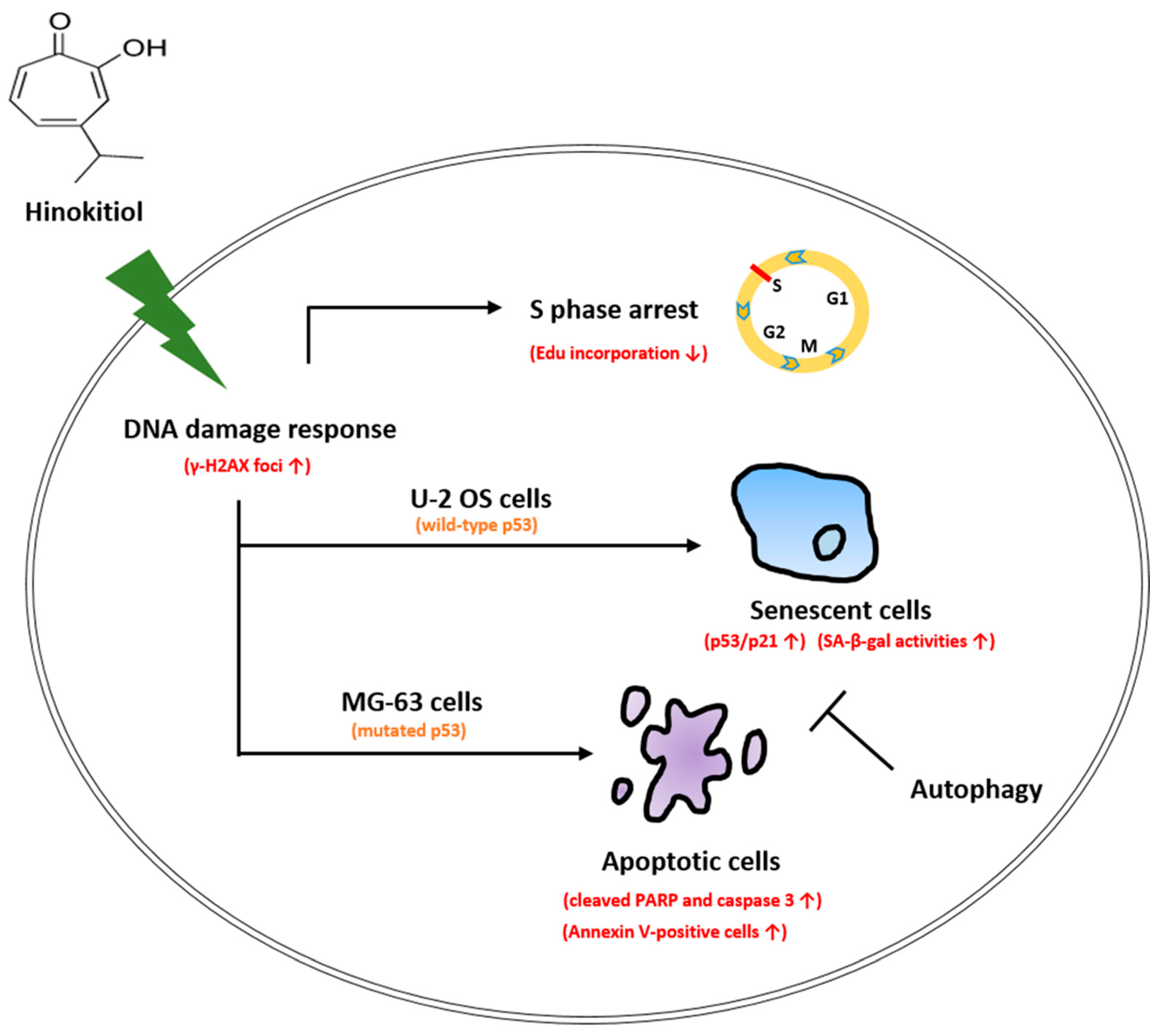
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Colony Formation Assay
4.3. Cell Viability Assay
4.4. Flow Cytometry Analysis
4.5. Apoptosis Assay
4.6. Cell Lysis and Immunoblotting
4.7. EdU Incorporation Assay
4.8. Immunofluorescence Staining
4.9. Senescence-Associated Beta-Galactosidase (SA-β-gal) Staining
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y.; Yang, J.; Zhao, N.; Wang, C.; Kamar, S.; Zhou, Y.; He, Z.; Yang, J.; Sun, B.; Shi, X.; et al. Progress in the chemotherapeutic treatment of osteosarcoma. Oncol. Lett. 2018, 16, 6228–6237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belayneh, R.; Fourman, M.S.; Bhogal, S.; Weiss, K.R. Update on Osteosarcoma. Curr. Oncol. Rep. 2021, 23, 71. [Google Scholar] [CrossRef] [PubMed]
- Domon, H.; Hiyoshi, T.; Maekawa, T.; Yonezawa, D.; Tamura, H.; Kawabata, S.; Yanagihara, K.; Kimura, O.; Kunitomo, E.; Terao, Y. Antibacterial activity of hinokitiol against both antibiotic-resistant and -susceptible pathogenic bacteria that predominate in the oral cavity and upper airways. Microbiol. Immunol. 2019, 63, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Suzuki, R.; Murata, I.; Nomura, H.; Isshiki, Y.; Kanamoto, I. Evaluation of Antibacterial Activity Expression of the Hinokitiol/Cyclodextrin Complex Against Bacteria. ACS Omega 2020, 5, 27180–27187. [Google Scholar] [CrossRef]
- Wei, K.C.; Chen, R.F.; Chen, Y.F.; Lin, C.H. Hinokitiol suppresses growth of B16 melanoma by activating ERK/MKP3/proteosome pathway to downregulate survivin expression. Toxicol. Appl. Pharmacol. 2019, 366, 35–45. [Google Scholar] [CrossRef]
- Jayakumar, T.; Liu, C.H.; Wu, G.Y.; Lee, T.Y.; Manubolu, M.; Hsieh, C.Y.; Yang, C.H.; Sheu, J.R. Hinokitiol Inhibits Migration of A549 Lung Cancer Cells via Suppression of MMPs and Induction of Antioxidant Enzymes and Apoptosis. Int. J. Mol. Sci. 2018, 19, 939. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Cheng, W.P.; Chiang, Y.F.; Hong, Y.H.; Ali, M.; Huang, T.C.; Wang, K.L.; Shieh, T.M.; Chang, H.Y.; Hsia, S.M. Hinokitiol Exhibits Antitumor Properties through Induction of ROS-Mediated Apoptosis and p53-Driven Cell-Cycle Arrest in Endometrial Cancer Cell Lines (Ishikawa, HEC-1A, KLE). Int. J. Mol. Sci. 2021, 22, 8268. [Google Scholar] [CrossRef]
- Zhang, G.; He, J.; Ye, X.; Zhu, J.; Hu, X.; Shen, M.; Ma, Y.; Mao, Z.; Song, H.; Chen, F. beta-Thujaplicin induces autophagic cell death, apoptosis, and cell cycle arrest through ROS-mediated Akt and p38/ERK MAPK signaling in human hepatocellular carcinoma. Cell Death Dis. 2019, 10, 255. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ko, J.; Kim, J.T.; Cho, J.S.; Qiu, S.; Kim, G.D.; Auh, J.H.; Lee, H.J. beta-Thujaplicin inhibits basal-like mammary tumor growth by regulating glycogen synthase kinase-3beta/beta-catenin signaling. Food Funct. 2019, 10, 2691–2700. [Google Scholar] [CrossRef]
