
Different Expression Levels of DLK2 Inhibit NOTCH Signaling and Inversely Modulate MDA-MB-231 Breast Cancer Tumor Growth In Vivo


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
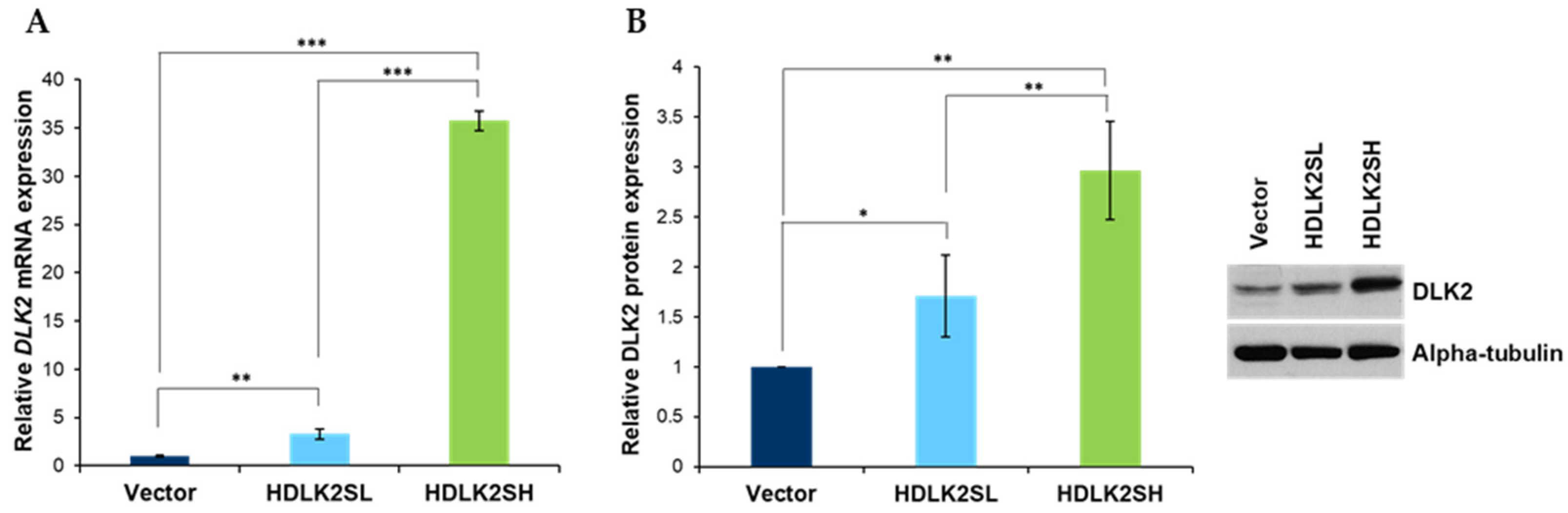
2.1. Generation of Transfected MDA-MB-231 Cells Stably Expressing Different Levels of Human DLK2 Protein
2.2. Different Levels of DLK2 Expression Inversely Modulate the Growth of MDA-MB-231 Cells
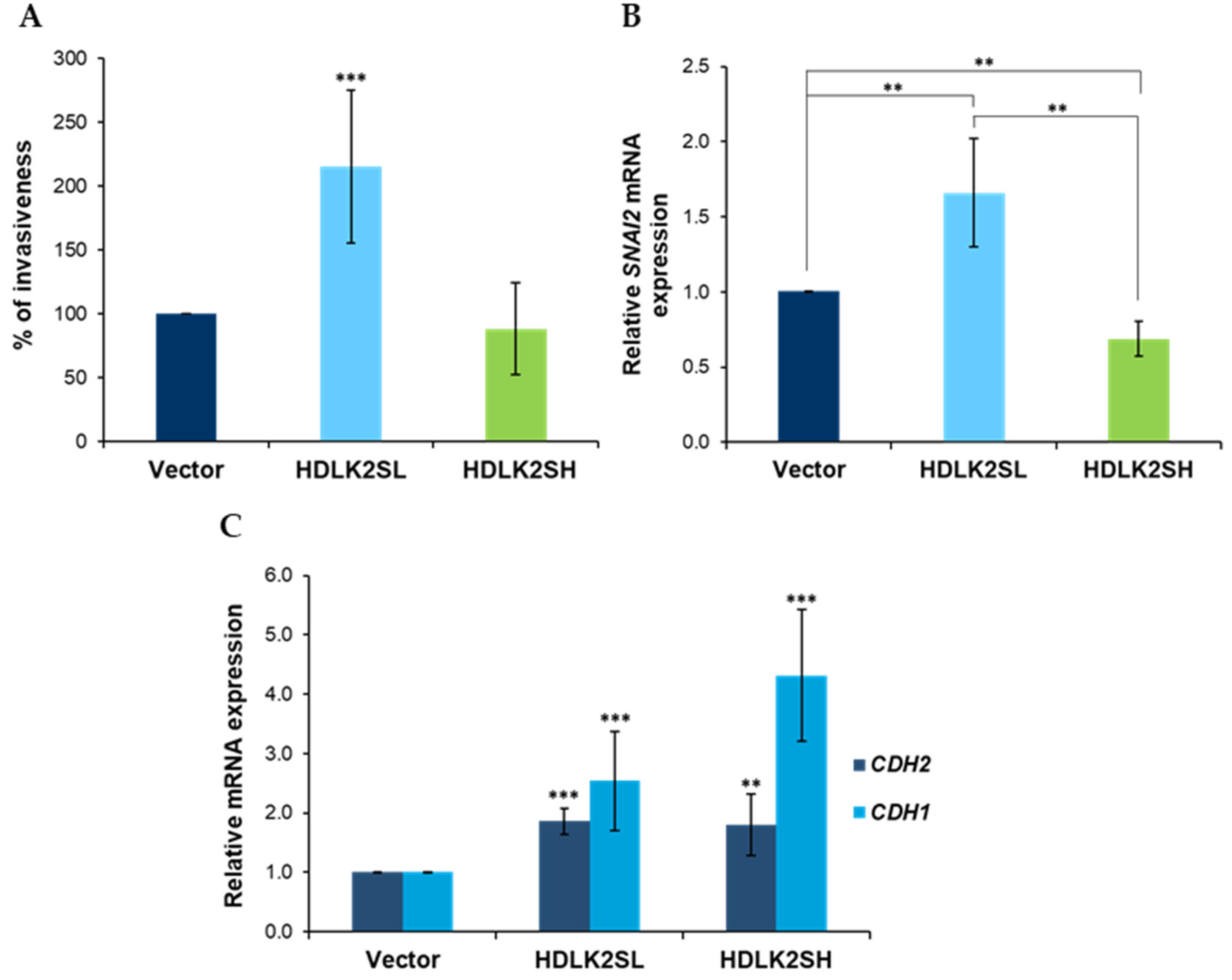
2.3. Invasion Features of DLK2-Overexpressing MDA-MB-231 Cells
2.4. DLK2 Inhibited NOTCH Activation in MDA-MB-231 Cells in a Dose-Dependent Manner
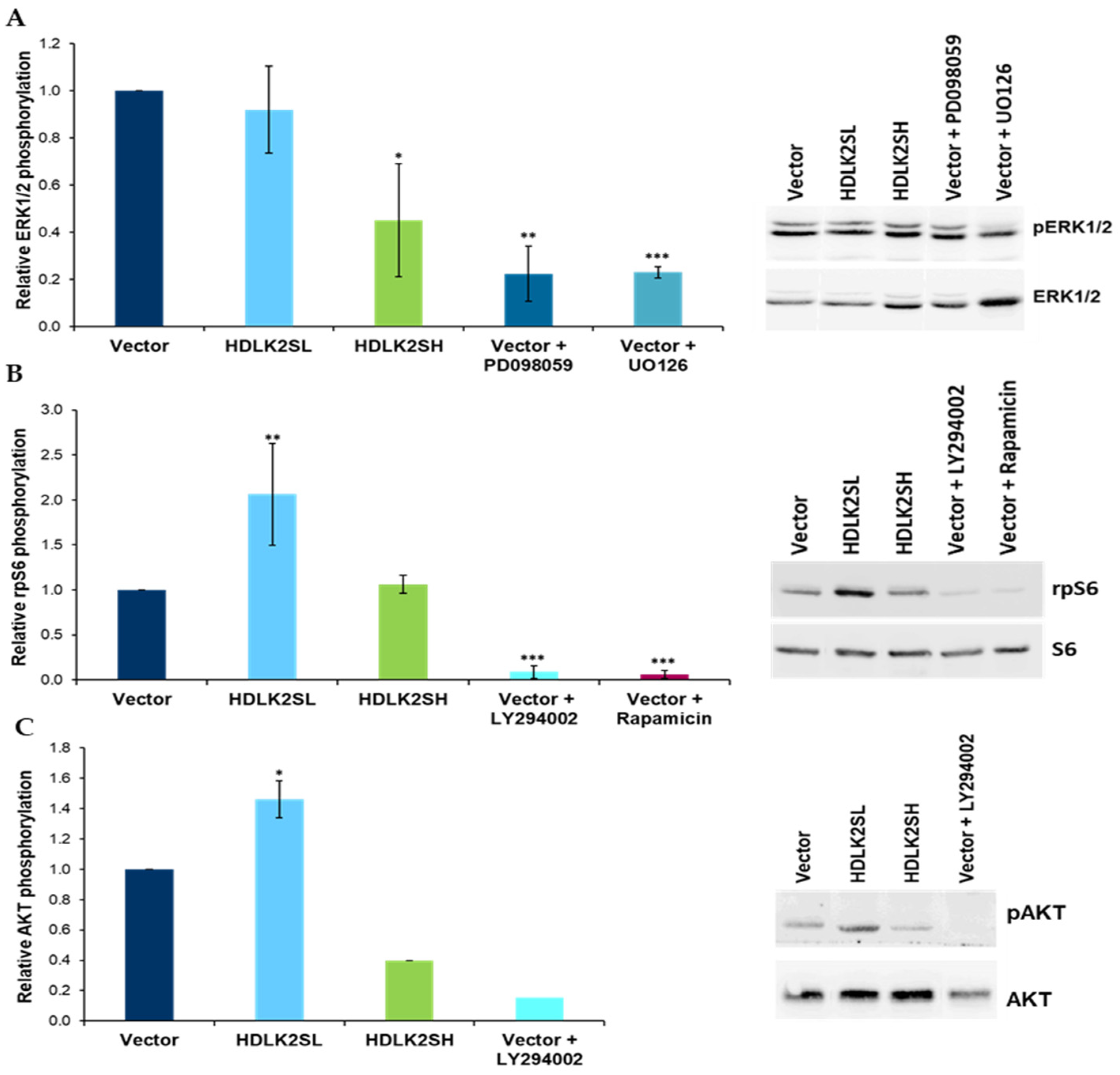
2.5. DLK2 Modulates Phosphorylation of ERK1/2 MAPK, AKT and rpS6 Kinases in MDA-MB-231 Cells
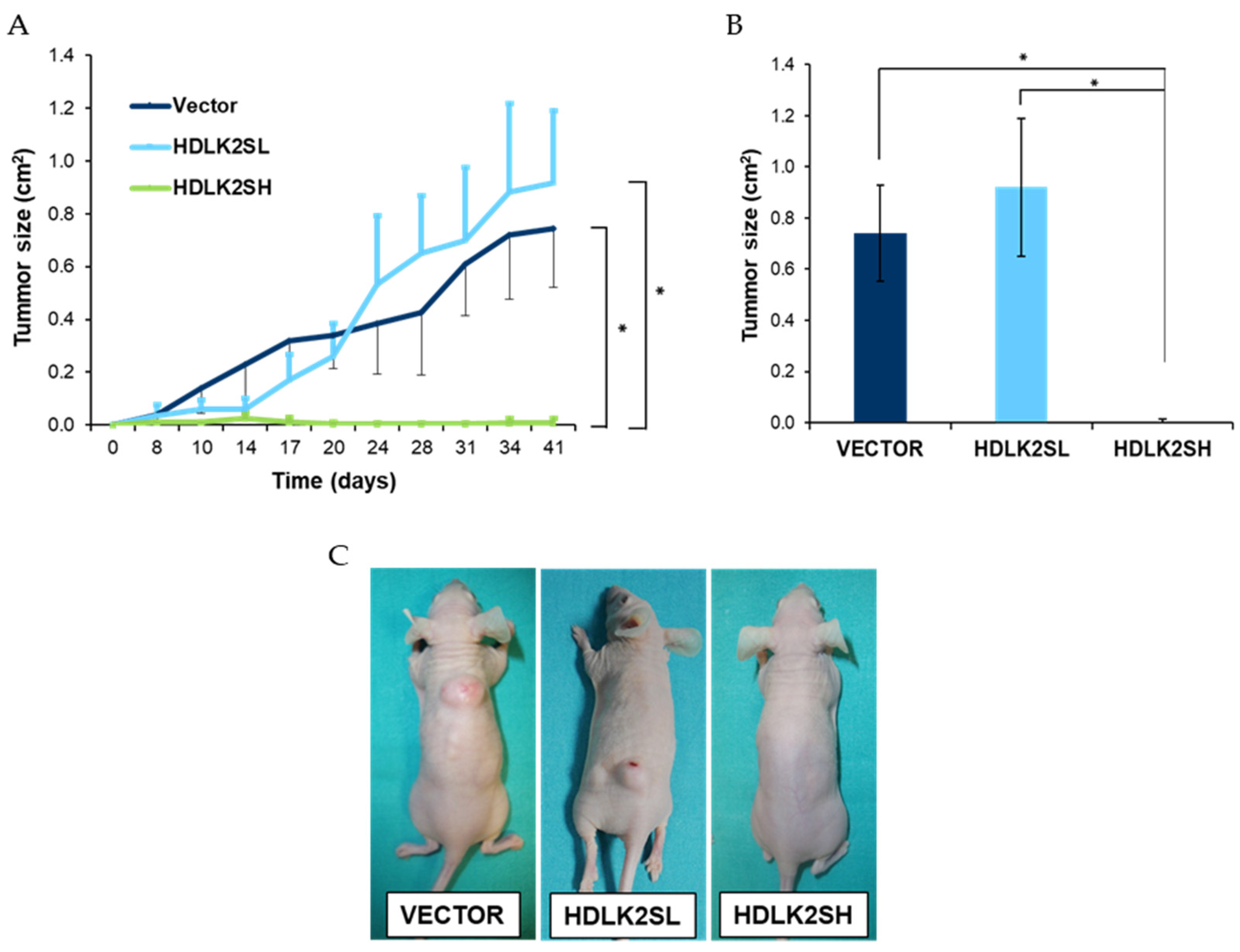
2.6. DLK2 Overexpression Affects MDA-MB-231 Tumor Development In Vivo
3. Discussion
4. Materials and Methods
4.1. Plasmids
4.2. Cell Culture and Transfection Conditions
4.3. Cell Growth Assays
4.4. Cell Cycle Assays
4.5. Annexin V Assay
4.6. Invasion Assays
4.7. RNA Extraction and RT-qPCR
4.8. Western Blot
4.9. Luciferase Assays
4.10. Animal Studies
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grishina, I.B. Mini-review: Does Notch promote or suppress cancer? New findings and old controversies. Am. J. Clin. Exp. Urol. 2015, 3, 24–27. [Google Scholar] [PubMed]
- Gil-Garcia, B.; Baladron, V. The complex role of NOTCH receptors and their ligands in the development of hepatoblastoma, cholangiocarcinoma and hepatocellular carcinoma. Biol. Cell 2016, 108, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Zhou, Y.; Cheng, A.S.; Yu, J.; To, K.F.; Kang, W. NOTCH receptors in gastric and other gastrointestinal cancers: Oncogenes or tumor suppressors? Mol. Cancer 2016, 15, 80. [Google Scholar] [CrossRef] [Green Version]
