
Rare Hereditary Gynecological Cancer Syndromes

 , , , and
, , , and
Abstract
:1. Introduction
2. Cowden Syndrome
2.1. Clinical Features
2.2. Gynecologic Cancers
2.3. Molecular Genetics
3. Peutz-Jeghers Syndrome
3.1. Clinical Features
3.2. Gynecologic Cancers
3.3. Molecular Genetics
4. DICER1 Syndrome
4.1. Clinical Features
4.2. Gynecologic Cancers
4.3. Molecular Genetics
5. Rhabdoid Tumor Predisposition Syndrome 2
5.1. Clinical Features
5.2. Gynecologic Cancers
5.3. Molecular Genetics
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garber, J.E.; Offit, K. Hereditary cancer predisposition syndromes. J. Clin. Oncol. 2005, 23, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Nagy, R.; Sweet, K.; Eng, C. Highly penetrant hereditary cancer syndromes. Oncogene 2004, 23, 6445–6470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinou, P.; Tischkowitz, M. Genetics of gynaecological cancers. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 42, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Ueki, A.; Hirasawa, A. Molecular Features and Clinical Management of Hereditary Gynecological Cancers. Int. J. Mol. Sci. 2020, 21, 9504. [Google Scholar] [CrossRef]
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E. ESMO Guidelines Committee. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. 2016, 27 (Suppl. S5), v103–v110. [Google Scholar] [CrossRef]
- Pavanello, M.; Chan, I.H.; Ariff, A.; Pharoah, P.D.; Gayther, S.A.; Ramus, S.J. Rare Germline Genetic Variants and the Risks of Epithelial Ovarian Cancer. Cancers 2020, 12, 3046. [Google Scholar] [CrossRef]
- Pietragalla, A.; Arcieri, M.; Marchetti, C.; Scambia, G.; Fagotti, A. Ovarian cancer predisposition beyond BRCA1 and BRCA2 genes. Int. J. Gynecol. Cancer 2020, 30, 1803–1810. [Google Scholar] [CrossRef]
- Patel, M.; Nowsheen, S.; Maraboyin, S.; Xia, F. The role of poly(ADP-ribose) polymerase inhibitors in the treatment of cancer and methods to overcome resistance: A review. Cell Biosci. 2020, 10, 35. [Google Scholar] [CrossRef]
- Therkildsen, C.; Jensen, L.H.; Rasmussen, M.; Bernstein, I. An Update on Immune Checkpoint Therapy for the Treatment of Lynch Syndrome. Clin. Exp. Gastroenterol. 2021, 14, 181–197. [Google Scholar] [CrossRef]
- Komiya, T.; Blumenthal, G.M.; DeChowdhury, R.; Fioravanti, S.; Ballas, M.S.; Morris, J.; Hornyak, T.J.; Wank, S.; Hewitt, S.M.; Morrow, B.; et al. A Pilot Study of Sirolimus in Subjects with Cowden Syndrome or Other Syndromes Characterized by Germline Mutations in PTEN. Oncologist 2019, 24, 1510-e1265. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, C.; Di Tommaso, S.; Loconte, D.C.; Grossi, V.; Sanese, P.; Bagnulo, R.; Susca, F.C.; Forte, G.; Peserico, A.; De Luisi, A.; et al. In vitro efficacy of ARQ 092, an allosteric AKT inhibitor, on primary fibroblast cells derived from patients with PIK3CA-related overgrowth spectrum (PROS). Neurogenetics 2018, 19, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Venot, Q.; Blanc, T.; Rabia, S.H.; Berteloot, L.; Ladraa, S.; Duong, J.P.; Blanc, E.; Johnson, S.C.; Hoguin, C.; Boccara, O.; et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 2018, 558, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Chen, J.; Cui, F.; Wang, H.; Wang, S.; Hang, W.; Zeng, Q.; Quan, C.S.; Zhai, Y.X.; Wang, J.W.; et al. LKB1 is a DNA damage response protein that regulates cellular sensitivity to PARP inhibitors. Oncotarget 2016, 7, 73389–73401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Valerio, M.; Cioce, M.; Mori, F.; Casadei, L.; Pulito, C.; Sacconi, A.; Biagioni, F.; Cortese, G.; Galanti, S.; et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat. Commun. 2012, 3, 865. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Y.; Huang, Y.; Chen, Y.; Wang, T.; Wu, S.; Tong, L.; Wang, Y.; Lin, L.; Hao, M.; et al. RNA-binding protein AUF1 suppresses miR-122 biogenesis by down-regulating Dicer1 in hepatocellular carcinoma. Oncotarget 2018, 9, 14815–14827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.D.; Martinez-Agosto, J.A. Hotspot Mutations in DICER1 Causing GLOW Syndrome-Associated Macrocephaly via Modulation of Specific microRNA Populations Result in the Activation of PI3K/ATK/mTOR Signaling. Microrna 2020, 9, 70–80. [Google Scholar] [CrossRef]
