
The Cumulative Formation of R-loop Interacts with Histone Modifications to Shape Cell Reprogramming


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
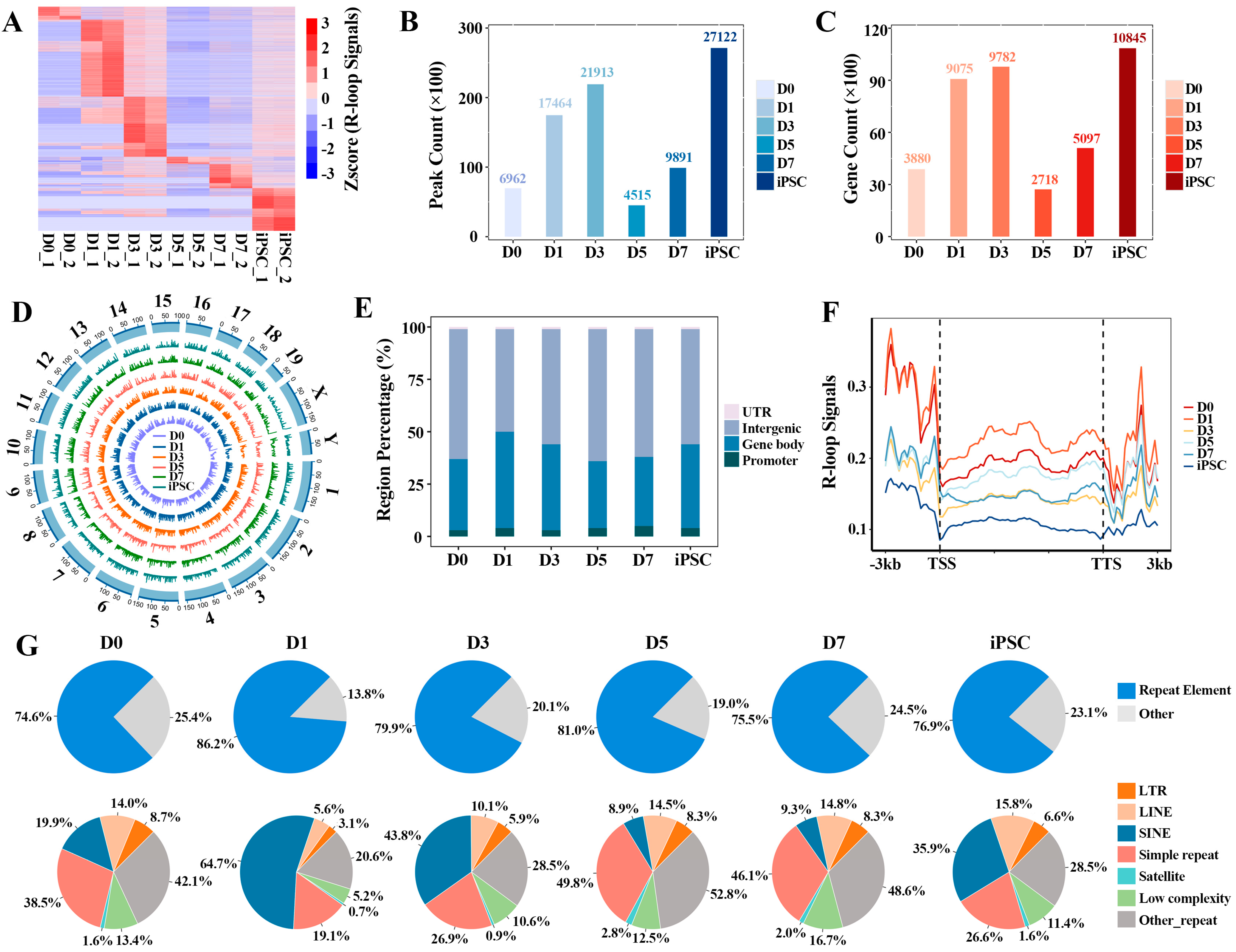
2.1. Stage-Specific R-loop Formation during the Reprograming Process
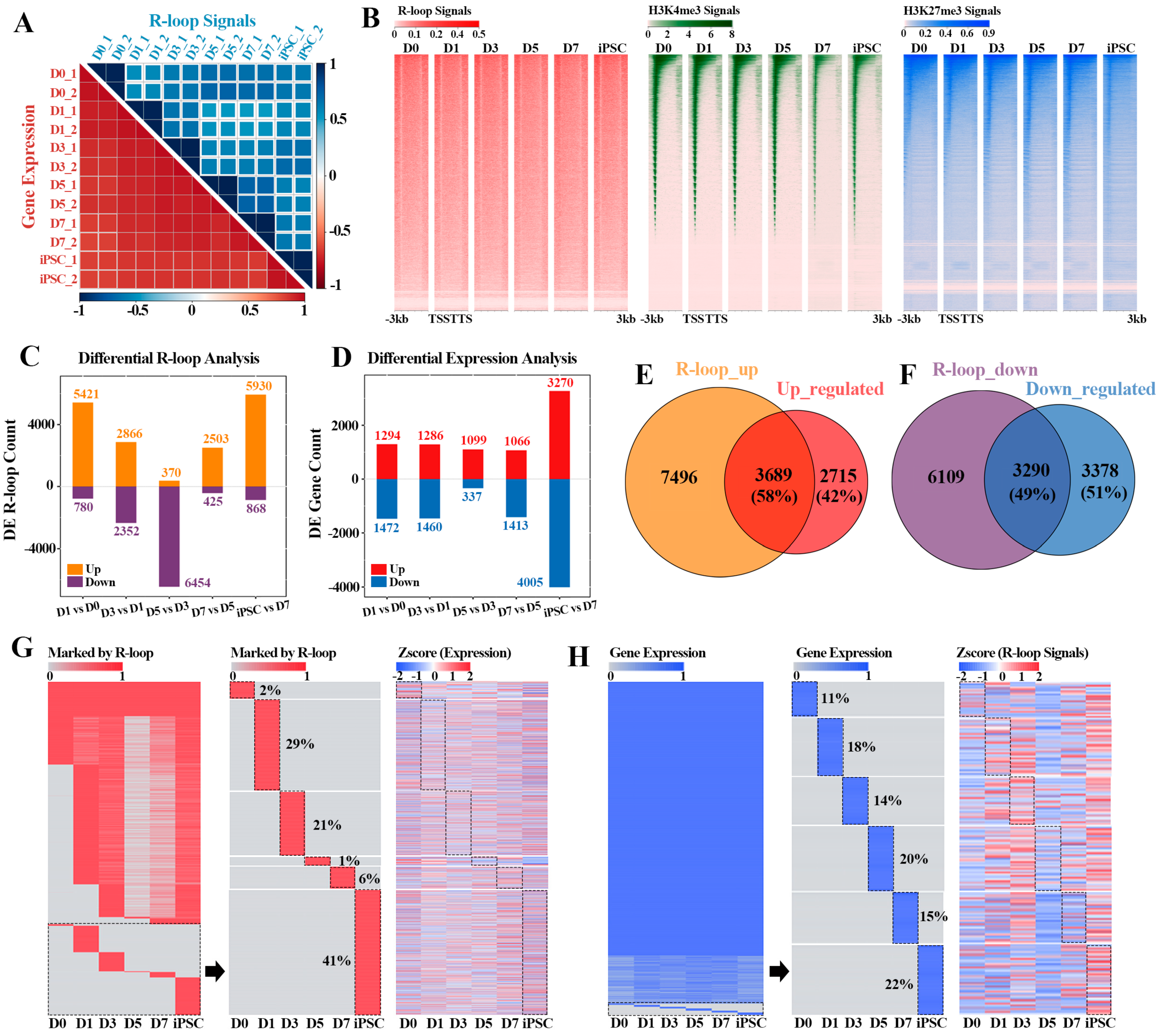
2.2. The Cumulative Effect of R-loops on Gene Expression Regulation in Reprogramming
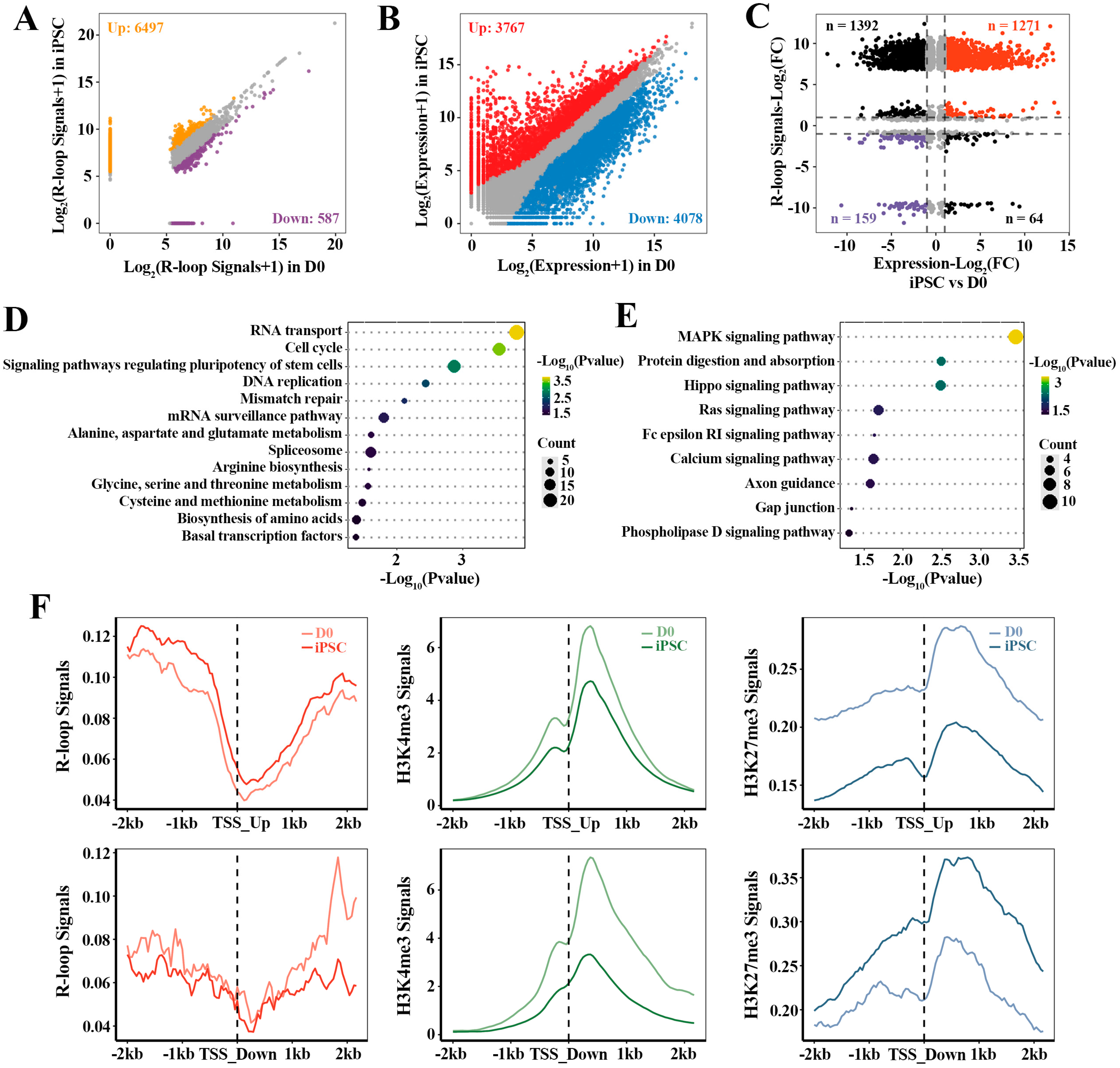
2.3. The Relationship between R-loop Dynamics and Gene Expression
2.4. Sequential Activation of Pluripotency Genes Linked to R-loops during Reprogramming
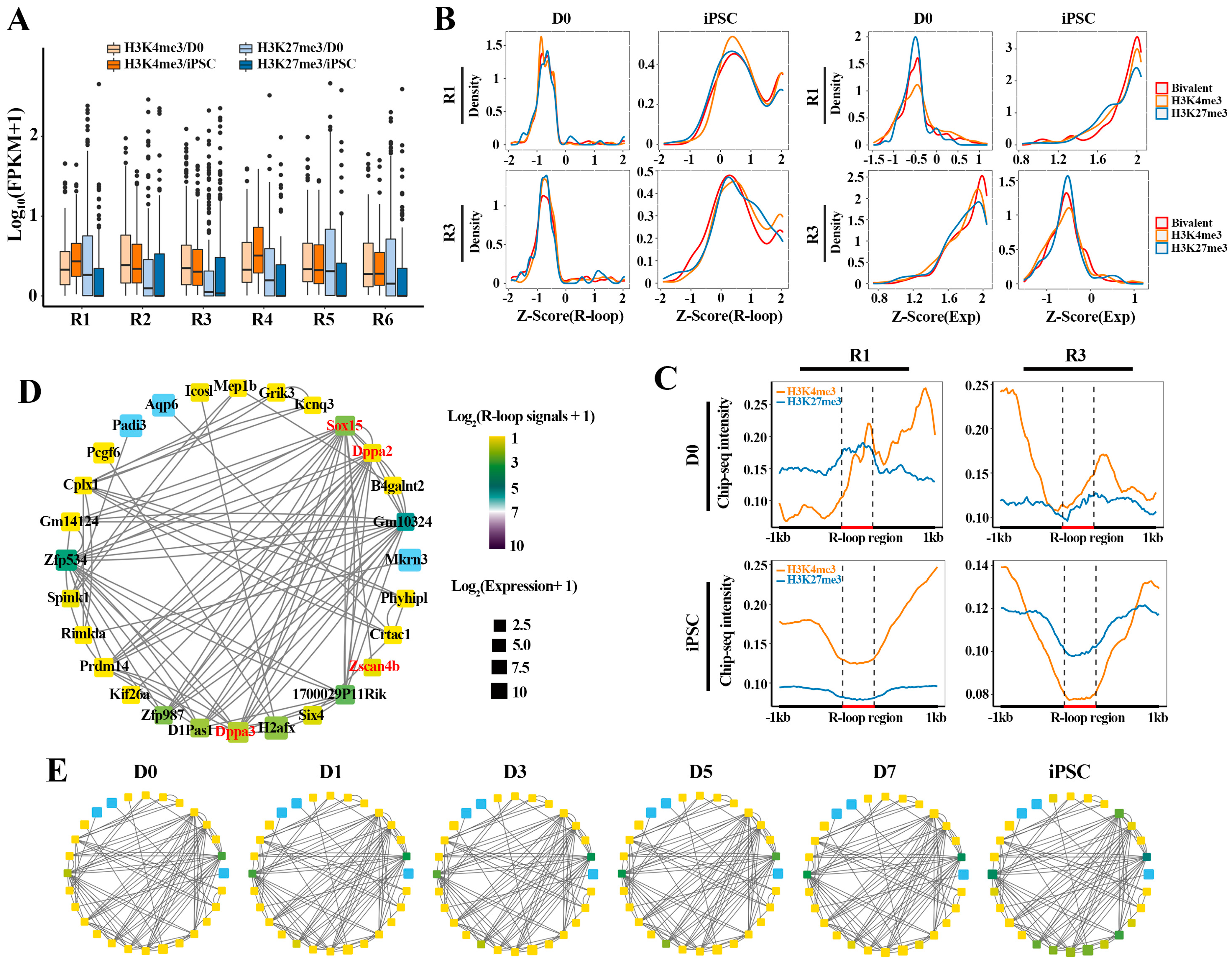
2.5. The Collaborative Regulation of R-loops and HMs Facilitates the Pluripotency Network Activation during Reprogramming
3. Discussion
4. Materials and Methods
4.1. Dataset Collection
4.2. ssDRIP-seq Data Processing
4.3. ChIP-seq Data Processing
4.4. RNA-seq Analysis
4.5. Identification of R-loop Peaks on Repetitive Elements and Transcription Factor (TF) Family
4.6. Differential Gene Expression Analysis
4.7. Fuzzy C-Means (FCM) Clustering Analysis
4.8. Transcriptional Regulatory Networks Analysis
4.9. Functional Enrichment and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milman, G.; Langridge, R.; Chamberlin, M.J. The structure of a DNA-rna hybrid. Proc. Natl. Acad. Sci. USA 1967, 57, 1804–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Davis, W. Hybridization of rna to double-stranded DNA: Formation of r-loops. Proc. Natl. Acad. Sci. USA 1976, 73, 2294–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginno, P.; Lott, P.; Christensen, H.; Korf, I.; Chédin, F. R-loop formation is a distinctive characteristic of unmethylated human cpg island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chedin, F. Gc