- Robbins, J.; Blondel, B.J.; Gallahan, D.; Callahan, R. Mouse mammary tumor gene int-3: A member of the notch gene family transforms mammary epithelial cells. J. Virol. 1992, 66, 2594–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.; Brennan, K. Notch Signalling in Breast Development and Cancer. Front. Cell Dev. Biol. 2021, 9, 692173. [Google Scholar] [CrossRef] [PubMed]
- Baladron, V.; Ruiz-Hidalgo, M.; Nueda, M.; Diaz-Guerra, M.; Garcia-Ramirez, J.; Bonvini, E.; Gubina, E.; Laborda, J. dlk acts as a negative regulator of Notch1 activation through interactions with specific EGF-like repeats. Exp. Cell Res. 2005, 303, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Nueda, M.; Baladrón, V.; Sánchez-Solana, B.; Ballesteros, M.; Laborda, J. The EGFlike protein dlk1 inhibits notch signaling and potentiates adipogenesis of mesenchymal cells. J. Mol. Biol. 2007, 367, 1281–1293. [Google Scholar] [CrossRef]
- Nueda, M.; González-Gómez, M.; Rodríguez-Cano, M.; Monsalve, E.; Díaz-Guerra, M.; Sánchez-Solana, B.; Laborda, J.; Baladrón, V. DLK proteins modulate NOTCH signaling to influence a brown or white 3T3-L1 adipocyte fate. Sci. Rep. 2018, 8, 16923. [Google Scholar] [CrossRef] [PubMed]
- Nueda, M.-L.; Naranjo, A.-I.; Baladron, V.; Laborda, J. The proteins DLK1 and DLK2 modulate NOTCH1-dependent proliferation and oncogenic potential of human SK-MEL-2 melanoma cells. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 2674–2684. [Google Scholar] [CrossRef] [Green Version]
