
MHY2245, a Sirtuin Inhibitor, Induces Cell Cycle Arrest and Apoptosis in HCT116 Human Colorectal Cancer Cells

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
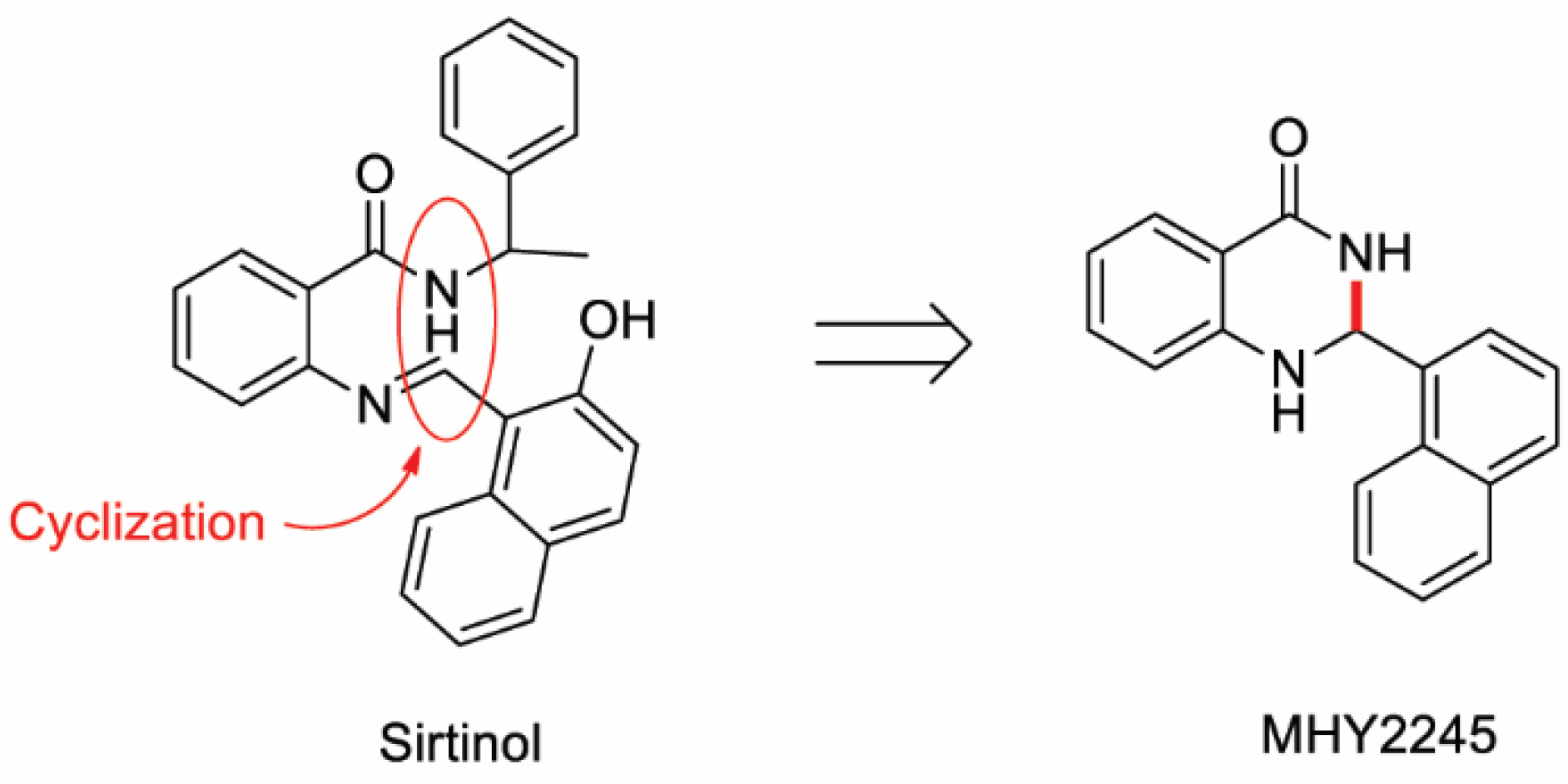
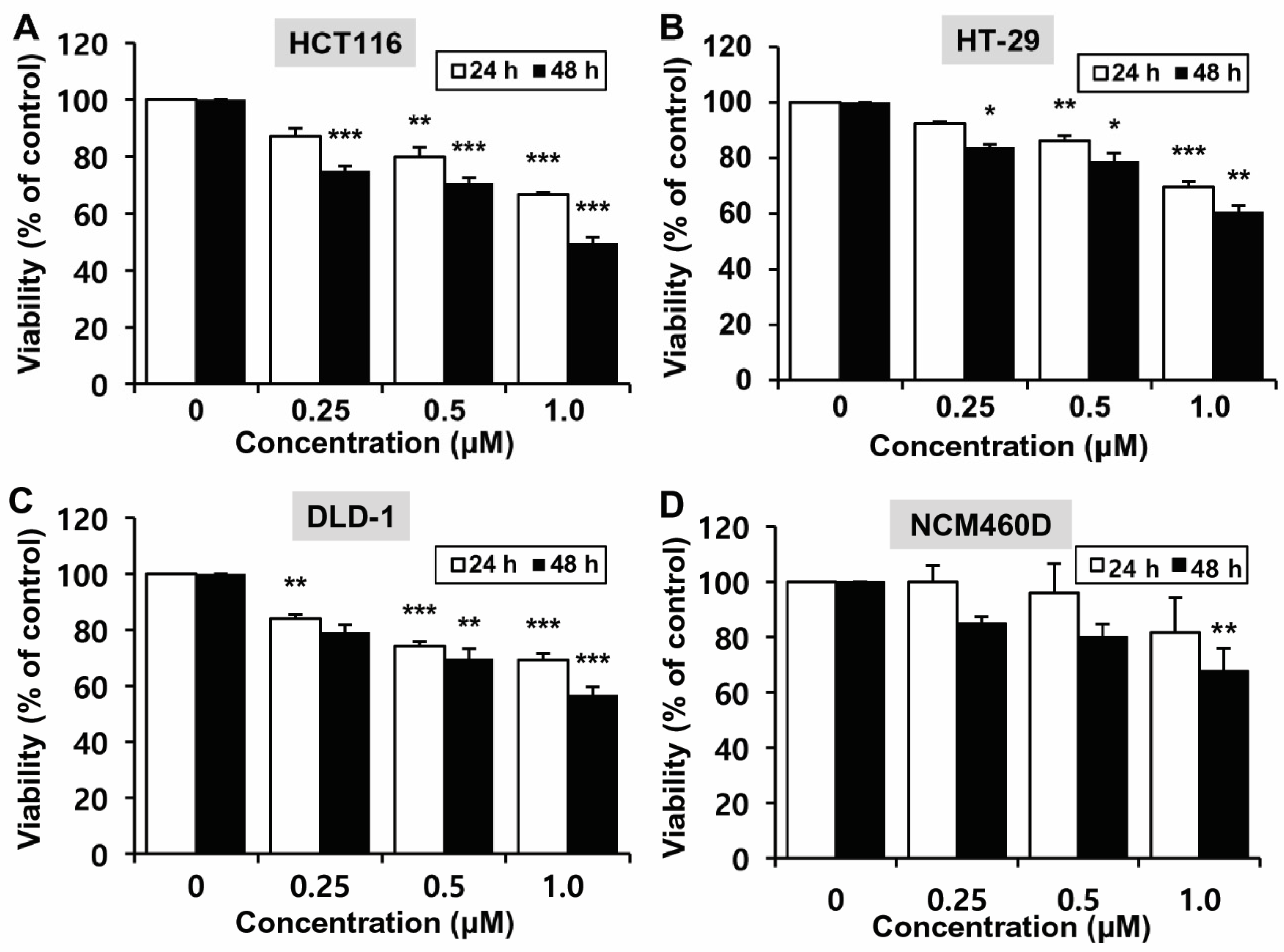
2.1. Effects of MHY2245 on the Growth of HCT116 Cells
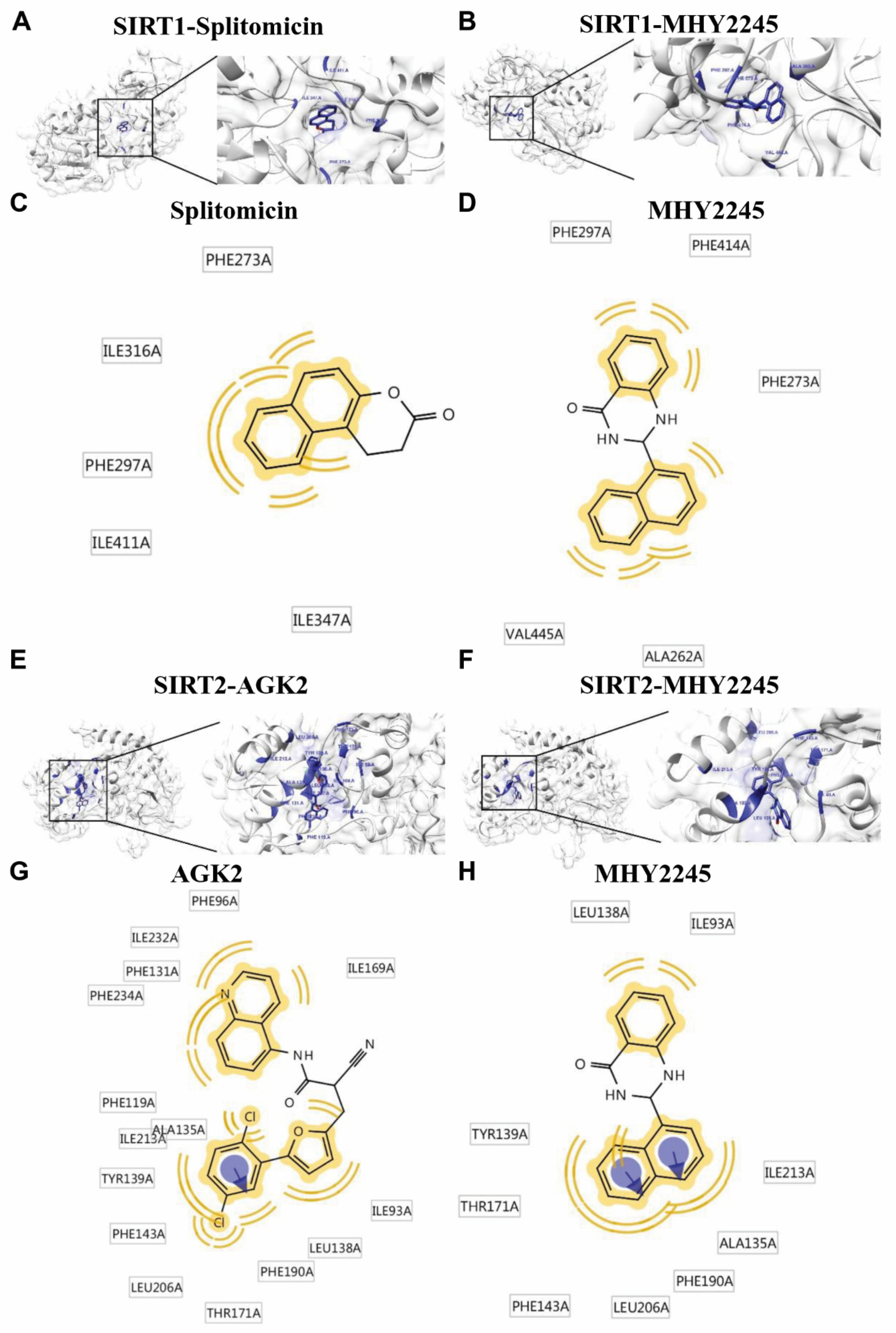
2.2. Molecular Docking Simulation of SIRT1/2 with MHY2245
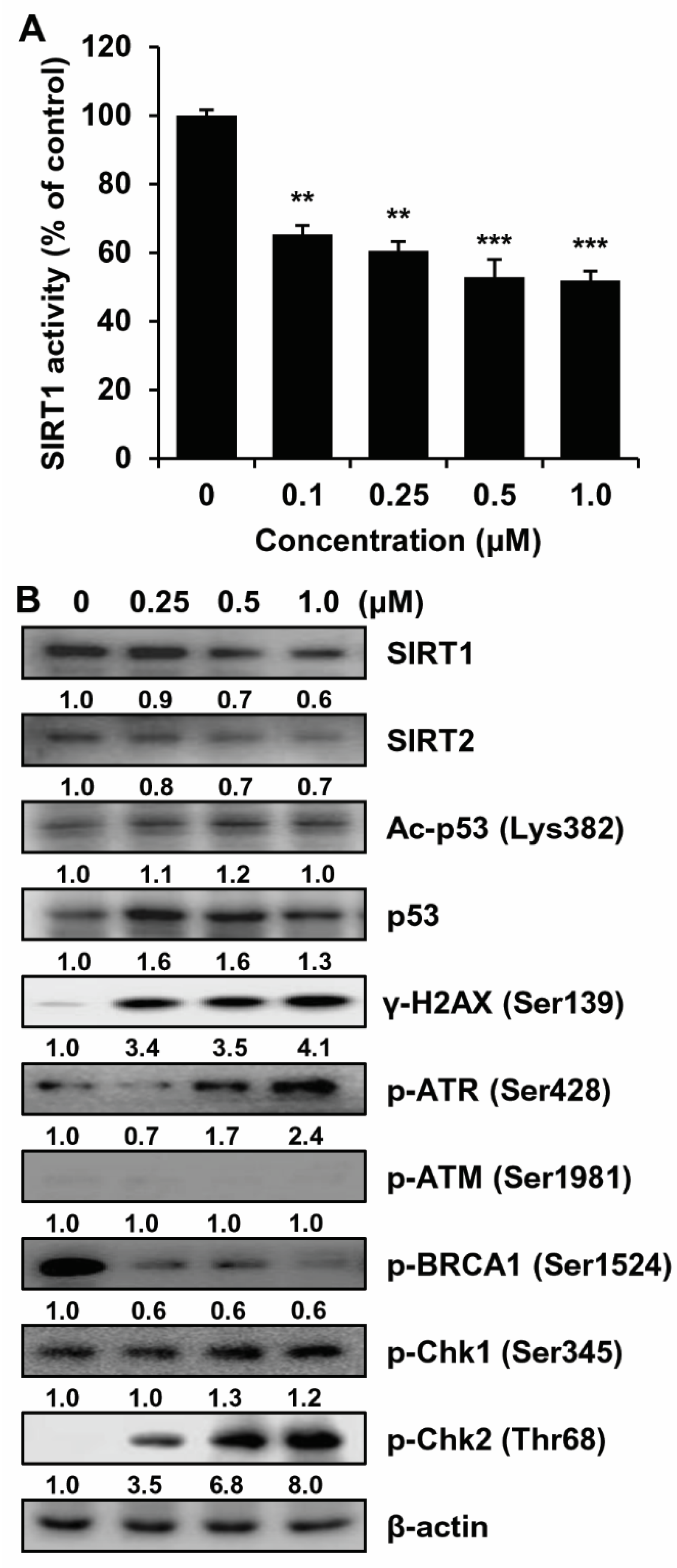
2.3. SIRT1/2 Inhibitory Effect and Induction of DNA Damage of MHY2245 in HCT116 Cells
2.4. Effects of MHY2245 on the Cell Cycle in HCT116 Cells
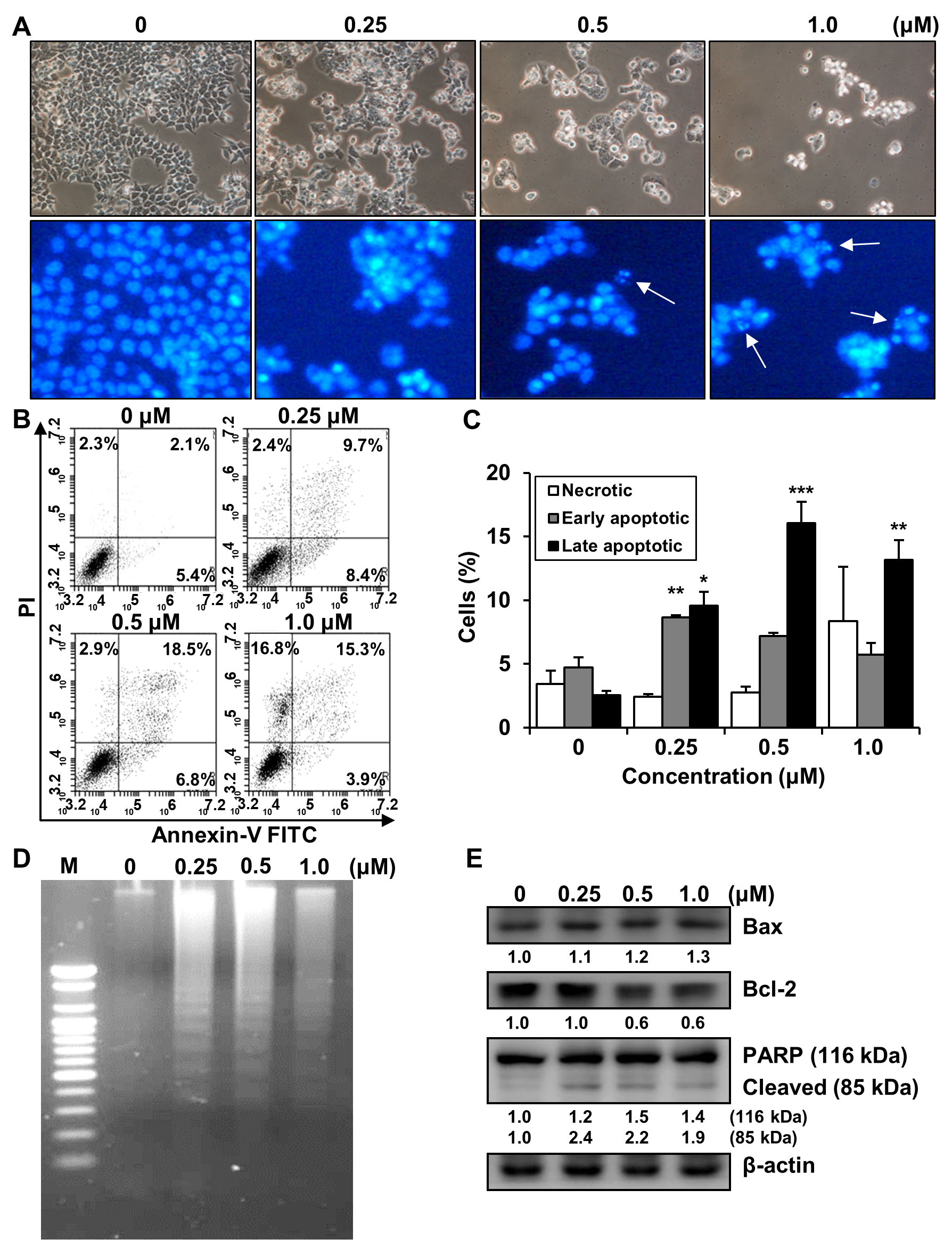
2.5. Effects of MHY2245 on the Induction of Apoptosis in HCT116 Cells
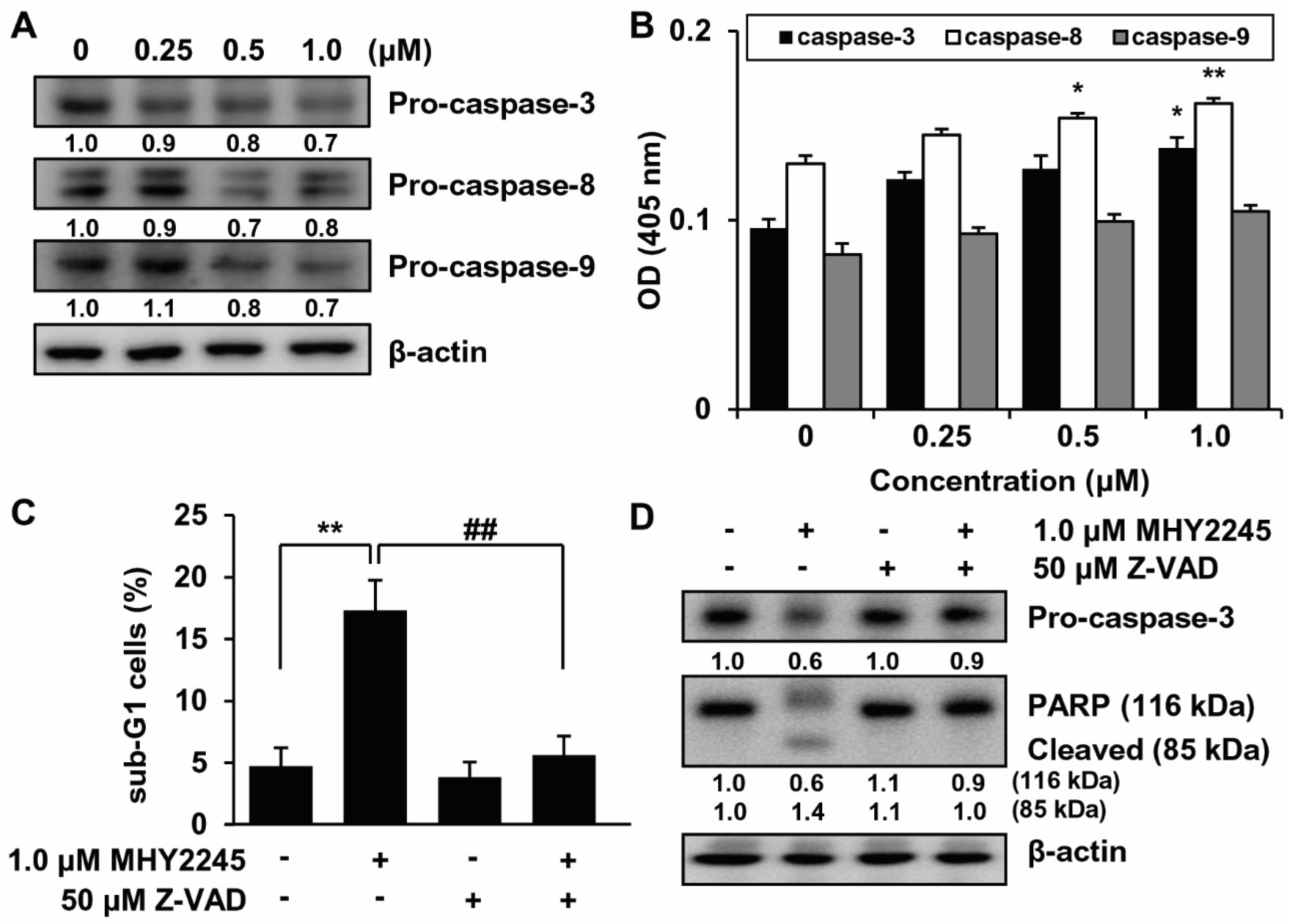
2.6. Effects of MHY2245 on Caspase Activation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Docking Simulation of SIRT1/2 and MHY2245
4.3. SIRT1 Deacetylase Activity Assay
4.4. Cell Culture and Viability Study
4.5. Cell Cycle Analysis
4.6. Nuclear Staining with Hoechst 33342
4.7. Annexin V-Fluorescein Isothiocyanate (FITC)/PI Double Staining
4.8. DNA Fragmentation Assay
4.9. Caspase Activity
4.10. Western Blot Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.W.; Won, Y.J.; Hong, S.; Kong, H.J.; Lee, E.S. Prediction of cancer incidence and mortality in Korea, 2020. Cancer Res. Treat. 2020, 52, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; Cassidy, J. Chemotherapy in metastatic colorectal cancer. Surg. Oncol. 2007, 16, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.C.; Shih, T.Y.; Kuo, C.L.; Ma, Y.S.; Yang, J.L.; Wu, P.P.; Huang, Y.P.; Lai, K.C.; Chung, J.G. Sulforaphane induces cell death through G2/M phase arrest and triggers apoptosis in HCT 116 human colon cancer cells. Am. J. Chin. Med. 2016, 44, 1289–1310. [Google Scholar] [CrossRef] [PubMed]