skew at the 5′ and 3′ ends of human genes links r-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Sanz, L.A.; Hartono, S.R.; Lim, Y.W.; Steyaert, S.; Rajpurkar, A.; Ginno, P.A.; Xu, X.; Chédin, F. Prevalent, dynamic, and conserved r-loop structures associate with specific epigenomic signatures in mammals. Mol. Cell 2016, 63, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, J.Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H. R-chip using inactive rnase h reveals dynamic coupling of r-loops with transcriptional pausing at gene promoters. Mol. Cell 2017, 68, 745–757.e5. [Google Scholar] [CrossRef] [Green Version]
- García-Muse, T.; Aguilera, A. R loops: From physiological to pathological roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates r-loop levels and activates distinct DNA damage responses-sciencedirect. Cell 2017, 170, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Wahba, L.; Gore, S.K.; Koshland, D. The homologous recombination machinery modulates the formation of rna-DNA hybrids and associated chromosome instability. eLife 2013, 2, e00505. [Google Scholar] [CrossRef] [PubMed]
- García-Pichardo, D.; Cañas, J.C.; García-Rubio, M.L.; Gómez-González, B.; Rondón, A.G.; Aguilera, A. Histone mutants separate r loop formation from genome instability induction. Mol. Cell 2017, 66, 597–609.e595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Dao, F.Y.; Zulfiqar, H.; Su, W.; Ding, H.; Liu, L.; Lin, H. A sequence-based deep learning approach to predict ctcf-mediated chromatin loop. Brief. Bioinform. 2021, 22, 1–13. [Google Scholar]
