
Molecular Aspects of Resistance to Immunotherapies—Advances in Understanding and Management of Diffuse Large B-Cell Lymphoma

 , , ,
, , ,
Abstract
1. Introduction
1.1. Diffuse Large B-Cell Lymphoma
1.2. Rituximab (RTX)
1.3. Mechanisms of Rituximab Cytotoxicity
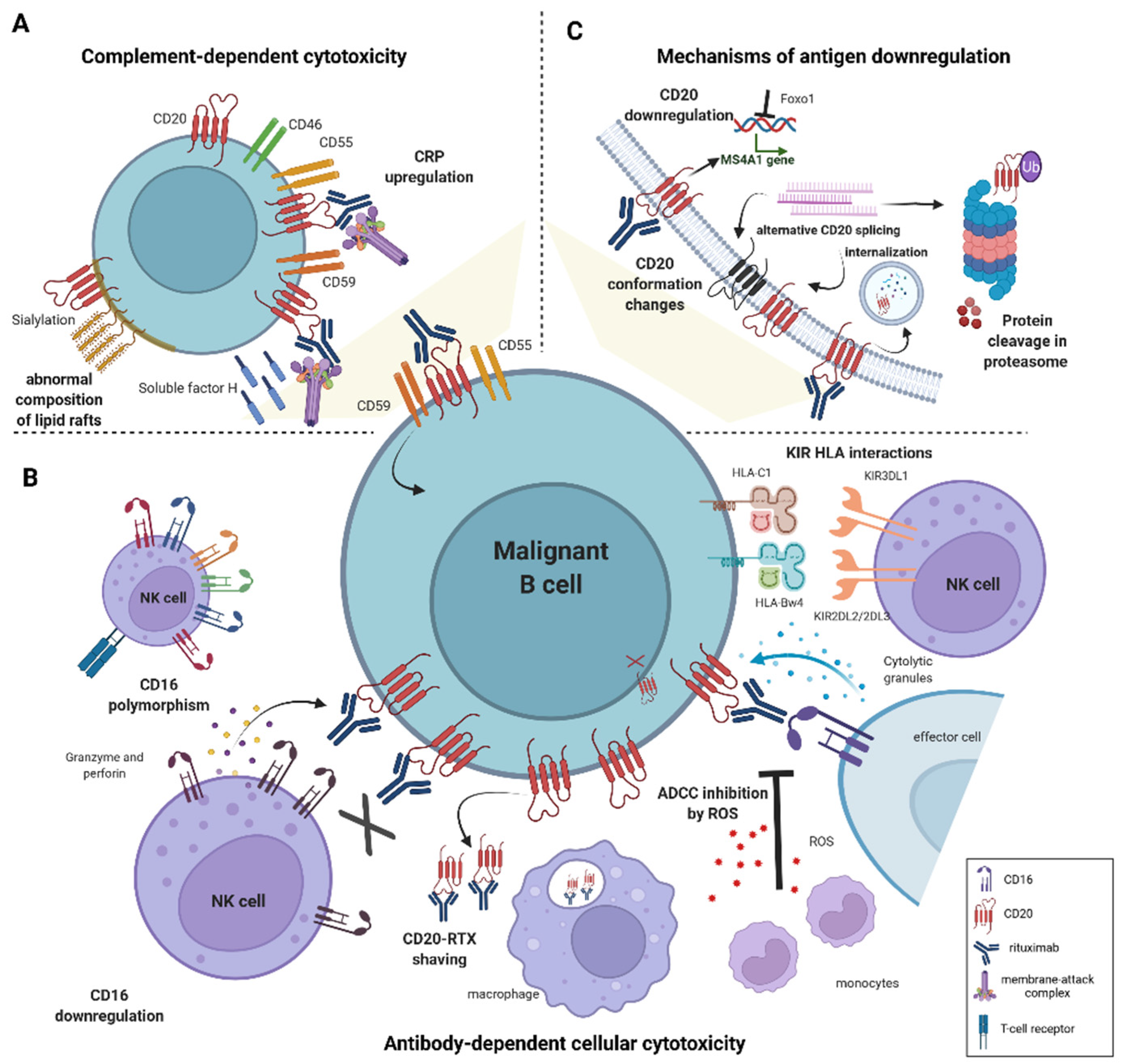
2. Resistance to Antibody-Dependent Cellular Cytotoxicity
2.1. Mechanisms of Primary Resistance to ADCC
2.1.1. Effector Cell Characteristics Influencing ADCC Efficacy
Genetic Polymorphism in FcγRIII Receptor
Low CD16 Expression
KIR/HLA Interactions
2.1.2. The Influence of the Microenvironment on ADCC Efficacy
2.1.3. Characteristics of Malignant B-Cells Contributing to Impaired ADCC
2.2. Studies on Acquired Resistance
2.2.1. ADCC-Resistance Models to Study Acquired Rituximab Resistance
2.2.2. RTX-Mediated Changes in NK Cells
2.3. Solutions to Improve ADCC Efficacy
2.3.1. Fc Receptor Engineering
2.3.2. Supplementation with Cytokines
3. Resistance to Complement-Dependent Cytotoxicity
3.1. The Role of the Cell Membrane Composition in Determining Primary Resistance to Rituximab
3.2. Rituximab-Resistant Cell Lines as a Tool to Study the Mechanisms of Acquired Resistance to CDC
3.3. CD20 Downregulation in Acquired Resistance to Rituximab
3.4. Hexamerization-Inducing Mutations as a Way to Improve CDC Efficacy
4. Mechanisms of Chemoresistance following Rituximab-Treatment
5. The Role of Tumor Microenvironment in Promoting Resistance to Rituximab-Containing Regimens
Targeting Immune Microenvironment as a Way to Improve Rituximab Efficacy
6. Other Treatment Modalities as Possible Solutions for Relapsed/Refractory Patients
6.1. Novel Targets for mAbs
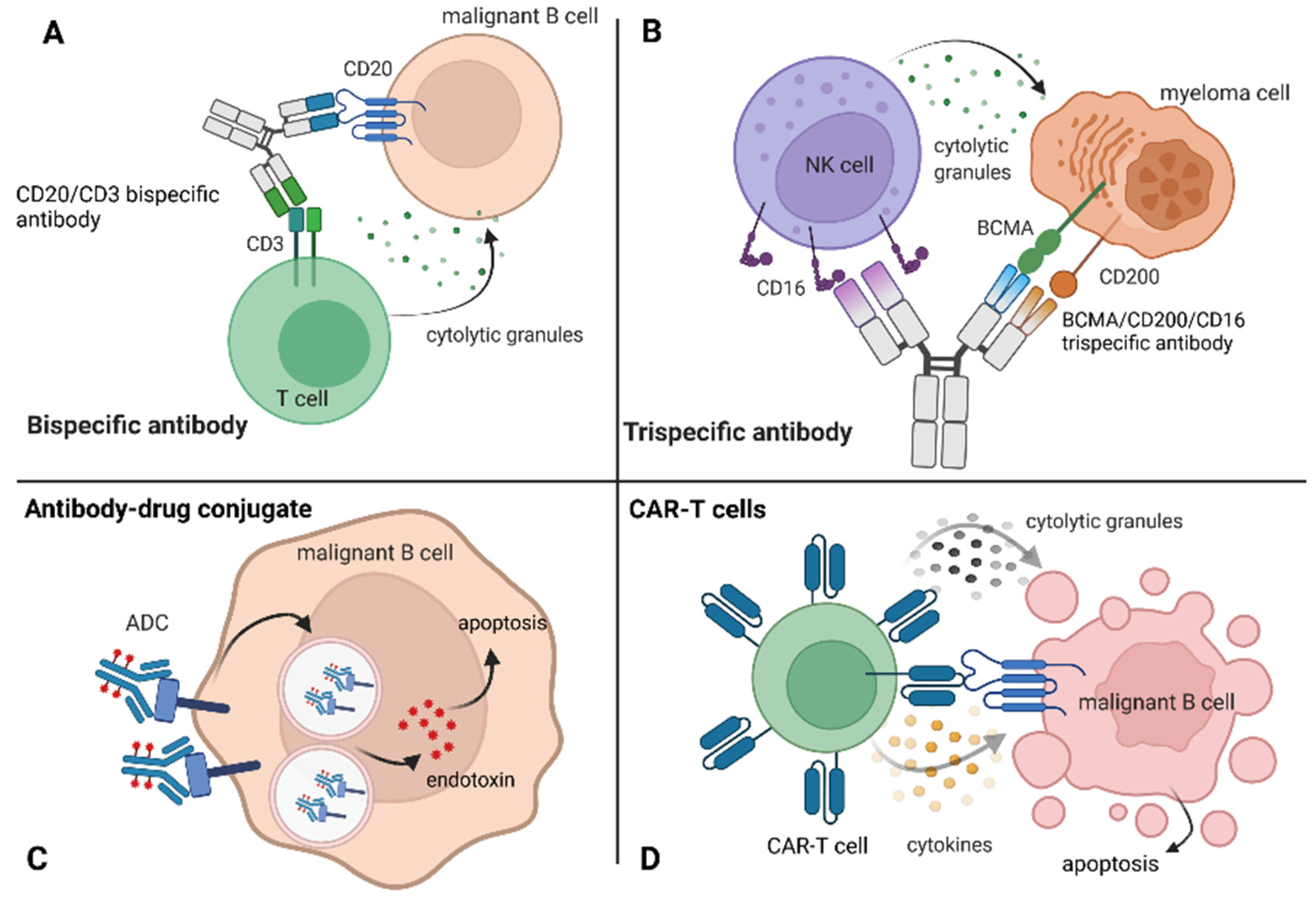
6.2. Bispecific Antibodies
6.3. Antibody–Drug Conjugates

{kind=link}
{kind=link}
| Name | Target Antigen | Cytotoxic Payload and its Mechanism of Action | Possible Application of ADC | Clinical Efficacy of ADC |
|---|---|---|---|---|
| Polatuzumab vedotin (PV) | CD79b | Monomethyl auristatin E. (MMAE) with a cleavable linker for disruption of microtubule network | DLBCL–registration | PV was combined with RTX and bendamustine (pola-BR) and compared to bendamustine and RTX treatment alone. Pola-BR patients had a significantly higher CR rate (40.0% vs. 17.5%; p = 0.026) and longer PFS [176]. |
| Brentuximab vedotin (BV) | CD30 | auristatin E. (MMAE) with a cleavable linker for disruption of microtubule network | HL–registration ALCL-registration | 75% and 86% OR rates in r/r HL and ALCL, consecutively [185]. |
| Inotuzumab ozogamicin (CMC-544) | CD22 | Calicheamicin with a cleavable (acid-labile) linker for disruption of double-stranded DNA in the nucleus | NHL B-ALL–registration | 39% ORR in r/r/B-ALL (NCT01134575) CMC-544 in combination with RTX resulted in longer survival in a disseminated B-cell lymphoma murine model [186]. |
| AGS67E | CD37 | Monomethyl auristatin E. (MMAE) with protease-cleavable linker for disruption of microtubule network | NHL CLL | Until now only one clinical trial was completed, the safety of AGS67E was demonstrated in patients with r/r lymphoid malignancies (NCT02175433) [187]. |
| Pinatuzumab vedotin | CD22 | Monomethyl auristatin E. (MMAE) with a cleavable linker (with sulfhydryl groups) for disruption of microtubule network | NHL | Objective responses were observed in DLBCL (9/25); CR in 2/8 patients treated with pinatuzumab vedotin and RTX [188]. |
| Naratuximab emtansine (IMGN529) | CD37 | Emtansine (DM1) with a non-cleavable linker for disruption of microtubule polymerization | r/r B-cell lymphomas | In a phase 1 study on B -NHL, a reduction in lymphocyte count was observed after the second dosing of IMGN529 [189]. |
| Denintuzumab mafodotin (SGN-CD19A) | CD19 | Monomethyl auristatin F with a cleavable linker for disruption of tubulin polymerization | NHL | In a phase 1 study SGN-CD19A exerted durable responses in heavily pre-treated NHL patients; 56% objective responses were achieved for relapsed patients with a CR rate of 40% [190]. |
| Coltuximab ravtansine (SAR3419) | CD19 | Ravtansine (DM4) with a cleavable linker (with disulfide groups) for disruption of tubulin polymerization | DLBCL | In a phase 2 study on r/r/DLBCL, ORR was achieved in 43% of patients (18/41), where PFS and OS were 4.4 and 9.2 months, respectively [191]. |
6.4. CAR-T Cells
7. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Padala, S.A.; Barsouk, A.; Rawla, P. Epidemiology of Non-Hodgkin’s Lymphoma. Med. Sci. 2021, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Harvey, C.; Richards, C.; Kim, C. Temporal Trends in Treatment and Survival of Older Adult Diffuse Large B-Cell Lymphoma Patients in the SEER-Medicare Linked Database. Leuk. Lymphoma 2019, 60, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- Rovira, J.; Valera, A.; Colomo, L.; Setoain, X.; Rodríguez, S.; Martínez-Trillos, A.; Giné, E.; Dlouhy, I.; Magnano, L.; Gaya, A.; et al. Prognosis of Patients with Diffuse Large B Cell Lymphoma Not Reaching Complete Response or Relapsing after Frontline Chemotherapy or Immunochemotherapy. Ann. Hematol. 2015, 94, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in Refractory Diffuse Large B-Cell Lymphoma: Results from the International SCHOLAR-1 Study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, G.S. Recently Approved Drugs Herald a New Era in Therapy for Diffuse Large B-Cell Lymphoma. Clin. Adv. Hematol. Oncol. 2021, 19, 284–287. [Google Scholar]
- Wang, L.; Li, L.; Young, K.H. New Agents and Regimens for Diffuse Large B Cell Lymphoma. J. Hematol. Oncol. 2020, 13, 175. [Google Scholar] [CrossRef]
- He, M.Y.; Kridel, R. Treatment Resistance in Diffuse Large B-Cell Lymphoma. Leukemia 2021, 35, 2151–2165. [Google Scholar] [CrossRef]
- Engelhard, M. Anti-CD20 Antibody Treatment of Non-Hodgkin Lymphomas. Clin. Immunol. 2016, 172, 101–104. [Google Scholar] [CrossRef]
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef]