- Henon, C.; Blay, J.Y.; Massard, C.; Mir, O.; Bahleda, R.; Dumont, S.; Postel-Vinay, S.; Adam, J.; Soria, J.C.; Le Cesne, A. Long lasting major response to pembrolizumab in a thoracic malignant rhabdoid-like SMARCA4-deficient tumor. Ann. Oncol. 2019, 30, 1401–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, T.; Umemura, S.; Nakamura, H.; Zenke, Y.; Udagawa, H.; Kirita, K.; Matsumoto, S.; Yoh, K.; Niho, S.; Motoi, N.; et al. Successful treatment with nivolumab for SMARCA4-deficient non-small cell lung carcinoma with a high tumor mutation burden: A case report. Thorac. Cancer 2019, 10, 1285–1288. [Google Scholar] [CrossRef]
- Takada, K.; Sugita, S.; Murase, K.; Kikuchi, T.; Oomori, G.; Ito, R.; Hayasaka, N.; Miyanishi, K.; Iyama, S.; Ikeda, H.; et al. Exceptionally rapid response to pembrolizumab in a SMARCA4-deficient thoracic sarcoma overexpressing PD-L1: A case report. Thorac. Cancer 2019, 10, 2312–2315. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Bandlamudi, C.; Lavery, J.A.; Montecalvo, J.; Namakydoust, A.; Rizvi, H.; Egger, J.; Concepcion, C.P.; Paul, S.; Arcila, M.E.; et al. The Genomic Landscape of SMARCA4 Alterations and Associations with Outcomes in Patients with Lung Cancer. Clin. Cancer Res. 2020, 26, 5701–5708. [Google Scholar] [CrossRef] [PubMed]
- Januario, T.; Ye, X.; Bainer, R.; Alicke, B.; Smith, T.; Haley, B.; Modrusan, Z.; Gould, S.; Yauch, R.L. PRC2-mediated repression of SMARCA2 predicts EZH2 inhibitor activity in SWI/SNF mutant tumors. Proc. Natl. Acad. Sci USA 2017, 114, 12249–12254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldi, R.; Ghosh Halder, T.; Weston, A.; Thode, T.; Drenner, K.; Lewis, R.; Kaadige, M.R.; Srivastava, S.; Daniel Ampanattu, S.; Rodriguez Del Villar, R.; et al. The novel reversible LSD1 inhibitor SP-2577 promotes anti-tumor immunity in SWItch/Sucrose-NonFermentable (SWI/SNF) complex mutated ovarian cancer. PLoS ONE 2020, 15, e0235705. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Fu, Z.; Wang, X.Q.D.; Fiset, P.O.; Rieker, R.; Levins, C.; Kong, T.; Zhu, X.; Morin, G.; et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 2019, 10, 557. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Macdonald, E.; Venneti, S.; Wang, X.Q.D.; Witkowski, L.; Jelinic, P.; Kong, T.; Martinez, D.; Morin, G.; et al. CDK4/6 inhibitors target SMARCA4-determined cyclin D1 deficiency in hypercalcemic small cell carcinoma of the ovary. Nat. Commun. 2019, 10, 558. [Google Scholar] [CrossRef]
- Shorstova, T.; Marques, M.; Su, J.; Johnston, J.; Kleinman, C.L.; Hamel, N.; Huang, S.; Alaoui-Jamali, M.A.; Foulkes, W.D.; Witcher, M. SWI/SNF-Compromised Cancers Are Susceptible to Bromodomain Inhibitors. Cancer Res. 2019, 79, 2761–2774. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.I.; An, G.Y.; Baek, M.; Yoo, E.; Chai, J.C.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. BET inhibitor suppresses migration of human hepatocellular carcinoma by inhibiting SMARCA4. Sci. Rep. 2021, 11, 11799. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, S.Y.; Colborne, S.; Lambert, G.; Shin, C.Y.; Santos, N.D.; Orlando, K.A.; Lang, J.D.; Hendricks, W.P.D.; Bally, M.B.; et al. Histone Deacetylase Inhibitors Synergize with Catalytic Inhibitors of EZH2 to Exhibit Antitumor Activity in Small Cell Carcinoma of the Ovary, Hypercalcemic Type. Mol. Cancer Ther. 2018, 17, 2767–2779. [Google Scholar] [CrossRef] [Green Version]
- Tagal, V.; Wei, S.; Zhang, W.; Brekken, R.A.; Posner, B.A.; Peyton, M.; Girard, L.; Hwang, T.; Wheeler, D.A.; Minna, J.D.; et al. SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat. Commun. 2017, 8, 14098. [Google Scholar] [CrossRef] [Green Version]
- Nelen, M.R.; Kremer, H.; Konings, I.B.; Schoute, F.; van Essen, A.J.; Koch, R.; Woods, C.G.; Fryns, J.P.; Hamel, B.; Hoefsloot, L.H.; et al. Novel PTEN mutations in patients with Cowden disease: Absence of clear genotype-phenotype correlations. Eur. J. Hum. Genet. 1999, 7, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.M., 2nd; Dennis, M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann. Intern. Med. 1963, 58, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Bubien, V.; Bonnet, F.; Brouste, V.; Hoppe, S.; Barouk-Simonet, E.; David, A.; Edery, P.; Bottani, A.; Layet, V.; Caron, O.; et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J. Med. Genet. 2013, 50, 255–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riegert-Johnson, D.L.; Gleeson, F.C.; Roberts, M.; Tholen, K.; Youngborg, L.; Bullock, M.; Boardman, L.A. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered. Cancer Clin. Pract. 2010, 8, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network (NCCN) in Oncology. Genetic/Familial High-Risk Assessment: Breast, Ovarian and Pancreatic. Version 1. 2022. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 9 November 2021).