- Olmos, Y.; Brosens, J.J.; Lam, E.W. Interplay between SIRT proteins and tumour suppressor transcription factors in chemotherapeutic resistance of cancer. Drug Resist. Updat. 2011, 14, 35–44. [Google Scholar] [CrossRef]
- Villalba, J.M.; Alcain, F.J. Sirtuin activators and inhibitors. Biofactors 2012, 38, 349–359. [Google Scholar] [CrossRef]
- Matsushita, N.; Takami, Y.; Kimura, M.; Tachiiri, S.; Ishiai, M.; Nakayama, T.; Takata, M. Role of NAD-dependent deacetylases SIRT1 and SIRT2 in radiation and cisplatin-induced cell death in vertebrate cells. Genes Cells 2005, 10, 321–332. [Google Scholar] [CrossRef]
- Peck, B.; Chen, C.Y.; Ho, K.K.; Di Fruscia, P.; Myatt, S.S.; Coombes, R.C.; Fuchter, M.J.; Hsiao, C.D.; Lam, E.W. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 2010, 9, 844–855. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Yuan, Z.; Li, Y.; Ling, H.; Izumi, V.; Fang, B.; Fukasawa, K.; Koomen, J.; Chen, J.; Seto, E. Ubiquitinated sirtuin 1 (SIRT1) function is modulated during DNA damage-induced cell death and survival. J. Biol. Chem. 2015, 290, 8904–8912. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.T.; Gu, W. SIRT1: Regulator of p53 deacetylation. Genes Cancer 2013, 4, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Solomon, J.M.; Pasupuleti, R.; Xu, L.; McDonagh, T.; Curtis, R.; DiStefano, P.S.; Huber, L.J. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 2006, 26, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhrmann, C.; Shayan, P.; Popper, B.; Goel, A.; Shakibaei, M. Sirt1 is required for resveratrol-mediated chemopreventive effects in colorectal cancer cells. Nutrients 2016, 8, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhrmann, C.; Shayan, P.; Goel, A.; Shakibaei, M. Resveratrol regulates colorectal cancer cell invasion by modulation of focal adhesion molecules. Nutrients 2017, 9, 1073. [Google Scholar] [CrossRef] [PubMed]