- Niehrs, C.; Luke, B. Regulatory r-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Nadel, J.; Athanasiadou, R.; Lemetre, C.; Wijetunga, N.A.; Broin, P.; Sato, H.; Zhang, Z.; Jeddeloh, J.; Montagna, C.; Golden, A. Rna:DNA hybrids in the human genome have distinctive nucleotide characteristics, chromatin composition, and transcriptional relationships. Epigenet. Chromatin 2015, 8, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Chin, M.H.; Pellegrini, M.; Plath, K.; Lowry, W.E. Molecular analyses of human induced pluripotent stem cells and embryonic stem cells. Cell Stem Cell 2010, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chen, X.; Li, M.; Liu, X.; Gao, Y.; Kou, X.; Zhao, Y.; Zheng, W.; Zhang, X.; Huo, Y.; et al. Hierarchical oct4 binding in concert with primed epigenetic rearrangements during somatic cell reprogramming. Cell Rep. 2016, 14, 1540–1554. [Google Scholar] [CrossRef] [Green Version]
- Gomes, K.M.; Costa, I.C.; Santos, J.F.; Dourado, P.M.; Forni, M.F.; Ferreira, J.C. Induced pluripotent stem cells reprogramming: Epigenetics and applications in the regenerative medicine. Rev. Assoc. Med. Bras. 2017, 63, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godini, R.; Lafta, H.Y.; Fallahi, H. Epigenetic modifications in the embryonic and induced pluripotent stem cells. Gene Expr. Patterns GEP 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ta, N.; Long, C.; Zhang, Q.; Li, S.; Liu, S.; Yang, L.; Zuo, Y. The spatial binding model of the pioneer factor oct4 with its target genes during cell reprogramming. Comput. Struct. Biotechnol. J. 2019, 17, 1226–1233. [Google Scholar] [CrossRef]
- Ying, Z.; Xiang, G.; Zheng, L.; Tang, H.; Duan, L.; Lin, X.; Zhao, Q.; Chen, K.; Wu, Y.; Xing, G.; et al. Short-term mitochondrial permeability transition pore opening modulates histone lysine methylation at the early phase of somatic cell reprogramming. Cell Metab. 