- Winiarska, M.; Glodkowska-Mrowka, E.; Bil, J.; Golab, J. Molecular Mechanisms of the Antitumor Effects of Anti-CD20 Antibodies. Front. Biosci. (Landmark Ed) 2011, 16, 277–306. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.; Glennie, M. The Mechanisms of Action of Rituximab in the Elimination of Tumor Cells. Semin. Oncol. 2003, 30, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Pavlasova, G.; Mraz, M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Alas, S.; Emmanouilides, C.; Bonavida, B. Inhibition of Interleukin 10 by Rituximab Results in Down-Regulation of Bcl-2 and Sensitization of B-Cell Non-Hodgkin’s Lymphoma to Apoptosis. Clin. Cancer Res. 2001, 7, 709–723. [Google Scholar]
- Jazirehi, A.R.; Bonavida, B. Cellular and Molecular Signal Transduction Pathways Modulated by Rituximab (Rituxan, Anti-CD20 MAb) in Non-Hodgkin’s Lymphoma: Implications in Chemosensitization and Therapeutic Intervention. Oncogene 2005, 24, 2121–2143. [Google Scholar] [CrossRef]
- Vega, M.I.; Baritaki, S.; Huerta-Yepez, S.; Martinez-Paniagua, M.A.; Bonavida, B. A Potential Mechanism of Rituximab-Induced Inhibition of Tumor Growth through Its Sensitization to Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Expressing Host Cytotoxic Cells. Leuk. Lymphoma 2011, 52, 108–121. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc Receptors Modulate in Vivo Cytoxicity against Tumor Targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Hamaguchi, Y.; Xiu, Y.; Komura, K.; Nimmerjahn, F.; Tedder, T.F. Antibody Isotype-Specific Engagement of Fcγ Receptors Regulates B Lymphocyte Depletion during CD20 Immunotherapy. J. Exp. Med. 2006, 203, 743–753. [Google Scholar] [CrossRef]
- Umana, P.; Moessner, E.; Bruenker, P.; Unsin, G.; Puentener, U.; Suter, T.; Grau, R.; Schmidt, C.; Gerdes, C.; Nopora, A.; et al. Novel 3rd Generation Humanized Type II CD20 Antibody with Glycoengineered Fc and Modified Elbow Hinge for Enhanced ADCC and Superior Apoptosis Induction. Blood 2006, 108, 229. [Google Scholar] [CrossRef]
- Boross, P.; Jansen, J.H.M.; de Haij, S.; Beurskens, F.J.; van der Poel, C.E.; Bevaart, L.; Nederend, M.; Golay, J.; van de Winkel, J.G.J.; Parren, P.W.H.I.; et al. The in Vivo Mechanism of Action of CD20 Monoclonal Antibodies Depends on Local Tumor Burden. Haematologica 2011, 96, 1822–1830. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of Natural Killer Cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The Biology of Human Natural Killer-Cell Subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Alderson, K.L.; Sondel, P.M. Clinical Cancer Therapy by NK Cells via Antibody-Dependent Cell-Mediated Cytotoxicity. J. Biomed. Biotechnol. 2011, 2011, 379123. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, K.; Liu, L.; Qu, Y.; Huang, Y.; Wu, Y.; Wei, J. Effects of Complement and Serum IgG on Rituximab-Dependent Natural Killer Cell-Mediated Cytotoxicity against Raji Cells. Oncol. Lett. 2019, 17, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Koene, H.R.; Kleijer, M.; Algra, J.; Roos, D.; von dem Borne, A.E.G.K.; de Haas, M. FcγRIIIa-158V/F Polymorphism Influences the Binding of IgG by Natural Killer Cell FcγRIIIa, Independently of the FcγRIIIa-48L/R/H Phenotype. Blood 1997, 90, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic Activity of Humanized Anti-CD20 Monoclonal Antibody and Polymorphism in IgG Fc Receptor FcγRIIIa Gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.-K.; Levy, R. Two Immunoglobulin G Fragment C Receptor Polymorphisms Independently Predict Response to Rituximab in Patients with Follicular Lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef]
- Kim, D.H.; Jung, H.D.; Kim, J.G.; Lee, J.-J.; Yang, D.-H.; Park, Y.H.; Do, Y.R.; Shin, H.J.; Kim, M.K.; Hyun, M.S.; et al. FCGR3A Gene Polymorphisms May Correlate with Response to Frontline R-CHOP Therapy for Diffuse Large B-Cell Lymphoma. Blood 2006, 108, 2720–2725. [Google Scholar] [CrossRef]
- Ahlgrimm, M.; Pfreundschuh, M.; Kreuz, M.; Regitz, E.; Preuss, K.-D.; Bittenbring, J. The Impact of Fc-γ Receptor Polymorphisms in Elderly Patients with Diffuse Large B-Cell Lymphoma Treated with CHOP with or without Rituximab. Blood 2011, 118, 4657–4662. [Google Scholar] [CrossRef]
- Büyükkurt, N.; Özcan, M.A.; Ergene, Ü.; Payzın, B.; Tunalı, S.; Demirkan, F.; Özsan, H.; Pişkin, Ö.; Ündar, B. The Effect of FcγRIIIA Gene Polymorphism on the Treatment of Diffuse Large B-Cell Non-Hodgkin Lymphoma: A Multicenter Prospective Observational Study. Turk. J. Haematol. 2015, 32, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ding, H.; Jin, X.; Ding, N.; Deng, L.; He, Y.; Zhu, J.; Song, Y. FCGR3A 158V/F Polymorphism and Response to Frontline R-CHOP Therapy in Diffuse Large B-Cell Lymphoma. DNA Cell Biol. 2014, 33, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Mitroviç, Z.; Aurer, I.; Radman, I.; Ajdukoviç, R.; Sertiç, J.; Labar, B. FCγRIIIA and FCγRIIA Polymorphisms Are Not Associated with Response to Rituximab and CHOP in Patients with Diffuse Large B-Cell Lymphoma. Haematologica 2007, 92, 998–999. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Váróczy, L.; Zilahi, E.; Gyetvai, A.; Kajtár, B.; Gergely, L.; Sipka, S.; Illés, A. Fc-Gamma-Receptor IIIa Polymorphism and Gene Expression Profile Do Not Predict the Prognosis in Diffuse Large B-Cell Lymphoma Treated with R-CHOP Protocol. Pathol. Oncol. Res. 2012, 18, 43–48. [Google Scholar] [CrossRef]
- Danielou-Lazareth, A.; Henry, G.; Geromin, D.; Khaznadar, Z.; Briere, J.; Tamouza, R.; Cayuela, J.-M.; Thieblemont, C.; Toubert, A.; Dulphy, N. At Diagnosis, Diffuse Large B-Cell Lymphoma Patients Show Impaired Rituximab-Mediated NK-Cell Cytotoxicity. Eur. J. Immunol. 2013, 43, 1383–1388. [Google Scholar] [CrossRef]
- Cox, M.C.; Battella, S.; La Scaleia, R.; Pelliccia, S.; Di Napoli, A.; Porzia, A.; Cecere, F.; Alma, E.; Zingoni, A.; Mainiero, F.; et al. Tumor-Associated and Immunochemotherapy-Dependent Long-Term Alterations of the Peripheral Blood NK Cell Compartment in DLBCL Patients. Oncoimmunology 2015, 4, e990773. [Google Scholar] [CrossRef]
- Borgerding, A.; Hasenkamp, J.; Engelke, M.; Burkhart, N.; Trümper, L.; Wienands, J.; Glass, B. B-Lymphoma Cells Escape Rituximab-Triggered Elimination by NK Cells through Increased HLA Class I Expression. Exp. Hematol. 2010, 38, 213–221. [Google Scholar] [CrossRef]
- Dębska-Zielkowska, J.; Moszkowska, G.; Zieliński, M.; Zielińska, H.; Dukat-Mazurek, A.; Trzonkowski, P.; Stefańska, K. KIR Receptors as Key Regulators of NK Cells Activity in Health and Disease. Cells 2021, 10, 1777. [Google Scholar] [CrossRef]