- Yehia, L.; Eng, C. 65 YEARS OF THE DOUBLE HELIX: One gene, many endocrine and metabolic syndromes: PTEN-opathies and precision medicine. Endocr. Relat. Cancer 2018, 25, T121–T140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin. Genet. 2021, 99, 219–225. [Google Scholar] [CrossRef]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.H.; Mester, J.; Peterson, C.; Yang, Y.; Chen, J.L.; Rybicki, L.A.; Milas, K.; Pederson, H.; Remzi, B.; Orloff, M.S.; et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet 2011, 88, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Crivelli, L.; Bubien, V.; Jones, N.; Chiron, J.; Bonnet, F.; Barouk-Simonet, E.; Couzigou, P.; Sevenet, N.; Caux, F.; Longy, M. Insertion of Alu elements at a PTEN hotspot in Cowden syndrome. Eur. J. Hum. Genet. 2017, 25, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Ngeow, J.; Eng, C. PTEN-opathies: From biological insights to evidence-based precision medicine. J. Clin. Investig. 2019, 129, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.P.; Waite, K.A.; Pilarski, R.; Hampel, H.; Fernandez, M.J.; Bos, C.; Dasouki, M.; Feldman, G.L.; Greenberg, L.A.; Ivanovich, J.; et al. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am. J. Hum. Genet. 2003, 73, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.J.; Romigh, T.; Sesock, K.; Eng, C. Characterization of cryptic splicing in germline PTEN intronic variants in Cowden syndrome. Hum. Mutat. 2017, 38, 1372–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mester, J.; Eng, C. When overgrowth bumps into cancer: The PTEN-opathies. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163C, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Orloff, M.S.; He, X.; Peterson, C.; Chen, F.; Chen, J.L.; Mester, J.L.; Eng, C. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am. J. Hum. Genet. 2013, 92, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, K.L.; Campbell, R.; Ganapathi, S.; Zhou, M.; Rini, B.; Ganapathi, R.; Neumann, H.P.; Eng, C. Germline and somatic DNA methylation and epigenetic regulation of KILLIN in renal cell carcinoma. Genes Chromosomes Cancer 2011, 50, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Nizialek, E.A.; Peterson, C.; Mester, J.L.; Downes-Kelly, E.; Eng, C. Germline and somatic KLLN alterations in breast cancer dysregulate G2 arrest. Hum. Mol. Genet. 2013, 22, 2451–2461. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.Y.; Andrews, K.A.; Challis, B.G.; Park, S.M.; Acerini, C.L.; Maher, E.R.; Casey, R.T. Clinical Practice Guidance: Surveillance for phaeochromocytoma and paraganglioma in paediatric succinate dehydrogenase gene mutation carriers. Clin. Endocrinol. 2019, 90, 499–505. [Google Scholar] [CrossRef]
- Ni, Y.; Zbuk, K.M.; Sadler, T.; Patocs, A.; Lobo, G.; Edelman, E.; Platzer, P.; Orloff, M.S.; Waite, K.A.; Eng, C. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am. J. Hum. Genet. 2008, 83, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.; Wang, B.; Liu, P. The Functional Role of SEC23 in Vesicle Transportation, Autophagy and Cancer. Int. J. Biol. Sci. 2019, 15, 2419–2426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peiling Yang, S.; Ngeow, J. Familial non-medullary thyroid cancer: Unraveling the genetic maze. Endocr. Relat. Cancer 2016, 23, R577–R595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalho-Carvalho, J.; Martins, J.B.; Cekaite, L.; Sveen, A.; Torres-Ferreira, J.; Graça, I.; Costa-Pinheiro, P.; Eilertsen, I.A.; Antunes, L.; Oliveira, J.; et al. Epigenetic disruption of miR-130a promotes prostate cancer by targeting SEC23B and DEPDC1. Cancer Lett. 2017, 385, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, N.; Li, X.; Lu, D.; Hou, Z.; Li, Y.; Jin, Y.; Gu, J.; Yin, Y. Mutations in the coat complex II component SEC23B promote colorectal cancer metastasis. Cell Death Dis. 2020, 11, 157. [Google Scholar] [CrossRef] [Green Version]
- Yehia, L.; Niazi, F.; Ni, Y.; Ngeow, J.; Sankunny, M.; Liu, Z.; Wei, W.; Mester, J.L.; Keri, R.A.; Zhang, B.; et al. Germline Heterozygous Variants in SEC23B Are Associated with Cowden Syndrome and Enriched in Apparently Sporadic Thyroid Cancer. Am. J. Hum. Genet. 2015, 97, 661–676. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Yu, J.; Lin, Z.; Feng, R.; Wang, Z.W.; Chen, G. The emerging role of WWP1 in cancer development and progression. Cell Death Discov. 2021, 7, 163. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef]
- Lee, Y.R.; Yehia, L.; Kishikawa, T.; Ni, Y.; Leach, B.; Zhang, J.; Panch, N.; Liu, J.; Wei, W.; Eng, C.; et al. WWP1 Gain-of-Function Inactivation of PTEN in Cancer Predisposition. N. Engl. J. Med. 2020, 382, 2103–2116. [Google Scholar] [CrossRef]
- Mahdi, H.; Mester, J.L.; Nizialek, E.A.; Ngeow, J.; Michener, C.; Eng, C. Germline PTEN, SDHB-D, and KLLN alterations in endometrial cancer patients with Cowden and Cowden-like syndromes: An international, multicenter, prospective study. Cancer 2015, 121, 688–696. [Google Scholar] [CrossRef] [Green Version]
- McGarrity, T.J.; Amos, C.A.; Baker, M.J. Peutz-Jeghers Syndrome. In GeneReviews. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1266/ (accessed on 9 November 2021).