- Brockmueller, A.; Sameri, S.; Liskova, A.; Zhai, K.; Varghese, E.; Samuel, S.M.; Büsselberg, D.; Kubatka, P.; Shakibaei, M. Resveratrol’s anti-cancer effects through the modulation of tumor glucose metabolism. Cancers 2021, 13, 188. [Google Scholar] [CrossRef] [PubMed]
- Bedalov, A.; Gatbonton, T.; Irvine, W.P.; Gottschling, D.E.; Simon, J.A. Identification of a small molecule inhibitor of Sir2p. Proc. Natl. Acad. Sci. USA 2001, 98, 15113–15118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaharski, A.J.; Rine, J.; Marshall, B.L.; Babiarz, J.; Zhang, L.; Verdin, E.; Smith, M.T. The flavoring agent dihydrocoumarin reverses epigenetic silencing and inhibits sirtuin deacetylases. PLoS Genet. 2005, 1, e77. [Google Scholar] [CrossRef]
- Napper, A.D.; Hixon, J.; McDonagh, T.; Keavey, K.; Pons, J.F.; Barker, J.; Yau, W.T.; Amouzegh, P.; Flegg, A.; Hamelin, E.; et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 2005, 48, 8045–8054. [Google Scholar] [CrossRef] [PubMed]
- Heltweg, B.; Gatbonton, T.; Schuler, A.D.; Posakony, J.; Li, H.; Goehle, S.; Kollipara, R.; Depinho, R.A.; Gu, Y.; Simon, J.A.; et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006, 66, 4368–4377. [Google Scholar] [CrossRef] [Green Version]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell. Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Chao, E.D.; Blackwell, H.E.; Moazed, D.; Schreiber, S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001, 276, 38837–38843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.C.; Liao, C.H.; Chang, Y.W.; Liou, J.T.; Day, Y.J. A new insight of anti-platelet effects of sirtinol in platelets aggregation via cyclic AMP phosphodiesterase. Biochem. Pharmacol. 