- Wu, Y.J.; Hsu, W.J.; Wu, L.H.; Liou, H.P.; Pangilinan, C.R.; Tyan, Y.C.; Lee, C.H. Hinokitiol reduces tumor metastasis by inhibiting heparanase via extracellular signal-regulated kinase and protein kinase B pathway. Int. J. Med. Sci. 2020, 17, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Choi, K.M.; Kim, W.; Jeon, Y.S.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; Yoo, H.S. Hinokitiol inhibits cell growth through induction of S-phase arrest and apoptosis in human colon cancer cells and suppresses tumor growth in a mouse xenograft experiment. J. Nat. Prod. 2013, 76, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.C.; Liao, Y.W.; Chen, P.N.; Lu, K.H.; Yu, C.C.; Hsieh, P.L. Hinokitiol suppresses cancer stemness and oncogenicity in glioma stem cells by Nrf2 regulation. Cancer Chemother. Pharmacol. 2017, 80, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Mongiardi, M.P.; Pellegrini, M.; Pallini, R.; Levi, A.; Falchetti, M.L. Cancer Response to Therapy-Induced Senescence: A Matter of Dose and Timing. Cancers 2021, 13, 484. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, L.; El-Deiry, W.S. Differential p53-Mediated Cellular Responses to DNA-Damaging Therapeutic Agents. Int. J. Mol. Sci. 2021, 22, 1828. [Google Scholar] [CrossRef] [PubMed]
- Synoradzki, K.J.; Bartnik, E.; Czarnecka, A.M.; Fiedorowicz, M.; Firlej, W.; Brodziak, A.; Stasinska, A.; Rutkowski, P.; Grieb, P. TP53 in Biology and Treatment of Osteosarcoma. Cancers 2021, 13, 4284. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Borrero, L.J.; El-Deiry, W.S. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Chang, M.D.; Shieh, S.Y. TTK/hMps1 mediates the p53-dependent postmitotic checkpoint by phosphorylating p53 at Thr18. Mol. Cell. Biol. 2009, 29, 2935–2944. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, J.; Zhang, K.; Guo, Y. TP53 Mutations and Survival in Osteosarcoma Patients: A Meta-Analysis of Published Data. Dis. Markers 2016, 2016, 4639575. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Guo, A.; Li, Q.; Wang, D. Meta-analysis of clinical significance of p53 protein expression in patients with osteosarcoma. Future Oncol. 