- Beggs, A.D.; Latchford, A.R.; Vasen, H.F.; Moslein, G.; Alonso, A.; Aretz, S.; Bertario, L.; Blanco, I.; Bülow, S.; Burn, J.; et al. Peutz-Jeghers syndrome: A systematic review and recommendations for management. Gut 2010, 59, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Giardiello, F.M.; Welsh, S.B.; Hamilton, S.R.; Offerhaus, G.J.; Gittelsohn, A.M.; Booker, S.V.; Krush, A.J.; Yardley, J.H.; Luk, G.D. Increased risk of cancer in the Peutz-Jeghers syndrome. N. Engl. J. Med. 1987, 316, 1511–1514. [Google Scholar] [CrossRef] [PubMed]
- van Lier, M.G.; Westerman, A.M.; Wagner, A.; Looman, C.W.; Wilson, J.H.; de Rooij, F.W.; Lemmens, V.E.; Kuipers, E.J.; Mathus-Vliegen, E.M.; van Leerdam, M.E. High cancer risk and increased mortality in patients with Peutz-Jeghers syndrome. Gut 2011, 60, 141–147. [Google Scholar] [CrossRef] [PubMed]
- NCCN Clinical Practice Guidelines in Oncology: Genetics/Familial High-Risk. Assessment: Colorectal. Version 1. 2021. Available online: http://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf (accessed on 9 November 2021).
- Benzel, J.; Fendrich, V. Familial Pancreatic Cancer. Oncol. Res. Treat. 2018, 41, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Young, R.H.; Welch, W.R.; Dickersin, G.R.; Scully, R.E. Ovarian sex cord tumor with annular tubules: Review of 74 cases including 27 with Peutz-Jeghers syndrome and four with adenoma malignum of the cervix. Cancer 1982, 50, 1384–1402. [Google Scholar] [CrossRef]
- Wade, K.S.; Estes, J.M.; Kline, R.C. Genetics and the Gynecologic Patient. Ochsner J. 2020, 20, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Banno, K.; Kisu, I.; Yanokura, M.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Hirasawa, A.; Aoki, D. Hereditary gynecological tumors associated with Peutz-Jeghers syndrome (Review). Oncol. Lett. 2013, 6, 1184–1188. [Google Scholar] [CrossRef]
- Connolly, D.C.; Katabuchi, H.; Cliby, W.A.; Cho, K.R. Somatic mutations in the STK11/LKB1 gene are uncommon in rare gynecological tumor types associated with Peutz-Jegher’s syndrome. Am. J. Pathol. 2000, 156, 339–345. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. American College of Gastroenterology. ACG clinical guideline. Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spoto, C.P.E.; Gullo, I.; Carneiro, F.; Montgomery, E.A.; Brosens, L.A.A. Hereditary gastrointestinal carcinomas and their precursors: An algorithm for genetic testing. Semin. Diagn. Pathol. 2018, 35, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Butel-Simoes, G.I.; Spigelman, A.D.; Scott, R.J.; Vilain, R.E. Low-level parental mosaicism in an apparent de novo case of Peutz-Jeghers syndrome. Fam. Cancer 2019, 18, 109–112. [Google Scholar] [CrossRef]
- McKay, V.; Cairns, D.; Gokhale, D.; Mountford, R.; Greenhalgh, L. First report of somatic mosaicism for mutations in STK11 in four patients with Peutz-Jeghers syndrome. Fam. Cancer 2016, 15, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cespedes, M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene 2007, 26, 7825–7832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.T.; Zhu, H.B. LKB1 and cancer: The dual role of metabolic regulation. Biomed. Pharmacother. 2020, 132, 110872. [Google Scholar] [CrossRef] [PubMed]
- Laderian, B.; Mundi, P.; Fojo, T.; Bates, S.E. Emerging Therapeutic Implications of STK11 Mutation: Case Series. Oncologist 2020, 25, 733–737. [Google Scholar] [CrossRef]
- Klümpen, H.J.; Queiroz, K.C.; Spek, C.A.; van Noesel, C.J.; Brink, H.C.; de Leng, W.W.; de Wilde, R.F.; Mathus-Vliegen, E.M.; Offerhaus, G.J.; Alleman, M.A.; et al. mTOR inhibitor treatment of pancreatic cancer in a patient with Peutz-Jeghers syndrome. J. Clin. Oncol. 2011, 29, e150–e153. [Google Scholar] [CrossRef] [Green Version]
- Trédan, O.; Treilleux, I.; Wang, Q.; Gane, N.; Pissaloux, D.; Bonnin, N.; Petit, T.; Cretin, J.; Bonichon-Lamichhane, N.; Priou, F.; et al. Predicting everolimus treatment efficacy in patients with advanced endometrial carcinoma: A GINECO group study. Target Oncol. 2013, 8, 243–251. [Google Scholar] [CrossRef]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Facchinetti, F.; Bluthgen, M.V.; Tergemina-Clain, G.; Faivre, L.; Pignon, J.P.; Planchard, D.; Remon, J.; Soria, J.C.; Lacroix, L.; Besse, B. LKB1/STK11 mutations in non-small cell lung cancer patients: Descriptive analysis and prognostic value. Lung Cancer 2017, 112, 62–68. [Google Scholar] [CrossRef]
- Skoulidis, F.; Arbour, K.C.; Hellmann, M.D.; Patil, P.D.; Marmarelis, M.E.; Awad, M.M.; Murray, J.C.; Hellyer, J.; Gainor, J.F.; Dimou, A.; et al. Association of STK11/LKB1 genomic alterations with lack of benefit from the addition of pembrolizumab to platinum doublet chemotherapy in non-squamous non-small cell lung cancer. J. Clin. Oncol. 2019, 37, 102. [Google Scholar] [CrossRef]
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK Pathway and Drug Response in Cancer: A Therapeutic Perspective. Oxid. Med. Cell Longev. 2019, 2019, 8730816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, D.R.; Best, A.F.; Williams, G.M.; Harney, L.A.; Carr, A.G.; Harris, A.K.; Kratz, C.P.; Dehner, L.P.; Messinger, Y.H.; Rosenberg, P.S.; et al. Neoplasm Risk Among Individuals with a Pathogenic Germline Variant in DICER1. J. Clin. Oncol. 2019, 37, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Caroleo, A.M.; De Ioris, M.A.; Boccuto, L.; Alessi, I.; Del Baldo, G.; Cacchione, A.; Agolini, E.; Rinelli, M.; Serra, A.; Carai, A.; et al. DICER1 Syndrome and Cancer Predisposition: From a Rare Pediatric Tumor to Lifetime Risk. Front. Oncol. 2021, 10, 614541. [Google Scholar] [CrossRef]
- Schultz, K.A.P.; Stewart, D.R.; Kamihara, J.; Bauer, A.J.; Merideth, M.A.; Stratton, P.; Huryn, L.A.; Harris, A.K.; Doros, L.; Field, A.; et al. DICER1 Tumor Predisposition. In GeneReviews. Available online: https://www.ncbi.nlm.nih.gov/books/NBK196157/ (accessed on 9 November 2021).