2009, 77, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.F.; Sheu, J.R.; Lin, C.H.; Yang, D.S.; Hsiao, G.; Ou, G.; Chiu, P.T.; Huang, Y.H.; Kuo, W.H.; Hsu, M.J. Trichostatin A and sirtinol suppressed survivin expression through AMPK and p38MAPK in HT29 colon cancer cells. Biochim. Biophys. Acta 2012, 1820, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Tokunaga, E.; Chang, K.; Hikasa, M.; Iijima, K.; Eto, M.; Kozaki, K.; Akishita, M.; Ouchi, Y.; Kaneki, M. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 2006, 25, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kim, T.H.; Ahn, M.Y.; Lee, J.; Jung, J.H.; Choi, W.S.; Lee, B.M.; Yoon, K.S.; Yoon, S.; Kim, H.S. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 1101–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozako, T.; Aikawa, A.; Shoji, T.; Fujimoto, T.; Yoshimitsu, M.; Shirasawa, S.; Tanaka, H.; Honda, S.; Shimeno, H.; Arima, N.; et al. High expression of the longevity gene product SIRT1 and apoptosis induction by sirtinol in adult T-cell leukemia cells. Int. J. Cancer 2012, 131, 2044–2055. [Google Scholar] [CrossRef]
- Fong, Y.; Lin, Y.C.; Wu, C.Y.; Wang, H.M.; Lin, L.L.; Chou, H.L.; Teng, Y.N.; Yuan, S.S.; Chiu, C.C. The antiproliferative and apoptotic effects of sirtinol, a sirtuin inhibitor on human lung cancer cells by modulating Akt/beta-catenin-Foxo3a axis. Sci. World J. 2014, 2014, 937051. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Massa, S.; Lavu, S.; Pezzi, R.; Simeoni, S.; Ragno, R.; Mariotti, F.R.; Chiani, F.; Camilloni, G.; Sinclair, D.A. Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J. Med. Chem. 2005, 48, 7789–7795. [Google Scholar] [CrossRef]
- Dar, B.A.; Sahu, A.K.; Patidar, P.; Sharma, P.R.; Mupparapu, N.; Vyas, D.; Maity, S.; Sharma, M.; Singh, B. Heteropolyacid-clay nano-composite as a novel heterogeneous catalyst for the synthesis of 2,3-dihydroquinazolinones. J. Ind. Eng. Chem. 2013, 19, 407–412. [Google Scholar] [CrossRef]