- Nueda, M.L.; Naranjo, A.I.; Baladron, V.; Laborda, J. Different expression levels of DLK1 inversely modulate the oncogenic potential of human MDA-MB-231 breast cancer cells through inhibition of NOTCH1 signaling. FASEB J. 2017, 31, 3484–3496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Solana, B.; Luisa Nueda, M.; Desamparados Ruvira, M.; Jose Ruiz-Hidalgo, M.; Maria Monsalve, E.; Rivero, S.; Javier Garcia-Ramirez, J.; Diaz-Guerra, M.J.M.; Baladron, V.; Laborda, J. The EGF-like proteins DLK1 and DLK2 function as inhibitory non-canonical ligands of NOTCH1 receptor that modulate each other’s activities. Biochim. Biophys. Acta-Mol. Cell Res. 2011, 1813, 1153–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traustadottir, G.A.; Jensen, C.H.; Thomassen, M.; Beck, H.C.; Mortensen, S.B.; Laborda, J.; Baladron, V.; Sheikh, S.P.; Andersen, D.C. Evidence of non-canonical NOTCH signaling: Delta-like 1 homolog (DLK1) directly interacts with the NOTCH1 receptor in mammals. Cell Signal 2016, 28, 246–254. [Google Scholar] [CrossRef]
- Rodriguez-Cano, M.M.; Gonzalez-Gomez, M.J.; Sanchez-Solana, B.; Monsalve, E.M.; Diaz-Guerra, M.M.; Laborda, J.; Nueda, M.L.; Baladron, V. NOTCH Receptors and DLK Proteins Enhance Brown Adipogenesis in Mesenchymal C3H10T1/2 Cells. Cells 2020, 9, 2032. [Google Scholar] [CrossRef] [PubMed]
- Nueda, M.-L.; Baladron, V.; Garcia-Ramirez, J.-J.; Sanchez-Solana, B.; Ruvira, M.-D.; Rivero, S.; Ballesteros, M.-A.; Monsalve, E.-M.; Diaz-Guerra, M.-J.M.; Ruiz-Hidalgo, M.-J.; et al. The novel gene EGFL9/Dlk2, highly homologous to Dlk1, functions as a modulator of adipogenesis. J. Mol. Biol. 2007, 367, 1270–1280. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Khaybullin, R.; Zhang, Y.; Xia, A.; Qi, X. Gene expression profiling leads to discovery of correlation of matrix metalloproteinase 11 and heparanase 2 in breast cancer progression. BMC Cancer 2015, 15, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Wu, L.; Dong, L.; Mitchell, A.V.; James Block, C.; Liu, J.; Zhang, H.; Lu, Q.; Song, W.M.; Zhang, B.; et al. EGFL9 promotes breast cancer metastasis by inducing cMET activation and metabolic reprogramming. Nat. Commun. 