- Robertson, J.C.; Jorcyk, C.L. Oxford JT. DICER1 Syndrome: DICER1 Mutations in Rare Cancers. Cancers 2018, 10, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenneman, M.; Field, A.; Yang, J.; Williams, G.; Doros, L.; Rossi, C.; Schultz, K.A.; Rosenberg, A.; Ivanovich, J.; Turner, J.; et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in pleuropulmonary blastoma / DICER1 syndrome: A unique variant of the two-hit tumor suppression model. F1000Res 2015, 4, 214. [Google Scholar] [CrossRef]
- Messinger, Y.H.; Stewart, D.R.; Priest, J.R.; Williams, G.M.; Harris, A.K.; Schultz, K.A.; Yang, J.; Doros, L.; Rosenberg, P.S.; Hill, D.A.; et al. Pleuropulmonary blastoma: A report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer 2015, 121, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.E.; Bauer, A.J.; Doros, L.; Schultz, K.A.; Decastro, R.M.; Harney, L.A.; Kase, R.G.; Carr, A.G.; Harris, A.K.; Williams, G.M.; et al. Macrocephaly associated with the DICER1 syndrome. Genet. Med. 2017, 19, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Heravi-Moussavi, A.; Anglesio, M.S.; Cheng, S.W.; Senz, J.; Yang, W.; Prentice, L.; Fejes, A.P.; Chow, C.; Tone, A.; Kalloger, S.E.; et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N. Engl. J. Med. 2012, 366, 234–242. [Google Scholar] [CrossRef]
- Goulvent, T.; Ray-Coquard, I.; Borel, S.; Haddad, V.; Devouassoux-Shisheboran, M.; Vacher-Lavenu, M.C.; Pujade-Laurraine, E.; Savina, A.; Maillet, D.; Gillet, G.; et al. DICER1 and FOXL2 mutations in ovarian sex cord-stromal tumours: A GINECO Group study. Histopathology 2016, 68, 279–285. [Google Scholar] [CrossRef]
- Kato, N.; Kusumi, T.; Kamataki, A.; Tsunoda, R.; Fukase, M.; Kurose, A. DICER1 hotspot mutations in ovarian Sertoli-Leydig cell tumors: A potential association with androgenic effects. Hum. Pathol. 2017, 59, 41–47. [Google Scholar] [CrossRef] [PubMed]
- De Paolis, E.; Paragliola, R.M.; Concolino, P. Spectrum of DICER1 Germline Pathogenic Variants in Ovarian Sertoli-Leydig Cell Tumor. J. Clin. Med. 2021, 10, 1845. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.A.; Lim, Y.K.; Chia, Y.N.; Yam, K.L. Sertoli-Leydig cell tumor of the ovary: Analysis of a single institution database. J. Obstet. Gynaecol. Res. 2013, 39, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Durmuş, Y.; Kılıç, Ç.; Çakır, C.; Yüksel, D.; Boran, N.; Karalök, A.; Boyraz, G.; Turan, A.T. Sertoli-Leydig cell tumor of the ovary: Analysis of a single institution database and review of the literature. J. Obstet. Gynaecol. Res. 2019, 45, 1311–1318. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, M.; Hong, Y.; Zhong, Y.; Cong, X.; Chen, C.; Liu, Z.; Man, Y.; Yang, L. Sertoli-Leydig cell tumor in two siblings with DICER1 syndrome: A case report and literature review. Medicine 2020, 99, e20806. [Google Scholar] [CrossRef]
- Yuan, Z.; Huo, X.; Jiang, D.; Yu, M.; Cao, D.; Wu, H.; Shen, K.; Yang, J.; Zhang, Y.; Zhou, H.; et al. Clinical Characteristics and Mutation Analyses of Ovarian Sertoli-Leydig Cell Tumors. Oncologist 2020, 25, e1396–e1405. [Google Scholar] [CrossRef]
- Khosla, D.; Gupta, R.; Srinivasan, R.; Patel, F.D.; Rajwanshi, A. Sarcomas of uterine cervix: Clinicopathological features, treatment, and outcome. Int. J. Gynecol. Cancer 2012, 22, 1026–1030. [Google Scholar] [CrossRef]
- Bennett, J.A.; Ordulu, Z.; Young, R.H.; Pinto, A.; Van de Vijver, K.; Burandt, E.; Wanjari, P.; Shah, R.; de Kock, L.; Foulkes, W.D.; et al. Embryonal rhabdomyosarcoma of the uterine corpus: A clinicopathological and molecular analysis of 21 cases highlighting a frequent association with DICER1 mutations. Mod. Pathol. 2021, 34, 1750–1762. [Google Scholar] [CrossRef]
- De Kock, L.; Yoon, J.Y.; Apellaniz-Ruiz, M.; Pelletier, D.; McCluggage, W.G.; Stewart, C.J.R.; Dickson, B.C.; Rouzbahman, M.; Clarke, B.A.; Foulkes, W.D. Significantly greater prevalence of DICER1 alterations in uterine embryonal rhabdomyosarcoma compared to adenosarcoma. Mod. Pathol. 2020, 33, 1207–1219. [Google Scholar] [CrossRef]
- Mercier, A.M.; Zorn, K.K.; Quick, C.M.; Huffman, L.B. Recurrent gynandroblastoma of the ovary with germline DICER1 mutation: A case report and review of the literature. Gynecol. Oncol. Rep. 2021, 37, 100806. [Google Scholar] [CrossRef]