- Makanga, D.R.; Jullien, M.; David, G.; Legrand, N.; Willem, C.; Dubreuil, L.; Walencik, A.; Touzeau, C.; Gastinne, T.; Tessoulin, B.; et al. Low Number of KIR Ligands in Lymphoma Patients Favors a Good Rituximab-Dependent NK Cell Response. Oncoimmunology 2021, 10, 1936392. [Google Scholar] [CrossRef]
- Beum, P.V.; Lindorfer, M.A.; Taylor, R.P. Within Peripheral Blood Mononuclear Cells, Antibody-Dependent Cellular Cytotoxicity of Rituximab-Opsonized Daudi Cells Is Promoted by NK Cells and Inhibited by Monocytes Due to Shaving. J. Immunol. 2008, 181, 2916–2924. [Google Scholar] [CrossRef]
- Beum, P.V.; Kennedy, A.D.; Williams, M.E.; Lindorfer, M.A.; Taylor, R.P. The Shaving Reaction: Rituximab/CD20 Complexes Are Removed from Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia Cells by THP-1 Monocytes. J. Immunol. 2006, 176, 2600–2609. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, A.E.; Jungersen, M.B.; Pedersen, C.D. Monocytes Mediate Shaving of B-Cell-Bound Anti-CD20 Antibodies. Immunology 2011, 133, 239–245. [Google Scholar] [CrossRef]
- Hansson, M.; Asea, A.; Ersson, U.; Hermodsson, S.; Hellstrand, K. Induction of Apoptosis in NK Cells by Monocyte-Derived Reactive Oxygen Metabolites. J. Immunol. 1996, 156, 42–47. Available online: https://www.jimmunol.org/content/156/1/42 (accessed on 30 October 2021). [PubMed]
- Klopotowska, M.; Bajor, M.; Graczyk-Jarzynka, A.; Kraft, A.; Pilch, Z.; Zhylko, A.; Firczuk, M.; Baranowska, I.; Lazniewski, M.; Plewczynski, D.; et al. PRDX-1 Supports the Survival and Antitumor Activity of Primary and CAR-Modified NK Cells under Oxidative Stress. Cancer Immunol. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Werlenius, O.; Riise, R.E.; Simpanen, M.; Aurelius, J.; Thorén, F.B. CD20 Antibodies Induce Production and Release of Reactive Oxygen Species by Neutrophils. Blood 2014, 123, 4001–4002. [Google Scholar] [CrossRef] [PubMed]
- Honeychurch, J.; Alduaij, W.; Azizyan, M.; Cheadle, E.J.; Pelicano, H.; Ivanov, A.; Huang, P.; Cragg, M.S.; Illidge, T.M. Antibody-Induced Nonapoptotic Cell Death in Human Lymphoma and Leukemia Cells Is Mediated through a Novel Reactive Oxygen Species-Dependent Pathway. Blood 2012, 119, 3523–3533. [Google Scholar] [CrossRef]
- Werlenius, O.; Aurelius, J.; Hallner, A.; Akhiani, A.A.; Simpanen, M.; Martner, A.; Andersson, P.-O.; Hellstrand, K.; Thorén, F.B. Reactive Oxygen Species Induced by Therapeutic CD20 Antibodies Inhibit Natural Killer Cell-Mediated Antibody-Dependent Cellular Cytotoxicity against Primary CLL Cells. Oncotarget 2016, 7, 32046–32053. [Google Scholar] [CrossRef]
- de Charette, M.; Houot, R. Hide or Defend, the Two Strategies of Lymphoma Immune Evasion: Potential Implications for Immunotherapy. Haematologica 2018, 103, 1256–1268. [Google Scholar] [CrossRef]
- Upadhyay, R.; Hammerich, L.; Peng, P.; Brown, B.; Merad, M.; Brody, J.D. Lymphoma: Immune Evasion Strategies. Cancers 2015, 7, 736–762. [Google Scholar] [CrossRef]
- Rimsza, L.M.; Roberts, R.A.; Miller, T.P.; Unger, J.M.; LeBlanc, M.; Braziel, R.M.; Weisenberger, D.D.; Chan, W.C.; Muller-Hermelink, H.K.; Jaffe, E.S.; et al. Loss of MHC Class II Gene and Protein Expression in Diffuse Large B-Cell Lymphoma Is Related to Decreased Tumor Immunosurveillance and Poor Patient Survival Regardless of Other Prognostic Factors: A Follow-up Study from the Leukemia and Lymphoma Molecular Profiling Project. Blood 2004, 103, 4251–4258. [Google Scholar] [CrossRef]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined Genetic Inactivation of Beta2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B-Cell Lymphoma. Cancer Cell 2011, 20, 728–740. [Google Scholar] [CrossRef]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Payer, Á.R.; Gonzalez, S.; López-Soto, A. Mechanisms of Apoptosis Resistance to NK Cell-Mediated Cytotoxicity in Cancer. Int. J. Mol. Sci. 2020, 21, 3726. [Google Scholar] [CrossRef]
- Matulis, S.M.; Boise, L.H. BCL2 Dependency in Diffuse Large B-Cell Lymphoma: It’s a Family Affair. Haematologica 2020, 105, 1993–1996. [Google Scholar] [CrossRef] [PubMed]
- Mestre-Escorihuela, C.; Rubio-Moscardo, F.; Richter, J.A.; Siebert, R.; Climent, J.; Fresquet, V.; Beltran, E.; Agirre, X.; Marugan, I.; Marín, M.; et al. Homozygous Deletions Localize Novel Tumor Suppressor Genes in B-Cell Lymphomas. Blood 2007, 109, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the Granzyme B/Perforin Pathway through Overexpression of the Serine Protease Inhibitor PI-9/SPI-6 Constitutes a Mechanism for Immune Escape by Tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar] [CrossRef] [PubMed]
- Czuczman, M.S.; Olejniczak, S.; Gowda, A.; Kotowski, A.; Binder, A.; Kaur, H.; Knight, J.; Starostik, P.; Deans, J.; Hernandez-Ilizaliturri, F.J. Acquirement of Rituximab Resistance in Lymphoma Cell Lines Is Associated with Both Global CD20 Gene and Protein Down-Regulation Regulated at the Pretranscriptional and Posttranscriptional Levels. Clin. Cancer Res. 2008, 14, 1561–1570. [Google Scholar] [CrossRef]
- Jazirehi, A.R.; Vega, M.I.; Bonavida, B. Development of Rituximab-Resistant Lymphoma Clones with Altered Cell Signaling and Cross-Resistance to Chemotherapy. Cancer Res. 2007, 67, 1270–1281. [Google Scholar] [CrossRef]
- Li, B.; Zhao, L.; Guo, H.; Wang, C.; Zhang, X.; Wu, L.; Chen, L.; Tong, Q.; Qian, W.; Wang, H.; et al. Characterization of a Rituximab Variant with Potent Antitumor Activity against Rituximab-Resistant B-Cell Lymphoma. Blood 2009, 114, 5007–5015. [Google Scholar] [CrossRef]
- Aldeghaither, D.S.; Zahavi, D.J.; Murray, J.C.; Fertig, E.J.; Graham, G.T.; Zhang, Y.-W.; O’Connell, A.; Ma, J.; Jablonski, S.A.; Weiner, L.M. A Mechanism of Resistance to Antibody-Targeted Immune Attack. Cancer Immunol. Res. 2019, 7, 230–243. [Google Scholar] [CrossRef]
- Dubrovsky, L.; Pankov, D.; Brea, E.J.; Dao, T.; Scott, A.; Yan, S.; O’Reilly, R.J.; Liu, C.; Scheinberg, D.A. A TCR-Mimic Antibody to WT1 Bypasses Tyrosine Kinase Inhibitor Resistance in Human BCR-ABL+ Leukemias. Blood 2014, 123, 3296–3304. [Google Scholar] [CrossRef]