- Hu, J.; Jing, H.; Lin, H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 2014, 6, 945–966. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Rassool, F.V. HDAC inhibitors: Roles of DNA damage and repair. Adv. Cancer Res. 2012, 116, 87–129. [Google Scholar] [CrossRef] [PubMed]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The role of SIRT1 on DNA damage response and epigenetic alterations in cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y. Caspase activation, inhibition, and reactivation: A mechanistic view. Protein Sci 2004, 13, 1979–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liszt, G.; Ford, E.; Kurtev, M.; Guarente, L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005, 280, 21313–21320. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Liu, P.Y.; Marshall, G.M. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009, 69, 1702–1705. [Google Scholar] [CrossRef] [Green Version]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef]
- Lim, C.S. Human SIRT1: A potential biomarker for tumorigenesis? Cell Biol. Int. 2007, 31, 636–637. [Google Scholar] [CrossRef]
- Kabra, N.; Li, Z.; Chen, L.; Li, B.; Zhang, X.; Wang, C.; Yeatman, T.; Coppola, D.; Chen, J. SirT1 is an inhibitor of proliferation and tumor formation in colon cancer. J. Biol. Chem. 2009, 284, 18210–18217. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Shen, Z.; Zhang, J.; Zhang, H.; Dong, J.; Yan, Y.; Liu, F.; Jiang, K.; Ye, Y.; Wang, S. Clinicopathological significance of SIRT1 expression in colorectal adenocarcinoma. Med Oncol. 2014, 31, 965. [Google Scholar] [CrossRef]
- Stunkel, W.; Peh, B.K.; Tan, Y.C.; Nayagam, V.M.; Wang, X.; Salto-Tellez, M.; Ni, B.; Entzeroth, M.; Wood, J. Function of the SIRT1 protein deacetylase in cancer. Biotechnol. J. 2007, 2, 1360–1368. [Google Scholar] [CrossRef]