- Wang, Y.; Karnezis, A.N.; Magrill, J.; Tessier-Cloutier, B.; Lum, A.; Senz, J.; Gilks, C.B.; McCluggage, W.G.; Huntsman, D.G.; Kommoss, F. DICER1 hot-spot mutations in ovarian gynandroblastoma. Histopathology 2018, 73, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.M.; Jacobs, M.F.; Anderson, B.; Rabah, R.; Wu, Y.M.; Else, T.; Mody, R.J. DICER1 Mutations in the Era of Expanding Integrative Clinical Sequencing in Pediatric Oncology. JCO Precis. Oncol. 2019, 3, PO.18.00172. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; Harris, A.K.; Finch, M.; Dehner, L.P.; Brown, J.B.; Gershenson, D.M.; Young, R.H.; Field, A.; Yu, W.; Turner, J.; et al. DICER1-related Sertoli-Leydig cell tumor and gynandroblastoma: Clinical and genetic findings from the International Ovarian and Testicular Stromal Tumor Registry. Gynecol. Oncol. 2017, 147, 521–527. [Google Scholar] [CrossRef] [PubMed]
- de Kock, L.; Wang, Y.C.; Revil, T.; Badescu, D.; Rivera, B.; Sabbaghian, N.; Wu, M.; Weber, E.; Sandoval, C.; Hopman, S.M.; et al. High-sensitivity sequencing reveals multi-organ somatic mosaicism causing DICER1 syndrome. J. Med. Genet. 2016, 53, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Field, A.; Schultz, K.A.P.; Hill, D.A.; Stewart, D.R. The prevalence of DICER1 pathogenic variation in population databases. Int. J. Cancer 2017, 141, 2030–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirshahi, U.L.; Kim, J.; Best, A.F.; Chen, Z.E.; Hu, Y.; Haley, J.S.; Golden, A.; Stahl, R.; Manickam, K.; Carr, A.G.; et al. A Genome-First Approach to Characterize DICER1 Pathogenic Variant Prevalence, Penetrance, and Phenotype. JAMA Netw. Open 2021, 4, e210112. [Google Scholar] [CrossRef]
- Rakheja, D.; Chen, K.S.; Liu, Y.; Shukla, A.A.; Schmid, V.; Chang, T.C.; Khokhar, S.; Wickiser, J.E.; Karandikar, N.J.; Malter, J.S.; et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat. Commun. 2014, 2, 4802. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef]
- Seki, M.; Yoshida, K.; Shiraishi, Y.; Shimamura, T.; Sato, Y.; Nishimura, R.; Okuno, Y.; Chiba, K.; Tanaka, H.; Kato, K.; et al. Biallelic DICER1 mutations in sporadic pleuropulmonary blastoma. Cancer Res. 2014, 74, 2742–2749. [Google Scholar] [CrossRef] [Green Version]
- Vedanayagam, J.; Chatila, W.K.; Aksoy, B.A.; Majumdar, S.; Skanderup, A.J.; Demir, E.; Schultz, N.; Sander, C.; Lai, E.C. Cancer-associated mutations in DICER1 RNase IIIa and IIIb domains exert similar effects on miRNA biogenesis. Nat. Commun. 2019, 10, 3682. [Google Scholar] [CrossRef]
- Klein, S.; Lee, H.; Ghahremani, S.; Kempert, P.; Ischander, M.; Teitell, M.A.; Nelson, S.F.; Martinez-Agosto, J.A. Expanding the phenotype of mutations in DICER1: Mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J. Med. Genet. 2014, 51, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemes, K.; Frühwald, M.C. Emerging therapeutic targets for the treatment of malignant rhabdoid tumors. Expert Opin. Ther. Targets 2018, 22, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Dho, Y.S.; Kim, S.K.; Cheon, J.E.; Park, S.H.; Wang, K.C.; Lee, J.Y.; Phi, J.H. Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nerv. Syst. 2015, 31, 1305–1311. [Google Scholar] [CrossRef]
- Del Baldo, G.; Carta, R.; Alessi, I.; Merli, P.; Agolini, E.; Rinelli, M.; Boccuto, L.; Milano, G.M.; Serra, A.; Carai, A.; et al. Rhabdoid Tumor Predisposition Syndrome: From Clinical Suspicion to General Management. Front. Oncol. 2021, 11, 586288. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Kamihara, J.; Evans, D.G.R.; Brugières, L.; Bourdeaut, F.; Molenaar, J.J.; Walsh, M.F.; Brodeur, G.M.; Diller, L. Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin. Cancer Res. 2017, 23, e62–e67. [Google Scholar] [CrossRef] [Green Version]
- Panwalkar, P.; Pratt, D.; Chung, C.; Dang, D.; Le, P.; Martinez, D.; Bayliss, J.M.; Smith, K.S.; Adam, M.; Potter, S.; et al. SWI/SNF complex heterogeneity is related to polyphenotypic differentiation, prognosis, and immune response in rhabdoid tumors. Neuro-Oncology 2020, 22, 785–796. [Google Scholar] [CrossRef]
- Nemes, K.; Bens, S.; Bourdeaut, F.; Hasselblatt, M.; Kool, M.; Johann, P.; Kordes, U.; Schneppenheim, R.; Siebert, R.; Frühwald, M.C. Rhabdoid Tumor Predisposition Syndrome. In GeneReviews. Available online: https://www.ncbi.nlm.nih.gov/books/NBK469816/ (accessed on 9 November 2021).