- Dubrovsky, L.; Brea, E.J.; Pankov, D.; Casey, E.; Dao, T.; Liu, C.; Scheinberg, D.A. Mechanisms of Leukemia Resistance to Antibody Dependent Cellular Cytotoxicity. Oncoimmunology 2016, 5, e1211221. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Merkt, W.; Lorenz, H.-M.; Watzl, C. Rituximab Induces Phenotypical and Functional Changes of NK Cells in a Non-Malignant Experimental Setting. Arthritis Res. Ther. 2016, 18, 206. [Google Scholar] [CrossRef] [PubMed]
- Capuano, C.; Romanelli, M.; Pighi, C.; Cimino, G.; Rago, A.; Molfetta, R.; Paolini, R.; Santoni, A.; Galandrini, R. Anti-CD20 Therapy Acts via FcγRIIIA to Diminish Responsiveness of Human Natural Killer Cells. Cancer Res. 2015, 75, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Wirt, T.; Rosskopf, S.; Rösner, T.; Eichholz, K.M.; Kahrs, A.; Lutz, S.; Kretschmer, A.; Valerius, T.; Klausz, K.; Otte, A.; et al. An Fc Double-Engineered CD20 Antibody with Enhanced Ability to Trigger Complement-Dependent Cytotoxicity and Antibody-Dependent Cell-Mediated Cytotoxicity. Transfus. Med. Hemother. 2017, 44, 292–300. [Google Scholar] [CrossRef]
- O’Nions, J.; Townsend, W. The Role of Obinutuzumab in the Management of Follicular Lymphoma. Future Oncol. 2019, 15, 3565–3578. [Google Scholar] [CrossRef]
- Terszowski, G.; Klein, C.; Stern, M. KIR/HLA Interactions Negatively Affect Rituximab- but Not GA101 (Obinutuzumab)-Induced Antibody-Dependent Cellular Cytotoxicity. J. Immunol. 2014, 192, 5618–5624. [Google Scholar] [CrossRef]
- Binyamin, L.; Alpaugh, R.K.; Hughes, T.L.; Lutz, C.T.; Campbell, K.S.; Weiner, L.M. Blocking NK Cell Inhibitory Self-Recognition Promotes Antibody-Dependent Cellular Cytotoxicity in a Model of Anti-Lymphoma Therapy. J. Immunol. 2008, 180, 6392–6401. [Google Scholar] [CrossRef]
- Sehn, L.H.; Martelli, M.; Trněný, M.; Liu, W.; Bolen, C.R.; Knapp, A.; Sahin, D.; Sellam, G.; Vitolo, U. A Randomized, Open-Label, Phase III Study of Obinutuzumab or Rituximab plus CHOP in Patients with Previously Untreated Diffuse Large B-Cell Lymphoma: Final Analysis of GOYA. J. Hematol. Oncol. 2020, 13, 71. [Google Scholar] [CrossRef]
- Le Gouill, S.; Ghesquières, H.; Oberic, L.; Morschhauser, F.; Tilly, H.; Ribrag, V.; Lamy, T.; Thieblemont, C.; Maisonneuve, H.; Gressin, R.; et al. Obinutuzumab vs Rituximab for Advanced DLBCL: A PET-Guided and Randomized Phase 3 Study by LYSA. Blood 2021, 137, 2307–2320. [Google Scholar] [CrossRef]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.-S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Wayne, J.L.; Campbell, M.-A.; Marulappa, S.; Jain, V. Abstract 3784: AME-133v, a Humanized, Fc-Engineered Anti-CD20 Monoclonal Antibody, Demonstrates Greater B Cell Depletion than Rituxan in Vitro. Cancer Res. 2012, 72, 3784. [Google Scholar] [CrossRef]
- Wayne, J.L.; Ganjoo, K.N.; Pohlman, B.L.; De Vos, S.; Flinn, I.W.; Dang, N.H.; Mapara, M.Y.; Smith, M.R.; O’Reilly, A.M.; Marulappa, S.Y.; et al. Efficacy of Ocaratuzumab (AME-133v) in Relapsed Follicular Lymphoma Patients Refractory to Prior Rituximab. J. Clin. Oncol. 2012, 30, 8081. [Google Scholar] [CrossRef]
- Forero-Torres, A.; de Vos, S.; Pohlman, B.L.; Pashkevich, M.; Cronier, D.M.; Dang, N.H.; Carpenter, S.P.; Allan, B.W.; Nelson, J.G.; Slapak, C.A.; et al. Results of a Phase 1 Study of AME-133v (LY2469298), an Fc-Engineered Humanized Monoclonal Anti-CD20 Antibody, in FcγRIIIa-Genotyped Patients with Previously Treated Follicular Lymphoma. Clin. Cancer Res. 2012, 18, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Ganjoo, K.N.; de Vos, S.; Pohlman, B.L.; Flinn, I.W.; Forero-Torres, A.; Enas, N.H.; Cronier, D.M.; Dang, N.H.; Foon, K.A.; Carpenter, S.P.; et al. Phase 1/2 Study of Ocaratuzumab, an Fc-Engineered Humanized Anti-CD20 Monoclonal Antibody, in Low-Affinity FcγRIIIa Patients with Previously Treated Follicular Lymphoma. Leuk. Lymphoma 2015, 56, 42–48. [Google Scholar] [CrossRef]
- Horton, H.M.; Bernett, M.J.; Pong, E.; Peipp, M.; Karki, S.; Chu, S.Y.; Richards, J.O.; Vostiar, I.; Joyce, P.F.; Repp, R.; et al. Potent In Vitro and In Vivo Activity of an Fc-Engineered Anti-CD19 Monoclonal Antibody against Lymphoma and Leukemia. Cancer Res. 2008, 68, 8049–8057. [Google Scholar] [CrossRef]
- Jurczak, W.; Zinzani, P.L.; Gaidano, G.; Goy, A.; Provencio, M.; Nagy, Z.; Robak, T.; Maddocks, K.; Buske, C.; Ambarkhane, S.; et al. Phase IIa Study of the CD19 Antibody MOR208 in Patients with Relapsed or Refractory B-Cell Non-Hodgkin’s Lymphoma. Ann. Oncol. 2018, 29, 1266–1272. [Google Scholar] [CrossRef]
- Rosario, M.; Bai, L.; Kong, L.; Collins, L.I.; Schneider, S.E.; Chen, X.; Han, K.; Jeng, E.K.; Rhode, P.R.; Leong, J.W.; et al. The IL-15-Based ALT-803 Complex Enhances FcγRIIIa-Triggered NK Cell Responses and in Vivo Clearance of B Cell Lymphomas. Clin. Cancer Res. 2016, 22, 596–608. [Google Scholar] [CrossRef]
- Bittenbring, J.T.; Neumann, F.; Altmann, B.; Achenbach, M.; Reichrath, J.; Ziepert, M.; Geisel, J.; Regitz, E.; Held, G.; Pfreundschuh, M. Vitamin D Deficiency Impairs Rituximab-Mediated Cellular Cytotoxicity and Outcome of Patients with Diffuse Large B-Cell Lymphoma Treated with but Not without Rituximab. J. Clin. Oncol. 2014, 32, 3242–3248. [Google Scholar] [CrossRef]
- Neumann, F.; Acker, F.; Schormann, C.; Pfreundschuh, M.; Bittenbring, J.T. Determination of Optimum Vitamin D3 Levels for NK Cell-Mediated Rituximab- and Obinutuzumab-Dependent Cellular Cytotoxicity. Cancer Immunol. Immunother. 2018, 67, 1709–1718. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab—The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Felberg, A.; Taszner, M.; Urban, A.; Majeranowski, A.; Jaskuła, K.; Jurkiewicz, A.; Stasiłojć, G.; Blom, A.M.; Zaucha, J.M.; Okrój, M. Monitoring of the Complement System Status in Patients With B-Cell Malignancies Treated with Rituximab. Front. Immunol. 2020, 11, 2956. [Google Scholar] [CrossRef] [PubMed]
- Bordron, A.; Bagacean, C.; Mohr, A.; Tempescul, A.; Bendaoud, B.; Deshayes, S.; Dalbies, F.; Buors, C.; Saad, H.; Berthou, C.; et al. Resistance to Complement Activation, Cell Membrane Hypersialylation and Relapses in Chronic Lymphocytic Leukemia Patients Treated with Rituximab and Chemotherapy. Oncotarget 2018, 9, 31590–31605. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Beum, P.V.; Solga, M.D.; DiLillo, D.J.; Lindorfer, M.A.; Hess, C.E.; Densmore, J.J.; Williams, M.E.; Taylor, R.P. Rituximab Infusion Promotes Rapid Complement Depletion and Acute CD20 Loss in Chronic Lymphocytic Leukemia. J. Immunol. 2004, 172, 3280–3288. [Google Scholar] [CrossRef]