- Lim, C.S. SIRT1: Tumor promoter or tumor suppressor? Med. Hypotheses 2006, 67, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Huffman, D.M.; Grizzle, W.E.; Bamman, M.M.; Kim, J.S.; Eltoum, I.A.; Elgavish, A.; Nagy, T.R. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007, 67, 6612–6618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tae, I.H.; Son, J.Y.; Lee, S.H.; Ahn, M.Y.; Yoon, K.; Yoon, S.; Moon, H.R.; Kim, H.S. A new SIRT1 inhibitor, MHY2245, induces autophagy and inhibits energy metabolism via PKM2/mTOR pathway in human ovarian cancer cells. Int. J. Biol. Sci. 2020, 16, 1901–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiyama, K.O.; Sakaguchi, K.; Kimura, S. DNA damage response in plants: Conserved and variable response compared to animals. Biology 2013, 2, 1338–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.J.; Baltimore, D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003, 17, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Friedenson, B. The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer 2007, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Sahu, R.P.; Batra, S.; Srivastava, S.K. Activation of ATM/Chk1 by curcumin causes cell cycle arrest and apoptosis in human pancreatic cancer cells. Br. J. Cancer 2009, 100, 1425–1433. [Google Scholar] [CrossRef] [Green Version]
- Karanika, S.; Karantanos, T.; Li, L.; Corn, P.G.; Thompson, T.C. DNA damage response and prostate cancer: Defects, regulation and therapeutic implications. Oncogene 2015, 34, 2815–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.H.; Huang, F.B.; Gao, J.P.; Liu, M.L.; Pang, W.B.; Li, W.; Yan, X.Y.; Huo, M.J.; Yang, X. Effects of fluoride on DNA damage and caspase-mediated apoptosis in the liver of rats. Biol. Trace Elem. Res. 2015, 166, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.S.; Lv, L.X.; Zhao, Z.; He, X.; You, L.; Liu, J.K.; Li, Y.Q. Cordycepol C induces caspase-independent apoptosis in human hepatocellular carcinoma HepG2 cells. Biol. Pharm. Bull. 2014, 37, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, K.L.; Park, J.Y.; Noh, E.J.; Hoe, K.L.; Lee, J.H.; Kim, J.H.; Nam, J.H. The effect of combined treatment with cisplatin and histone deacetylase inhibitors on HeLa cells. J. Gynecol. Oncol. 