2018, 28, 935–945.e935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, S.M.; Puri, M.C.; Tonge, P.D.; Benevento, M.; Corso, A.J.; Clancy, J.L.; Mosbergen, R.; Li, M.; Lee, D.S.; Cloonan, N.; et al. Genome-wide characterization of the routes to pluripotency. Nature 2014, 516, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.R.; Liu, D.Y.; Xu, B.F.; Tian, R.X.; Zuo, Y.C. Modular arrangements of sequence motifs determine the functional diversity of kdm proteins. Brief. Bioinform. 2021, 22, bbaa215. [Google Scholar] [CrossRef]
- Li, Y.; Song, Y. R-loops coordinate with sox2 in regulating reprogramming to pluripotency. Sci. Adv. 2020, 6, eaba0777. [Google Scholar] [CrossRef]
- Yan, P.; Liu, Z.; Song, M.; Wu, Z.; Liu, G.H. Genome-wide r-loop landscapes during cell differentiation and reprogramming. Cell Rep. 2020, 32, 107870. [Google Scholar] [CrossRef]
- Gaillard, H.; Aguilera, A. Transcription as a threat to genome integrity. Annu. Rev. Biochem. 2016, 85, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.; Liu, W.; Li, J.; Li, C.; Kou, X.; Chen, J.; Zhao, Y.; Gao, H.; Wang, H.; et al. Distinct features of h3k4me3 and h3k27me3 chromatin domains in pre-implantation embryos. Nature 2016, 537, 558–562. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, C.; Hou, Z.; Yang, Y.; Bi, Y.; Wang, H.; Zhang, Y.; Gao, S. Unique molecular events during reprogramming of human somatic cells to induced pluripotent stem cells (ipscs) at naïve state. eLife 2018, 7, e29518. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Song, M.M.; Yang, W.R.T.; Cao, P.B.; Zheng, L.; Zuo, Y.C. A comparative analysis of single-cell transcriptome identifies reprogramming driver factors for efficiency improvement. Mol. Ther.-Nucl. Acids 2020, 19, 1053–1064. [Google Scholar] [CrossRef]