- Füst, G.; Czink, E.; Minh, D.; Miszlay, Z.; Varga, L.; Hollán, S.R. Depressed Classical Complement Pathway Activities in Chronic Lymphocytic Leukaemia. Clin. Exp. Immunol. 1985, 60, 489–495. [Google Scholar] [PubMed]
- Füst, G.; Miszlay, Z.s.; Czink, E.; Varga, L.; Pálóczi, K.; Szegedi, G.; Hollán, S.R. C1 and C4 Abnormalities in Chronic Lymphocytic Leukaemia and Their Significance. Immunol. Lett. 1987, 14, 255–259. [Google Scholar] [CrossRef]
- Heath, M.E.; Cheson, B.D. Defective Complement Activity in Chronic Lymphocytic Leukemia. Am. J. Hematol. 1985, 19, 63–73. [Google Scholar] [CrossRef]
- Middleton, O.; Cosimo, E.; Dobbin, E.; McCaig, A.M.; Clarke, C.; Brant, A.M.; Leach, M.T.; Michie, A.M.; Wheadon, H. Complement Deficiencies Limit CD20 Monoclonal Antibody Treatment Efficacy in CLL. Leukemia 2015, 29, 107–114. [Google Scholar] [CrossRef]
- Klepfish, A.; Rachmilewitz, E.A.; Kotsianidis, I.; Patchenko, P.; Schattner, A. Adding Fresh Frozen Plasma to Rituximab for the Treatment of Patients with Refractory Advanced CLL. QJM Int. J. Med. 2008, 101, 737–740. [Google Scholar] [CrossRef]
- Golay, J.; Lazzari, M.; Facchinetti, V.; Bernasconi, S.; Borleri, G.; Barbui, T.; Rambaldi, A.; Introna, M. CD20 Levels Determine the in Vitro Susceptibility to Rituximab and Complement of B-Cell Chronic Lymphocytic Leukemia: Further Regulation by CD55 and CD59. Blood 2001, 98, 3383–3389. [Google Scholar] [CrossRef]
- Winkler, M.T.; Bushey, R.T.; Gottlin, E.B.; Campa, M.J.; Guadalupe, E.S.; Volkheimer, A.D.; Weinberg, J.B.; Patz, E.F. Enhanced CDC of B Cell Chronic Lymphocytic Leukemia Cells Mediated by Rituximab Combined with a Novel Anti-Complement Factor H Antibody. PLoS ONE 2017, 12, e0179841. [Google Scholar] [CrossRef]
- Gaetano, N.D.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement Activation Determines the Therapeutic Activity of Rituximab In Vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Ou, Q.; Ye, S.; Lee, W.P.; Cornelius, J.; Diehl, L.; Lin, W.Y.; Hu, Z.; Lu, Y.; Chen, Y.; et al. Importance of Cellular Microenvironment and Circulatory Dynamics in B Cell Immunotherapy. J. Immunol. 2005, 174, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Song, G.; Ni, H.; Gu, L.; Liu, H.; Chen, B.; He, B.; Pan, Y.; Wang, S.; Cho, W.C. Deregulated Expression of MiR-224 and Its Target Gene: CD59 Predicts Outcome of Diffuse Large B-Cell Lymphoma Patients Treated with R-CHOP. Curr. Cancer Drug Targets 2014, 14, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Masuho, Y. CD59: Its Role in Complement Regulation and Potential for Therapeutic Use. Immunotechnology 1995, 1, 157–168. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Veeramani, S.; Racila, E.; Cagley, J.; Fritzinger, D.C.; Vogel, C.-W.; St John, W.; Weiner, G.J. Depletion of the C3 Component of Complement Enhances the Ability of Rituximab-Coated Target Cells to Activate Human NK Cells and Improves the Efficacy of Monoclonal Antibody Therapy in an in Vivo Model. Blood 2009, 114, 5322–5330. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Y.; Racila, E.; Taylor, R.P.; Weiner, G.J. NK-Cell Activation and Antibody-Dependent Cellular Cytotoxicity Induced by Rituximab-Coated Target Cells Is Inhibited by the C3b Component of Complement. Blood 2008, 111, 1456–1463. [Google Scholar] [CrossRef]
- Racila, E.; Link, B.K.; Weng, W.-K.; Witzig, T.E.; Ansell, S.; Maurer, M.J.; Huang, J.; Dahle, C.; Halwani, A.; Levy, R.; et al. A Polymorphism in the Complement Component C1qA Correlates with Prolonged Response Following Rituximab Therapy of Follicular Lymphoma. Clin. Cancer Res. 2008, 14, 6697–6703. [Google Scholar] [CrossRef]
- Jin, X.; Ding, H.; Ding, N.; Fu, Z.; Song, Y.; Zhu, J. Homozygous A Polymorphism of the Complement C1qA276correlates with Prolonged Overall Survival in Patients with Diffuse Large B Cell Lymphoma Treated with R-CHOP. J. Hematol. Oncol. 2012, 5, 51. [Google Scholar] [CrossRef]
- Deans, J.P.; Robbins, S.M.; Polyak, M.J.; Savage, J.A. Rapid Redistribution of CD20 to a Low Density Detergent-Insoluble Membrane Compartment. J. Biol. Chem. 1998, 273, 344–348. [Google Scholar] [CrossRef]
- Rezvani, A.R.; Maloney, D.G. Rituximab Resistance. Best Pract. Res. Clin. Haematol. 2011, 24, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Lipid Rafts as Signaling Hubs in Cancer Cell Survival/Death and Invasion: Implications in Tumor Progression and Therapy: Thematic Review Series: Biology of Lipid Rafts. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef]
- Hammadi, M.; Youinou, P.; Tempescul, A.; Tobón, G.; Berthou, C.; Bordron, A.; Pers, J.-O. Membrane Microdomain Sphingolipids Are Required for Anti-CD20-Induced Death of Chronic Lymphocytic Leukemia B Cells. Haematologica 2012, 97, 288–296. [Google Scholar] [CrossRef][Green Version]
- Meyer zum Büschenfelde, C.; Feuerstacke, Y.; Götze, K.S.; Scholze, K.; Peschel, C. GM1 Expression of Non-Hodgkin’s Lymphoma Determines Susceptibility to Rituximab Treatment. Cancer Res. 2008, 68, 5414–5422. [Google Scholar] [CrossRef] [PubMed]
- Meri, S. Self-Nonself Discrimination by the Complement System. FEBS Lett. 2016, 590, 2418–2434. [Google Scholar] [CrossRef]
- Boyd, R.S.; Jukes-Jones, R.; Walewska, R.; Brown, D.; Dyer, M.J.S.; Cain, K. Protein Profiling of Plasma Membranes Defines Aberrant Signaling Pathways in Mantle Cell Lymphoma. Mol. Cell Proteom. 2009, 8, 1501–1515. [Google Scholar] [CrossRef] [PubMed]
- Hirpara, J.L.; Loh, T.; Ng, S.B.; Chng, W.J.; Pervaiz, S. Aberrant Localization of Apoptosis Protease Activating Factor-1 in Lipid Raft Sub-Domains of Diffuse Large B Cell Lymphomas. Oncotarget 2016, 7, 83964–83975. [Google Scholar] [CrossRef]
- Winiarska, M.; Bil, J.; Wilczek, E.; Wilczynski, G.M.; Lekka, M.; Engelberts, P.J.; Mackus, W.J.M.; Gorska, E.; Bojarski, L.; Stoklosa, T.; et al. Statins Impair Antitumor Effects of Rituximab by Inducing Conformational Changes of CD20. PLoS Med. 2008, 5, e64. [Google Scholar] [CrossRef]
- Gisselbrecht, C.; Glass, B.; Mounier, N.; Singh Gill, D.; Linch, D.C.; Trneny, M.; Bosly, A.; Ketterer, N.; Shpilberg, O.; Hagberg, H.; et al. Salvage Regimens With Autologous Transplantation for Relapsed Large B-Cell Lymphoma in the Rituximab Era. J. Clin. Oncol. 2010, 28, 4184–4190. [Google Scholar] [CrossRef]