2019, 10, 5033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royer-Bertrand, B.; Torsello, M.; Rimoldi, D.; El Zaoui, I.; Cisarova, K.; Pescini-Gobert, R.; Raynaud, F.; Zografos, L.; Schalenbourg, A.; Speiser, D.; et al. Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet. 2016, 99, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef] [Green Version]
- Mallini, P.; Lennard, T.; Kirby, J.; Meeson, A. Epithelial-to-mesenchymal transition: What is the impact on breast cancer stem cells and drug resistance. Cancer Treat. Rev. 2014, 40, 341–348. [Google Scholar] [CrossRef]
- Huang, Z.; Yu, P.; Tang, J. Characterization of Triple-Negative Breast Cancer MDA-MB-231 Cell Spheroid Model. OncoTargets Ther. 2020, 13, 5395–5405. [Google Scholar] [CrossRef] [PubMed]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar] [PubMed]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P.; Yamada, K.M.; Thiery, J.P. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J. Cell Biol. 1997, 137, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Hair, A.; Razin, S.V.; Vasetskii, E.S. Changes in chromatin organization during early development and carcinogenesis. Ontogenez 2002, 33, 85–89. [Google Scholar] [PubMed]
- Fender, A.W.; Nutter, J.M.; Fitzgerald, T.L.; Bertrand, F.E.; Sigounas, G. Notch-1 promotes stemness and epithelial to mesenchymal transition in colorectal cancer. J. Cell Biochem. 2015, 116, 2517–2527. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, H.; Yoshiura, S.; Ohtsuka, T.; Bessho, Y.; Harada, T.; Yoshikawa, K.; Kageyama, R. Oscillatory expression of the bHLH factor Hes1 regulated by a negative feedback loop. Science 2002, 298, 840–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukazawa, H.; Noguchi, K.; Murakami, Y.; Uehara, Y. Mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK) inhibitors restore anoikis sensitivity in human breast cancer cell lines with a constitutively activated extracellular-regulated kinase (ERK) pathway. Mol. Cancer Ther. 2002, 1, 303–309. [Google Scholar]
- Chen, L.; Qanie, D.; Jafari, A.; Taipaleenmaki, H.; Jensen, C.H.; Saamanen, A.M.; Sanz, M.L.; Laborda, J.; Abdallah, B.M.; Kassem, M. Delta-like 1/fetal antigen-1 (Dlk1/FA1) is a novel regulator of chondrogenic cell differentiation via inhibition of the Akt kinase-dependent pathway. J. Biol. Chem. 2011, 286, 32140–32149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Noohr, J.; Jensen, C.H.; Petersen, R.K.; Bachmann, E.; Teisner, B.; Larsen, L.K.; Mandrup, S.; Kristiansen, K. Insulin-like growth factor-1/insulin bypasses Pref-1/FA1-mediated inhibition of adipocyte differentiation. J. Biol. Chem. 2003, 278, 20906–20914. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Hidalgo, M.; Gubina, E.; Tull, L.; Baladron, V.; Laborda, J. dlk modulates mitogen-activated protein kinase signaling to allow or prevent differentiation. Exp. Cell Res. 2002, 274, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Rivero, S.; Diaz-Guerra, M.J.; Monsalve, E.M.; Laborda, J.; Garcia-Ramirez, J.J. DLK2 is a transcriptional target of KLF4 in the early stages of adipogenesis. J. Mol. Biol. 2012, 417, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, Y.; Zhao, H.; Fan, B.; Guo, K.; Cai, M.; Zhang, S. Delta-like 2 negatively regulates chondrogenic differentiation. J. Cell. Physiol. 2018, 233, 6574–6582. [Google Scholar] [CrossRef] [PubMed]