- Chen, C.; Khaleel, S.S.; Huang, H.; Wu, C.H. Software for pre-processing illumina next-generation sequencing short read sequences. Source Code Biol. Med. 2014, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Liu, T.; Zhang, Y. Using macs to identify peaks from chip-seq data. Curr. Protoc. Bioinform. 2011, 2, 2.14.1–2.14.14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Iaco, A.; Coudray, A.; Duc, J.; Trono, D. Dppa2 and dppa4 are necessary to establish a 2c-like state in mouse embryonic stem cells. EMBO Rep. 2019, 20, e47382. [Google Scholar] [CrossRef]
- Li, H.; Long, C.; Xiang, J.; Liang, P.; Li, X.; Zuo, Y. Dppa2/4 as a trigger of signaling pathways to promote zygote genome activation by binding to cg-rich region. Brief. Bioinform. 2020, 22, bbaa342. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; He, Q.Y. Chipseeker: An r/bioconductor package for chip peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Shao, N.; Liu, X.; Nestler, E. Ngs.Plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genom. 2014, 15, 284. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.; Leek, J.; Salzberg, S. Transcript-level expression analysis of rna-seq experiments with hisat, stringtie and ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. Stringtie enables improved reconstruction of a transcriptome from rna-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. Htseq—a python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using repeatmasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef] [PubMed]