2010, 21, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.G.; Kim, D.H.; Lee, H.R.; Lee, J.S.; Jin, S.J.; Lee, H.T. Sirtuin inhibition leads to autophagy and apoptosis in porcine preimplantation blastocysts. Biochem. Biophys. Res. Commun. 2017, 488, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Allison, D.; Condon, B.; Zhang, F.; Gheyi, T.; Zhang, A.; Ashok, S.; Russell, M.; MacEwan, I.; Qian, Y.; et al. The 2.5 A crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J. Med. Chem. 2013, 56, 963–969. [Google Scholar] [CrossRef]
- Yang, L.L.; Wang, H.L.; Zhong, L.; Yuan, C.; Liu, S.Y.; Yu, Z.J.; Liu, S.; Yan, Y.H.; Wu, C.; Wang, Y.; et al. X-ray crystal structure guided discovery of new selective, substrate-mimicking sirtuin 2 inhibitors that exhibit activities against non-small cell lung cancer cells. Eur. J. Med. Chem. 2018, 155, 806–823. [Google Scholar] [CrossRef]
- Kim, M.J.; Kang, Y.J.; Sung, B.; Jang, J.Y.; Ahn, Y.R.; Oh, H.J.; Choi, H.; Choi, I.; Im, E.; Moon, H.R.; et al. Novel SIRT inhibitor, MHY2256, induces cell cycle arrest, apoptosis, and autophagic cell death in HCT116 human colorectal cancer cells. Biomol. Ther. 2020, 28, 561–568. [Google Scholar] [CrossRef]
- Jang, J.Y.; Kang, Y.J.; Sung, B.; Kim, M.J.; Park, C.; Kang, D.; Moon, H.R.; Chung, H.Y.; Kim, N.D. MHY440, a novel topoisomerase Ι inhibitor, induces cell cycle arrest and apoptosis via a ROS-dependent DNA damage signaling pathway in AGS human gastric cancer cells. Molecules 2018, 24, 96. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Binding Energy (kcal/mol) a | No. of Van der Waals Bond Interaction Residues b | ||