- Giuli, M.V.; Giuliani, E.; Screpanti, I.; Bellavia, D.; Checquolo, S. Notch Signaling Activation as a Hallmark for Triple-Negative Breast Cancer Subtype. J. Oncol. 2019, 2019, 8707053. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, I.; Onishi, H.; Matsushita, S.; Kubo, M.; Kai, M.; Imaizumi, A.; Nakano, K.; Hattori, M.; Oda, Y.; Tanaka, M.; et al. NOTCH4 is a potential therapeutic target for triple-negative breast cancer. Anticancer. Res. 2014, 34, 69–80. [Google Scholar]
- Dou, X.W.; Liang, Y.K.; Lin, H.Y.; Wei, X.L.; Zhang, Y.Q.; Bai, J.W.; Chen, C.F.; Chen, M.; Du, C.W.; Li, Y.C.; et al. Notch3 Maintains Luminal Phenotype and Suppresses Tumorigenesis and Metastasis of Breast Cancer via Trans-Activating Estrogen Receptor-alpha. Theranostics 2017, 7, 4041–4056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.F.; Dou, X.W.; Liang, Y.K.; Lin, H.Y.; Bai, J.W.; Zhang, X.X.; Wei, X.L.; Li, Y.C.; Zhang, G.J. Notch3 overexpression causes arrest of cell cycle progression by inducing Cdh1 expression in human breast cancer cells. Cell Cycle 2016, 15, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, M.; Selfors, L.M.; Albeck, J.; Overholtzer, M.; Sale, S.; Carroll, D.L.; Pandya, D.; Lu, Y.; Mills, G.B.; Aster, J.C.; et al. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5012–5017. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Paranjape, A.N.; Rangarajan, A.; Dighe, R.R. A monoclonal antibody against human Notch1 ligand-binding domain depletes subpopulation of putative breast cancer stem-like cells. Mol. Cancer Ther. 2012, 11, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Inukai, T.; Inoue, A.; Kurosawa, H.; Goi, K.; Shinjyo, T.; Ozawa, K.; Mao, M.; Inaba, T.; Look, A.T. SLUG, a ces-1-related zinc finger transcription factor gene with antiapoptotic activity, is a downstream target of the E2A-HLF oncoprotein. Mol. Cell 1999, 4, 343–352. [Google Scholar] [CrossRef]