- Mathelier, A.; Fornes, O.; Arenillas, D.J.; Chen, C.Y.; Denay, G.; Lee, J.; Shi, W.; Shyr, C.; Tan, G.; Worsley-Hunt, R.; et al. Jaspar 2016: A major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016, 44, D110–D115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Long, C.; Li, W.; Liang, P.; Liu, S.; Zuo, Y. Transcriptome comparisons of multi-species identify differential genome activation of mammals embryogenesis. IEEE Access 2019, 7, 7794–7802. [Google Scholar] [CrossRef]
- Liu, W.; Liu, X.; Wang, C.; Gao, Y.; Gao, R.; Kou, X.; Zhao, Y.; Li, J.; Wu, Y.; Xiu, W.; et al. Identification of key factors conquering developmental arrest of somatic cell cloned embryos by combining embryo biopsy and single-cell sequencing. Cell Discov. 2016, 2, 16010. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. Rna-seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Zheng, L.; Long, C.; Yang, W.; Yang, L.; Zuo, Y. Helpredictor models single-cell transcriptome to predict human embryo lineage allocation. Brief. Bioinform. 2021, 22, bbab196. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Damian, S.; Morris, J.H.; Helen, C.; Michael, K.; Stefan, W.; Milan, S.; Alberto, S.; Doncheva, N.T.; Alexander, R.; Peer, B. The string database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Li, H.; Li, X.; Yang, W.; Zuo, Y. Nuclear transfer arrest embryos show massive dysregulation of genes involved in transcription pathways. Int. J. Mol. Sci. 2021, 22, 8187. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An r package for comparing biological themes among gene clusters. Omics-A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ahlgren, P.; Jarneving, B.; Rousseau, R. Requirements for a cocitation similarity measure, with special reference to pearson’s correlation coefficient. J. Am. Soc. Inf. Sci. Technol. 2003, 54, 550–560. [Google Scholar] [CrossRef]