- Martín, A.; Conde, E.; Arnan, M.; Canales, M.A.; Deben, G.; Sancho, J.M.; Andreu, R.; Salar, A.; García-Sanchez, P.; Vázquez, L.; et al. R-ESHAP as Salvage Therapy for Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma: The Influence of Prior Exposure to Rituximab on Outcome. A GEL/TAMO Study. Haematologica 2008, 93, 1829–1836. [Google Scholar] [CrossRef]
- Barth, M.J.; Hernandez-Ilizaliturri, F.J.; Mavis, C.; Tsai, P.-C.; Gibbs, J.F.; Deeb, G.; Czuczman, M.S. Ofatumumab Demonstrates Activity against Rituximab-Sensitive and -Resistant Cell Lines, Lymphoma Xenografts and Primary Tumour Cells from Patients with B-Cell Lymphoma. Br. J. Haematol. 2012, 156, 490–498. [Google Scholar] [CrossRef]
- Takei, K.; Yamazaki, T.; Sawada, U.; Ishizuka, H.; Aizawa, S. Analysis of Changes in CD20, CD55, and CD59 Expression on Established Rituximab-Resistant B-Lymphoma Cell Lines. Leuk. Res. 2006, 30, 625–631. [Google Scholar] [CrossRef]
- Tsai, P.-C.; Hernandez-Ilizaliturri, F.J.; Bangia, N.; Olejniczak, S.H.; Czuczman, M.S. Regulation of CD20 in Rituximab-Resistant Cell Lines and B-Cell Non-Hodgkin Lymphoma. Clin. Cancer Res. 2012, 18, 1039–1050. [Google Scholar] [CrossRef]
- Golay, J.; Zaffaroni, L.; Vaccari, T.; Lazzari, M.; Borleri, G.M.; Bernasconi, S.; Tedesco, F.; Rambaldi, A.; Introna, M. Biologic Response of B Lymphoma Cells to Anti-CD20 Monoclonal Antibody Rituximab in Vitro: CD55 and CD59 Regulate Complement-Mediated Cell Lysis. Blood 2000, 95, 3900–3908. [Google Scholar] [CrossRef]
- Hiraga, J.; Tomita, A.; Sugimoto, T.; Shimada, K.; Ito, M.; Nakamura, S.; Kiyoi, H.; Kinoshita, T.; Naoe, T. Down-Regulation of CD20 Expression in B-Cell Lymphoma Cells after Treatment with Rituximab-Containing Combination Chemotherapies: Its Prevalence and Clinical Significance. Blood 2009, 113, 4885–4893. [Google Scholar] [CrossRef]
- Davis, T.A.; Czerwinski, D.K.; Levy, R. Therapy of B-Cell Lymphoma with Anti-CD20 Antibodies Can Result in the Loss of CD20 Antigen Expression. Clin. Cancer Res. 1999, 5, 611–615. [Google Scholar]
- Haidar, J.H.; Shamseddine, A.; Salem, Z.; Mrad, Y.A.; Nasr, M.R.; Zaatari, G.; Bazarbachi, A. Loss of CD20 Expression in Relapsed Lymphomas after Rituximab Therapy. Eur. J. Haematol. 2003, 70, 330–332. [Google Scholar] [CrossRef]
- Rawal, Y.B.; Nuovo, G.J.; Frambach, G.E.; Porcu, P.; Baiocchi, R.A.; Magro, C.M. The Absence of CD20 Messenger RNA in Recurrent Cutaneous B-Cell Lymphoma Following Rituximab Therapy. J. Cutan. Pathol. 2005, 32, 616–621. [Google Scholar] [CrossRef]
- Duman, B.B.; Sahin, B.; Ergin, M.; Guvenc, B. Loss of CD20 Antigen Expression after Rituximab Therapy of CD20 Positive B Cell Lymphoma (Diffuse Large B Cell Extranodal Marginal Zone Lymphoma Combination): A Case Report and Review of the Literature. Med. Oncol. 2012, 29, 1223–1226. [Google Scholar] [CrossRef]
- van Meerten, T.; Hagenbeek, A. CD20-Targeted Therapy: A Breakthrough in the Treatment of Non-Hodgkin’s Lymphoma. Neth. J. Med. 2009, 67, 251–259. [Google Scholar]
- Bobrowicz, M.; Dwojak, M.; Pyrzynska, B.; Stachura, J.; Muchowicz, A.; Berthel, E.; Dalla-Venezia, N.; Kozikowski, M.; Siernicka, M.; Miazek, N.; et al. HDAC6 Inhibition Upregulates CD20 Levels and Increases the Efficacy of Anti-CD20 Monoclonal Antibodies. Blood 2017, 130, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ayer, L.M.; Lytton, J.; Deans, J.P. Store-Operated Cation Entry Mediated by CD20 in Membrane Rafts. J. Biol. Chem. 2003, 278, 42427–42434. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, V.; Ledererova, A.; Ladungova, A.; Peschelova, H.; Janovska, P.; Slusarczyk, A.; Domagala, J.; Kopcil, P.; Vakulova, V.; Oppelt, J.; et al. CD20 Is Dispensable for B-Cell Receptor Signaling but Is Required for Proper Actin Polymerization, Adhesion and Migration of Malignant B Cells. PLoS ONE 2020, 15, e0229170. [Google Scholar] [CrossRef] [PubMed]
- Pyrzynska, B.; Dwojak, M.; Zerrouqi, A.; Morlino, G.; Zapala, P.; Miazek, N.; Zagozdzon, A.; Bojarczuk, K.; Bobrowicz, M.; Siernicka, M.; et al. FOXO1 Promotes Resistance of Non-Hodgkin Lymphomas to Anti-CD20-Based Therapy. Oncoimmunology 2018, 7, e1423183. [Google Scholar] [CrossRef]
- Winiarska, M.; Bojarczuk, K.; Pyrzynska, B.; Bil, J.; Siernicka, M.; Dwojak, M.; Bobrowicz, M.; Miazek, N.; Zapala, P.; Zagozdzon, A.; et al. Inhibitors of SRC Kinases Impair Antitumor Activity of Anti-CD20 Monoclonal Antibodies. mAbs 2014, 6, 1300–1313. [Google Scholar] [CrossRef]
- Winiarska, M.; Nowis, D.; Bil, J.; Glodkowska-Mrowka, E.; Muchowicz, A.; Wanczyk, M.; Bojarczuk, K.; Dwojak, M.; Firczuk, M.; Wilczek, E.; et al. Prenyltransferases Regulate CD20 Protein Levels and Influence Anti-CD20 Monoclonal Antibody-Mediated Activation of Complement-Dependent Cytotoxicity. J. Biol. Chem. 2012, 287, 31983–31993. [Google Scholar] [CrossRef]
- Bil, J.; Winiarska, M.; Nowis, D.; Bojarczuk, K.; Dabrowska-Iwanicka, A.; Basak, G.W.; Sułek, K.; Jakobisiak, M.; Golab, J. Bortezomib Modulates Surface CD20 in B-Cell Malignancies and Affects Rituximab-Mediated Complement-Dependent Cytotoxicity. Blood 2010, 115, 3745–3755. [Google Scholar] [CrossRef]
- Winiarska, M.; Bil, J.; Nowis, D.; Golab, J. Proteolytic Pathways Involved in Modulation of CD20 Levels. Autophagy 2010, 6, 810–812. [Google Scholar] [CrossRef][Green Version]
- Manshouri, T.; Do, K.; Wang, X.; Giles, F.J.; O’Brien, S.M.; Saffer, H.; Thomas, D.; Jilani, I.; Kantarjian, H.M.; Keating, M.J.; et al. Circulating CD20 Is Detectable in the Plasma of Patients with Chronic Lymphocytic Leukemia and Is of Prognostic Significance. Blood 2003, 101, 2507–2513. [Google Scholar] [CrossRef]
- Aung, T.; Chapuy, B.; Vogel, D.; Wenzel, D.; Oppermann, M.; Lahmann, M.; Weinhage, T.; Menck, K.; Hupfeld, T.; Koch, R.; et al. Exosomal Evasion of Humoral Immunotherapy in Aggressive B-Cell Lymphoma Modulated by ATP-Binding Cassette Transporter A3. Proc. Natl. Acad. Sci. USA 2011, 108, 15336–15341. [Google Scholar] [CrossRef]