2017, 13, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Xu, Y.F.; Lou, L.X.; Jin, K.; Miao, Q.; Ye, X.; Xi, Y. Anti-inflammatory effects of hinokitiol on human corneal epithelial cells: An in vitro study. Eye (Lond.) 2015, 29, 964–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.H.; Wu, P.; Lee, J.Y.; Li, P.R.; Hsieh, W.Y.; Ho, C.C.; Ho, C.L.; Chen, W.J.; Wang, C.C.; Yen, M.Y.; et al. Hinokitiol induces DNA damage and autophagy followed by cell cycle arrest and senescence in gefitinib-resistant lung adenocarcinoma cells. PLoS ONE 2014, 9, e104203. [Google Scholar] [CrossRef]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Wang, W.K.; Lin, S.T.; Chang, W.W.; Liu, L.W.; Li, T.Y.; Kuo, C.Y.; Hsieh, J.L.; Lee, C.H. Hinokitiol induces autophagy in murine breast and colorectal cancer cells. Environ. Toxicol. 2016, 31, 77–84. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Shih, Y.H.; Lin, D.J.; Chang, K.W.; Hsia, S.M.; Ko, S.Y.; Lee, S.Y.; Hsue, S.S.; Wang, T.H.; Chen, Y.L.; Shieh, T.M. Evaluation physical characteristics and comparison antimicrobial and anti-inflammation potentials of dental root canal sealers containing hinokitiol in vitro. PLoS ONE 2014, 9, e94941. [Google Scholar] [CrossRef] [Green Version]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-Inflammatory Drugs as Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.T.; Huang, Y.F.; Hsieh, C.P.; Wu, C.C.; Shen, T.S. JNK Inactivation Induces Polyploidy and Drug-Resistance in Coronarin D-Treated Osteosarcoma Cells. Molecules 2018, 23, 2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.S.; Hsu, Y.K.; Huang, Y.F.; Chen, H.Y.; Hsieh, C.P.; Chen, C.L. Licochalcone A Suppresses the Proliferation of Osteosarcoma Cells through Autophagy and ATM-Chk2 Activation. Molecules 2019, 24, 2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.-C.; Chen, H.-Y.; Chuang, W.-L.; Wang, H.-C.; Hsieh, C.-P.; Huang, Y.-F. Different Cell Responses to Hinokitiol Treatment Result in Senescence or Apoptosis in Human Osteosarcoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 1632. https://doi.org/10.3390/ijms23031632
Yang S-C, Chen H-Y, Chuang W-L, Wang H-C, Hsieh C-P, Huang Y-F. Different Cell Responses to Hinokitiol Treatment Result in Senescence or Apoptosis in Human Osteosarcoma Cell Lines. International Journal of Molecular Sciences. 2022; 23(3):1632. https://doi.org/10.3390/ijms23031632
Chicago/Turabian StyleYang, Shun-Cheng, Hsuan-Ying Chen, Wan-Ling Chuang, Hui-Chun Wang, Cheng-Pu Hsieh, and Yi-Fu Huang. 2022. "Different Cell Responses to Hinokitiol Treatment Result in Senescence or Apoptosis in Human Osteosarcoma Cell Lines" International Journal of Molecular Sciences 23, no. 3: 1632. https://doi.org/10.3390/ijms23031632
APA StyleYang, S.-C., Chen, H.-Y., Chuang, W.-L., Wang, H.-C., Hsieh, C.-P., & Huang, Y.-F. (2022). Different Cell Responses to Hinokitiol Treatment Result in Senescence or Apoptosis in Human Osteosarcoma Cell Lines. International Journal of Molecular Sciences, 23(3), 1632. https://doi.org/10.3390/ijms23031632