- Tang, Q.; Nie, F.; Kang, J.; Chen, W. Mrnalocater: Enhance the prediction accuracy of eukaryotic mrna subcellular localization by using model fusion strategy. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 2617–2623. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Mallik, S.; Mukhopadhyay, A. A survey and comparative study of statistical tests for identifying differential expression from microarray data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 95–115. [Google Scholar] [CrossRef]
- Greenland, S.; Senn, S.J.; Rothman, K.J.; Carlin, J.B.; Poole, C.; Goodman, S.N.; Altman, D.G. Statistical tests, p values, confidence intervals, and power: A guide to misinterpretations. Eur. J. Epidemiol. 2016, 31, 337–350. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Long, C.; Hong, Y.; Chao, L.; Peng, Y.; Zuo, Y. The Cumulative Formation of R-loop Interacts with Histone Modifications to Shape Cell Reprogramming. Int. J. Mol. Sci. 2022, 23, 1567. https://doi.org/10.3390/ijms23031567
Li H, Long C, Hong Y, Chao L, Peng Y, Zuo Y. The Cumulative Formation of R-loop Interacts with Histone Modifications to Shape Cell Reprogramming. International Journal of Molecular Sciences. 2022; 23(3):1567. https://doi.org/10.3390/ijms23031567
Chicago/Turabian StyleLi, Hanshuang, Chunshen Long, Yan Hong, Lemuge Chao, Yong Peng, and Yongchun Zuo. 2022. "The Cumulative Formation of R-loop Interacts with Histone Modifications to Shape Cell Reprogramming" International Journal of Molecular Sciences 23, no. 3: 1567. https://doi.org/10.3390/ijms23031567
APA StyleLi, H., Long, C., Hong, Y., Chao, L., Peng, Y., & Zuo, Y. (2022). The Cumulative Formation of R-loop Interacts with Histone Modifications to Shape Cell Reprogramming. International Journal of Molecular Sciences, 23(3), 1567. https://doi.org/10.3390/ijms23031567