|---|---|---|---|---|
| AutoDock Vina | AutoDock 4 | Dock 6 | ||
| Splitomicin (SIRT1 inhibitor) | −9.6 | −7.63 | −28.46 | 5 |
| MHY2245 | −9.7 | −9.7 | −33.47 | 5 |
| Compounds | Binding Energy (kcal/mol) a | No. of Van der Waals Bond Interaction Residues b | No. of Aromatic Interaction Residues b | ||
|---|---|---|---|---|---|
| AutoDock Vina | AutoDock 4 | Dock 6 | |||
| AGK2 (SIRT2 inhibitor) | −10.7 | −12.7 | −36.66 | 15 | 1 |
| MHY2245 | −10.9 | −9.99 | −32.00 | 9 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, Y.J.; Jang, J.Y.; Kwon, Y.H.; Lee, J.H.; Lee, S.; Park, Y.; Jung, Y.-S.; Im, E.; Moon, H.R.; Chung, H.Y.; et al. MHY2245, a Sirtuin Inhibitor, Induces Cell Cycle Arrest and Apoptosis in HCT116 Human Colorectal Cancer Cells. Int. J. Mol. Sci. 2022, 23, 1590. https://doi.org/10.3390/ijms23031590
Kang YJ, Jang JY, Kwon YH, Lee JH, Lee S, Park Y, Jung Y-S, Im E, Moon HR, Chung HY, et al. MHY2245, a Sirtuin Inhibitor, Induces Cell Cycle Arrest and Apoptosis in HCT116 Human Colorectal Cancer Cells. International Journal of Molecular Sciences. 2022; 23(3):1590. https://doi.org/10.3390/ijms23031590
Chicago/Turabian StyleKang, Yong Jung, Jung Yoon Jang, Young Hoon Kwon, Jun Ho Lee, Sanggwon Lee, Yujin Park, Young-Suk Jung, Eunok Im, Hyung Ryong Moon, Hae Young Chung, and et al. 2022. "MHY2245, a Sirtuin Inhibitor, Induces Cell Cycle Arrest and Apoptosis in HCT116 Human Colorectal Cancer Cells" International Journal of Molecular Sciences 23, no. 3: 1590. https://doi.org/10.3390/ijms23031590
APA StyleKang, Y. J., Jang, J. Y., Kwon, Y. H., Lee, J. H., Lee, S., Park, Y., Jung, Y.-S., Im, E., Moon, H. R., Chung, H. Y., & Kim, N. D. (2022). MHY2245, a Sirtuin Inhibitor, Induces Cell Cycle Arrest and Apoptosis in HCT116 Human Colorectal Cancer Cells. International Journal of Molecular Sciences, 23(3), 1590. https://doi.org/10.3390/ijms23031590