- Young, R.H.; Oliva, E.; Scully, R.E. Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases. Am. J. Surg. Pathol. 1994, 18, 1102–1116. [Google Scholar] [CrossRef]
- Lin, D.I.; Chudnovsky, Y.; Duggan, B.; Zajchowski, D.; Greenbowe, J.; Ross, J.S.; Gay, L.M.; Ali, S.M.; Elvin, J.A. Comprehensive genomic profiling reveals inactivating SMARCA4 mutations and low tumor mutational burden in small cell carcinoma of the ovary, hypercalcemic-type. Gynecol. Oncol. 2017, 147, 626–633. [Google Scholar] [CrossRef]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B.; et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef]
- Lu, B.; Shi, H. An In-Depth Look at Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT): Clinical Implications from Recent Molecular Findings. J. Cancer 2019, 10, 223–237. [Google Scholar] [CrossRef]
- Witkowski, L.; Goudie, C.; Ramos, P.; Boshari, T.; Brunet, J.S.; Karnezis, A.N.; Longy, M.; Knost, J.A.; Saloustros, E.; McCluggage, W.G.; et al. The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecol. Oncol. 2016, 141, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Lavrut, P.M.; Le Loarer, F.; Normand, C.; Grosos, C.; Dubois, R.; Buenerd, A.; Conter, C.; Dijoud, F.; Blay, J.Y.; Collardeau-Frachon, S. Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Report of a Bilateral Case in a Teenager Associated with SMARCA4 Germline Mutation. Pediatr. Dev. Pathol. 2016, 19, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Donini, N.; Byler-Dann, R.; Knost, J.A.; Albrecht, S.; Berchuck, A.; McCluggage, W.G.; Hasselblatt, M.; Foulkes, W.D. The hereditary nature of small cell carcinoma of the ovary, hypercalcemic type: Two new familial cases. Fam. Cancer 2017, 16, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkowski, L.; Goudie, C.; Foulkes, W.D.; McCluggage, W.G. Small-Cell Carcinoma of the Ovary of Hypercalcemic Type (Malignant Rhabdoid Tumor of the Ovary): A Review with Recent Developments on Pathogenesis. Surg. Pathol. Clin. 2016, 9, 215–226. [Google Scholar] [CrossRef]
- Kolin, D.L.; Dong, F.; Baltay, M.; Lindeman, N.; MacConaill, L.; Nucci, M.R.; Crum, C.P.; Howitt, B.E. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): A clinicopathologic entity distinct from undifferentiated carcinoma. Mod. Pathol. 2018, 31, 1442–1456. [Google Scholar] [CrossRef]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer—biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef]
- Yoshida, A.; Kobayashi, E.; Kubo, T.; Kodaira, M.; Motoi, T.; Motoi, N.; Yonemori, K.; Ohe, Y.; Watanabe, S.I.; Kawai, A.; et al. Clinicopathological and molecular characterization of SMARCA4-deficient thoracic sarcomas with comparison to potentially related entities. Mod. Pathol. 2017, 30, 797–809. [Google Scholar] [CrossRef] [Green Version]
- Wanior, M.; Krämer, A.; Knapp, S.; Joerger, A.C. Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene 2021, 40, 3637–3654. [Google Scholar] [CrossRef]
- Holsten, T.; Bens, S.; Oyen, F.; Nemes, K.; Hasselblatt, M.; Kordes, U.; Siebert, R.; Frühwald, M.C.; Schneppenheim, R.; Schüller, U. Germline variants in SMARCB1 and other members of the BAF chromatin-remodeling complex across human disease entities: A meta-analysis. Eur. J. Hum. Genet. 2018, 26, 1083–1093. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Tsurusaki, Y.; Okamoto, N.; Teik, K.W.; Mizuno, S.; Suzumura, H.; Isidor, B.; Ong, W.P.; Haniffa, M.; White, S.M.; et al. Genetic abnormalities in a large cohort of Coffin-Siris syndrome patients. J. Hum. Genet. 2019, 64, 1173–1186. [Google Scholar] [CrossRef]
- Bögershausen, N.; Wollnik, B. Mutational Landscapes and Phenotypic Spectrum of SWI/SNF-Related Intellectual Disability Disorders. Front. Mol. Neurosci. 2018, 11, 252. [Google Scholar] [CrossRef]