- Bolos, V.; Mira, E.; Martinez-Poveda, B.; Luxan, G.; Canamero, M.; Martinez, A.C.; Manes, S.; de la Pompa, J.L. Notch activation stimulates migration of breast cancer cells and promotes tumor growth. Breast Cancer Res. 2013, 15, R54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tan, J.; Zhang, Y.; Han, N.; Di, X.; Xiao, T.; Cheng, S.; Gao, Y.; Liu, Y. DLK1 promotes lung cancer cell invasion through upregulation of MMP9 expression depending on Notch signaling. PLoS ONE 2014, 9, e91509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.H.; Sung, I.C.; Fang, W.L.; Chi, C.W.; Yeh, T.S.; Lee, H.C.; Yin, P.H.; Li, A.F.; Wu, C.W.; Shyr, Y.M.; et al. Correlation between HGF/c-Met and Notch1 signaling pathways in human gastric cancer cells. Oncol. Rep. 2018, 40, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, N.; Zhang, Y.; Cai, Y.; Liu, Y.; Qu, X.; Li, Z.; Li, D.; Hou, K.; Kang, J.; et al. Coexpression of c-Met and Notch-1 correlates with poor prognosis in resected non-small-cell lung cancer. Tumour Biol. 2015, 36, 7053–7059. [Google Scholar] [CrossRef]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, M.A.; Oza, G.; Sharma, A.; Arriaga, L.G.; Hernandez Hernandez, J.M.; Rotello, V.M.; Ramirez, J.T. Triple-Negative Breast Cancer: A Review of Conventional and Advanced Therapeutic Strategies. Int. J. Environ. Res. Public Health 2020, 17, 2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, S.; Sharma, A.; Balaji, S.A.; Gowda, M.C.; Dighe, R.R.; Kumar, R.V.; Rangarajan, A. Coordinate hyperactivation of Notch1 and Ras/MAPK pathways correlates with poor patient survival: Novel therapeutic