- Marshall, M.J.E.; Stopforth, R.J.; Cragg, M.S. Therapeutic Antibodies: What Have We Learnt from Targeting CD20 and Where Are We Going? Front. Immunol. 2017, 8, 1245. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.R.W.; Visser, L.; Huls, G.; Diepstra, A.; van Vugt, M.; Ammatuna, E.; van Rijn, R.S.; Vellenga, E.; van den Berg, A.; Fehrmann, R.S.N.; et al. Identification of Relevant Drugable Targets in Diffuse Large B-Cell Lymphoma Using a Genome-Wide Unbiased CD20 Guilt-by Association Approach. PLoS ONE 2018, 13, e0193098. [Google Scholar] [CrossRef] [PubMed]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.R.; et al. Complement Is Activated by IgG Hexamers Assembled at the Cell Surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- de Jong, R.N.; Beurskens, F.J.; Verploegen, S.; Strumane, K.; van Kampen, M.D.; Voorhorst, M.; Horstman, W.; Engelberts, P.J.; Oostindie, S.C.; Wang, G.; et al. A Novel Platform for the Potentiation of Therapeutic Antibodies Based on Antigen-Dependent Formation of IgG Hexamers at the Cell Surface. PLoS Biol. 2016, 14, e1002344. [Google Scholar] [CrossRef]
- Olejniczak, S.H.; Hernandez-Ilizaliturri, F.J.; Clements, J.L.; Czuczman, M.S. Acquired Resistance to Rituximab Is Associated with Chemotherapy Resistance Resulting from Decreased Bax and Bak Expression. Clin. Cancer Res. 2008, 14, 1550–1560. [Google Scholar] [CrossRef]
- Bonavida, B. Rituximab-Induced Inhibition of Antiapoptotic Cell Survival Pathways: Implications in Chemo/Immunoresistance, Rituximab Unresponsiveness, Prognostic and Novel Therapeutic Interventions. Oncogene 2007, 26, 3629–3636. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, J.J.; Yang, L.; Tsai, P.-C.; Guo, Y.; Xue, K.; Xia, Z.; Liu, X.; Lv, F.; Cao, J.; et al. The Adhesion Molecule ICAM-1 in Diffuse Large B-Cell Lymphoma Post-Rituximab Era: Relationship with Prognostic Importance and Rituximab Resistance. Aging (Albany NY) 2020, 13, 181–193. [Google Scholar] [CrossRef]
- Gu, J.J.; Singh, A.; Xue, K.; Mavis, C.; Barth, M.; Yanamadala, V.; Lenz, P.; Grau, M.; Lenz, G.; Czuczman, M.S.; et al. Up-Regulation of Hexokinase II Contributes to Rituximab-Chemotherapy Resistance and Is a Clinically Relevant Target for Therapeutic Development. Oncotarget 2018, 9, 4020–4033. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- .Sircar, A.; Chowdhury, S.M.; Hart, A.; Bell, W.C.; Singh, S.; Sehgal, L.; Epperla, N. Impact and Intricacies of Bone Marrow Microenvironment in B-Cell Lymphomas: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 904. [Google Scholar] [CrossRef]
- Marquez, M.-E.; Hernández-Uzcátegui, O.; Cornejo, A.; Vargas, P.; Da Costa, O. Bone Marrow Stromal Mesenchymal Cells Induce down Regulation of CD20 Expression on B-CLL: Implications for Rituximab Resistance in CLL. Br. J. Haematol. 2015, 169, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Mraz, M.; Zent, C.S.; Church, A.K.; Jelinek, D.F.; Wu, X.; Pospisilova, S.; Ansell, S.M.; Novak, A.J.; Kay, N.E.; Witzig, T.E.; et al. Bone Marrow Stromal Cells Protect Lymphoma B-Cells from Rituximab-Induced Apoptosis and Targeting Integrin α-4-β-1 (VLA-4) with Natalizumab Can Overcome This Resistance. Br. J. Haematol. 2011, 155, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Zhu, Z.; Xu, X.; Zhang, H.; Xiong, H.; Li, Q.; Wei, Y. Human Bone Marrow-Derived Mesenchymal Stem Cells Promote the Growth and Drug-Resistance of Diffuse Large B-Cell Lymphoma by Secreting IL-6 and Elevating IL-17A Levels. J. Exp. Clin. Cancer Res. 2019, 38, 73. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Li, Q. Rituximab or Irradiation Promotes IL-17 Secretion and Thereby Induces Resistance to Rituximab or Irradiation. Cell. Mol. Immunol. 2017, 14, 1020–1022. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abès, R.; Gélizé, E.; Fridman, W.H.; Teillaud, J.-L. Long-Lasting Antitumor Protection by Anti-CD20 Antibody through Cellular Immune Response. Blood 2010, 116, 926–934. [Google Scholar] [CrossRef]
- Deligne, C.; Metidji, A.; Fridman, W.-H.; Teillaud, J.-L. Anti-CD20 Therapy Induces a Memory Th1 Response through the IFN-γ/IL-12 Axis and Prevents Protumor Regulatory T-Cell Expansion in Mice. Leukemia 2015, 29, 947–957. [Google Scholar] [CrossRef]
- Opinto, G.; Vegliante, M.C.; Negri, A.; Skrypets, T.; Loseto, G.; Pileri, S.A.; Guarini, A.; Ciavarella, S. The Tumor Microenvironment of DLBCL in the Computational Era. Front. Oncol. 2020, 10, 351. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal Gene Signatures in Large-B-Cell Lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef]
- Marchesi, F.; Cirillo, M.; Bianchi, A.; Gately, M.; Olimpieri, O.M.; Cerchiara, E.; Renzi, D.; Micera, A.; Balzamino, B.O.; Bonini, S.; et al. High Density of CD68+/CD163+ Tumour-Associated Macrophages (M2-TAM) at Diagnosis Is Significantly Correlated to Unfavorable Prognostic Factors and to Poor Clinical Outcomes in Patients with Diffuse Large B-Cell Lymphoma. Hematol. Oncol. 2015, 33, 110–112. [Google Scholar] [CrossRef]
- Desai, M.; Newberry, K.; Ou, Z.; Wang, M.; Zhang, L. Lenalidomide in Relapsed or Refractory Mantle Cell Lymphoma: Overview and Perspective. Ther. Adv. Hematol. 2014, 5, 91–101. [Google Scholar] [CrossRef]
- Witzig, T.E.; Vose, J.M.; Zinzani, P.L.; Reeder, C.B.; Buckstein, R.; Polikoff, J.A.; Bouabdallah, R.; Haioun, C.; Tilly, H.; Guo, P.; et al. An International Phase II Trial of Single-Agent Lenalidomide for Relapsed or Refractory Aggressive B-Cell Non-Hodgkin’s Lymphoma. Ann. Oncol. 2011, 22, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Fowler, N.H.; Davis, R.E.; Rawal, S.; Nastoupil, L.; Hagemeister, F.B.; McLaughlin, P.; Kwak, L.W.; Romaguera, J.E.; Fanale, M.A.; Fayad, L.E.; et al. Safety, Activity, and Immune Effects of Lenalidomide and Rituximab in Untreated Indolent Lymphoma. Lancet Oncol. 2014, 15, 1311–1318. [Google Scholar] [CrossRef]
- Gribben, J.G.; Fowler, N.; Morschhauser, F. Mechanisms of Action of Lenalidomide in B-Cell Non-Hodgkin Lymphoma. J. Clin. Oncol. 2015, 33, 2803–2811. [Google Scholar] [CrossRef] [PubMed]
- Walewski, J. Novel Monoclonal Antibodies for Diffuse Large B-Cell Lymphoma. Acta Haematol. Pol. 2021, 52, 329–333. [Google Scholar] [CrossRef]
- Bhat, S.A.; Czuczman, M.S. Novel Antibodies in the Treatment of Non-Hodgkin’s Lymphoma. Neth. J. Med. 2009, 67, 311–321. [Google Scholar]