- Filatova, A.; Rey, L.K.; Lechler, M.B.; Schaper, J.; Hempel, M.; Posmyk, R.; Szczaluba, K.; Santen, G.W.E.; Wieczorek, D.; Nuber, U.A. Mutations in SMARCB1 and in other Coffin-Siris syndrome genes lead to various brain midline defects. Nat. Commun. 2019, 10, 2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Ahrens-Nicklas, R.C.; Baker, J.; Bhambhani, V.; Calhoun, A.; Cohen, J.S.; Deardorff, M.A.; Fernández-Jaén, A.; Kamien, B.; Jain, M.; et al. The variability of SMARCA4-related Coffin-Siris syndrome: Do nonsense candidate variants add to milder phenotypes? Am. J. Med. Genet. A 2020, 182, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Vasko, A.; Drivas, T.G.; Schrier Vergano, S.A. Genotype-Phenotype Correlations in 208 Individuals with Coffin-Siris Syndrome. Genes 2021, 12, 937. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Mardinian, K.; Adashek, J.J.; Botta, G.P.; Kato, S.; Kurzrock, R. SMARCA4: Implications of an altered chromatin-remodeling gene for cancer development and therapy. Mol. Cancer Ther. 2021, 20, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.; Huang, S.; Banerjee, S.; Hague, J.; Hendricks, W.P.D.; Huntsman, D.G.; Lang, J.D.; Orlando, K.A.; Oza, A.M.; Pautier, P.; et al. Small-Cell Carcinoma of the Ovary, Hypercalcemic Type-Genetics, New Treatment Targets, and Current Management Guidelines. Clin. Cancer Res. 2020, 26, 3908–3917. [Google Scholar] [CrossRef] [Green Version]
- Shah, R.R. Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [Google Scholar] [CrossRef]
- Rao, V.; Bauer, F.; Vredenburgh, J.J. Refractory Small Cell Carcinoma of the Ovary—Hypercalcemic Type (SCCOHT) Treated with Romidepsin and Topotecan: A Case Report and Review of the Literature. Conn. Med. 2016, 80, 529–532. [Google Scholar]
- Chen, Y.; Zhang, H.; Xu, Z.; Tang, H.; Geng, A.; Cai, B.; Su, T.; Shi, J.; Jiang, C.; Tian, X.; et al. A PARP1-BRG1-SIRT1 axis promotes HR repair by reducing nucleosome density at DNA damage sites. Nucleic Acids Res. 2019, 47, 8563–8580. [Google Scholar]
- Kurashima, K.; Kashiwagi, H.; Shimomura, I.; Suzuki, A.; Takeshita, F.; Mazevet, M.; Harata, M.; Yamashita, T.; Yamamoto, Y.; Kohno, T.; et al. SMARCA4 deficiency-associated heterochromatin induces intrinsic DNA replication stress and susceptibility to ATR inhibition in lung adenocarcinoma. NAR Cancer 2020, 2, zcaa005. [Google Scholar] [CrossRef] [PubMed]

| Hereditary Cancer Syndromes | Related Cancers 1 (%) | Affected Genes | Potential Targets | Drugs | Available Clinical Trials | References | |
|---|---|---|---|---|---|---|---|
| 2Cowden | Breast (77–85) | PTEN (~85%) | |||||
| Thyroid (21–38) | KLLN | 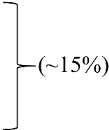 | mTOR | Everolimus, Sirolimus | NCT02461446 | [10] | |
| Kidney (15–34) | SDHx | AKT | ARQ092 | [11] | |||
| Colon (9–16) | SEC23B | PIK3CA | BYL719 | [12] | |||
| Endometrium (19–28) | WWP1 | ||||||
| Peutz-Jeghers | Breast (45–50) | STK11 | |||||
| Colon (36) | |||||||
| Gastric (29) | |||||||
| Small intestine (13) | PARP | Olaparib, Talazoparib | NCT03375307, NCT04173507 | [13] | |||
| Pancreas (11–36) | Glutaminase | CB-839 HCl | NCT03872427 | [14] | |||
| Lung (15–17) | |||||||
| SCTAT (36) | |||||||
| MDA (15–30) | |||||||
| DICER1 | PPB (70) | DICER1 | Metformin | Metformin | [15,16,17] | ||
| SLCT (60) | PI3K/ATK/mTOR | Rapamycin, TORIN-1 | [18] | ||||
| ERMS (~50) | |||||||
| RTPS2 | SMARC4A | PD-1 | Pembrolizumab | NCT03012620 | [19,20,21,22] | ||
| EZH2 | Tazemetostat | NCT02601950 | [23] | ||||
| CNS (65) | LSD1 | Seclidemstat | NCT04611139 | [24] | |||
| Kidney (~7) | CDK4/6 | Abemaciclib, Palbociclib | NCT02644460, NCT03297606 | [25,26] | |||
| SCCOHT (~40) | BET | JQ1, OTX-015 | [27,28] | ||||
| HDAC | Quisinostat | [29] | |||||
| AURKA | VX-680 | [30] | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, T.; Soeda, S.; Endo, Y.; Okabe, C.; Sato, T.; Kamo, N.; Ueda, M.; Kojima, M.; Furukawa, S.; Nishigori, H.; et al. Rare Hereditary Gynecological Cancer Syndromes. Int. J. Mol. Sci. 2022, 23, 1563. https://doi.org/10.3390/ijms23031563
Watanabe T, Soeda S, Endo Y, Okabe C, Sato T, Kamo N, Ueda M, Kojima M, Furukawa S, Nishigori H, et al. Rare Hereditary Gynecological Cancer Syndromes. International Journal of Molecular Sciences. 2022; 23(3):1563. https://doi.org/10.3390/ijms23031563
Chicago/Turabian StyleWatanabe, Takafumi, Shu Soeda, Yuta Endo, Chikako Okabe, Tetsu Sato, Norihito Kamo, Makiko Ueda, Manabu Kojima, Shigenori Furukawa, Hidekazu Nishigori, and et al. 2022. "Rare Hereditary Gynecological Cancer Syndromes" International Journal of Molecular Sciences 23, no. 3: 1563. https://doi.org/10.3390/ijms23031563
APA StyleWatanabe, T., Soeda, S., Endo, Y., Okabe, C., Sato, T., Kamo, N., Ueda, M., Kojima, M., Furukawa, S., Nishigori, H., Takahashi, T., & Fujimori, K. (2022). Rare Hereditary Gynecological Cancer Syndromes. International Journal of Molecular Sciences, 23(3), 1563. https://doi.org/10.3390/ijms23031563