strategy for aggressive breast cancers. Mol. Cancer Ther. 2014, 13, 3198–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izrailit, J.; Berman, H.K.; Datti, A.; Wrana, J.L.; Reedijk, M. High throughput kinase inhibitor screens reveal TRB3 and MAPK-ERK/TGFbeta pathways as fundamental Notch regulators in breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 1714–1719. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophys. Acta 2011, 1815, 197–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naranjo, A.-I.; González-Gómez, M.-J.; Baladrón, V.; Laborda, J.; Nueda, M.-L. Different Expression Levels of DLK2 Inhibit NOTCH Signaling and Inversely Modulate MDA-MB-231 Breast Cancer Tumor Growth In Vivo. Int. J. Mol. Sci. 2022, 23, 1554. https://doi.org/10.3390/ijms23031554
Naranjo A-I, González-Gómez M-J, Baladrón V, Laborda J, Nueda M-L. Different Expression Levels of DLK2 Inhibit NOTCH Signaling and Inversely Modulate MDA-MB-231 Breast Cancer Tumor Growth In Vivo. International Journal of Molecular Sciences. 2022; 23(3):1554. https://doi.org/10.3390/ijms23031554
Chicago/Turabian StyleNaranjo, Ana-Isabel, María-Julia González-Gómez, Victoriano Baladrón, Jorge Laborda, and María-Luisa Nueda. 2022. "Different Expression Levels of DLK2 Inhibit NOTCH Signaling and Inversely Modulate MDA-MB-231 Breast Cancer Tumor Growth In Vivo" International Journal of Molecular Sciences 23, no. 3: 1554. https://doi.org/10.3390/ijms23031554
APA StyleNaranjo, A.-I., González-Gómez, M.-J., Baladrón, V., Laborda, J., & Nueda, M.-L. (2022). Different Expression Levels of DLK2 Inhibit NOTCH Signaling and Inversely Modulate MDA-MB-231 Breast Cancer Tumor Growth In Vivo. International Journal of Molecular Sciences, 23(3), 1554. https://doi.org/10.3390/ijms23031554