- Patriarca, A.; Gaidano, G. Investigational Drugs for the Treatment of Diffuse Large B-Cell Lymphoma. Expert Opin. Investig. Drugs 2021, 30, 25–38. [Google Scholar] [CrossRef]
- Bobrowicz, M.; Kubacz, M.; Slusarczyk, A.; Winiarska, M. CD37 in B Cell Derived Tumors—More than Just a Docking Point for Monoclonal Antibodies. Int. J. Mol. Sci. 2020, 21, 9531. [Google Scholar] [CrossRef]
- Capuano, C.; Pighi, C.; Battella, S.; De Federicis, D.; Galandrini, R.; Palmieri, G. Harnessing CD16-Mediated NK Cell Functions to Enhance Therapeutic Efficacy of Tumor-Targeting MAbs. Cancers 2021, 13, 2500. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.-E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 Study of the Bispecific T-Cell Engager (BiTE) Antibody Blinatumomab in Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Viardot, A.; Hess, G.; Bargou, R.C.; Morley, N.J.; Gritti, G.; Goebeler, M.-E.; Iskander, K.; Cohan, D.; Zhang, A.; Franklin, J.; et al. Durability of Complete Response after Blinatumomab Therapy for Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Leuk. Lymphoma 2020, 61, 2767–2770. [Google Scholar] [CrossRef]
- Dufner, V.; Sayehli, C.M.; Chatterjee, M.; Hummel, H.D.; Gelbrich, G.; Bargou, R.C.; Goebeler, M.-E. Long-Term Outcome of Patients with Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma Treated with Blinatumomab. Blood Adv. 2019, 3, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, H.J.; de Jonge, A.V.; Hiemstra, I.H.; Gelderloos, A.T.; Berry, D.R.A.I.; Hijmering, N.J.; van Essen, H.F.; de Jong, D.; Chamuleau, M.E.D.; Zweegman, S.; et al. Epcoritamab Induces Potent Anti-Tumor Activity against Malignant B-Cells from Patients with DLBCL, FL and MCL, Irrespective of Prior CD20 Monoclonal Antibody Treatment. Blood Cancer J. 2021, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Van Der Horst, H.J.; de Jonge, A.V.; Hiemstra, I.H.; Gelderloos, A.T.; Berry, D.R.; Hijmering, N.J.; de Jong, D.; Chamuleau, M.E.D.; Zweegman, S.; Breij, E.C.; et al. Duobody-CD3xCD20 Induces Potent Anti-Tumor Activity in Malignant Lymph Node B Cells from Patients with DLBCL, FL and MCL Ex Vivo, Irrespective of Prior Treatment with CD20 Monoclonal Antibodies. Blood 2019, 134, 4066. [Google Scholar] [CrossRef]
- Hutchings, M.; Mous, R.; Clausen, M.R.; Johnson, P.; Linton, K.M.; Chamuleau, M.E.D.; Lewis, D.J.; Balari, A.S.; Cunningham, D.; Oliveri, R.S.; et al. Dose Escalation of Subcutaneous Epcoritamab in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma: An Open-Label, Phase 1/2 Study. Lancet 2021, 398, 1157–1169. [Google Scholar] [CrossRef]
- Glorius, P.; Baerenwaldt, A.; Kellner, C.; Staudinger, M.; Dechant, M.; Stauch, M.; Beurskens, F.J.; Parren, P.W.H.I.; van de Winkel, J.G.J.; Valerius, T.; et al. The Novel Tribody [(CD20)(2)XCD16] Efficiently Triggers Effector Cell-Mediated Lysis of Malignant B Cells. Leukemia 2013, 27, 190–201. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef]
- Buatois, V.; Johnson, Z.; Salgado-Pires, S.; Papaioannou, A.; Hatterer, E.; Chauchet, X.; Richard, F.; Barba, L.; Daubeuf, B.; Cons, L.; et al. Preclinical Development of a Bispecific Antibody That Safely and Effectively Targets CD19 and CD47 for the Treatment of B-Cell Lymphoma and Leukemia. Mol. Cancer Ther. 2018, 17, 1739–1751. [Google Scholar] [CrossRef]
- Zeng, J.; Liu, R.; Wang, J.; Fang, Y. A Bispecific Antibody Directly Induces Lymphoma Cell Death by Simultaneously Targeting CD20 and HLA-DR. J. Cancer Res. Clin. Oncol. 2015, 141, 1899–1907. [Google Scholar] [CrossRef]
- Tuscano, J.M.; Ma, Y.; Martin, S.M.; Kato, J.; O’Donnell, R.T. The Bs20x22 Anti-CD20-CD22 Bispecific Antibody Has More Lymphomacidal Activity than Do the Parent Antibodies Alone. Cancer Immunol. Immunother. 2011, 60, 771–780. [Google Scholar] [CrossRef]
- Qu, Z.; Goldenberg, D.M.; Cardillo, T.M.; Shi, V.; Hansen, H.J.; Chang, C.-H. Bispecific Anti-CD20/22 Antibodies Inhibit B-Cell Lymphoma Proliferation by a Unique Mechanism of Action. Blood 2008, 111, 2211–2219. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Senter, P.D. Potent Antibody Drug Conjugates for Cancer Therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Gerber, H.-P.; Sapra, P.; Loganzo, F.; May, C. Combining Antibody–Drug Conjugates and Immune-Mediated Cancer Therapy: What to Expect? Biochem. Pharmacol. 2016, 102, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.-W.; Polson, A. Antibody–Drug Conjugates for the Treatment of B-Cell Non-Hodgkin’s Lymphoma and Leukemia. Future Oncol. 2013, 9, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Zinzani, P.L.; Sasse, S.; Radford, J.; Shonukan, O.; Bonthapally, V. Experience of Brentuximab Vedotin in Relapsed/Refractory Hodgkin Lymphoma and Relapsed/Refractory Systemic Anaplastic Large-Cell Lymphoma in the Named Patient Program: Review of the Literature. Crit. Rev. Oncol. Hematol. 2015, 95, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of Resistance to Antibody–Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the Potential of Antibody–Drug Conjugates for Cancer Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting Multidrug Resistance in Cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Chen, R.; Herrera, A.F.; Hou, J.; Chen, L.; Wu, J.; Guo, Y.; Synold, T.W.; Ngo, V.N.; Puverel, S.; Mei, M.; et al. Inhibition of MDR1 Overcomes Resistance to Brentuximab Vedotin in Hodgkin Lymphoma. Clin. Cancer Res. 2020, 26, 1034–1044. [Google Scholar] [CrossRef]
- Qing, K.; Jin, Z.; Xu, Z.; Wang, W.; Li, X.; Zhang, Y.; Wang, L.; Zhu, H.; Xiang, R.; Wu, S.; et al. Dysregulated MDR1 by PRDM1/Blimp1 Is Involved in the Doxorubicin Resistance of Non-Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. Chemotherapy 2021, 1–12. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody–Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Buecheler, J.W.; Winzer, M.; Weber, C.; Gieseler, H. Alteration of Physicochemical Properties for Antibody-Drug Conjugates and Their Impact on. Stability. J. Pharm. Sci. 2020, 109, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.M.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired Resistance to Antibody-Drug Conjugates. Cancers 2019, 11, 394. [Google Scholar] [CrossRef]
- Vaklavas, C.; Forero-Torres, A. Safety and Efficacy of Brentuximab Vedotin in Patients with Hodgkin Lymphoma or Systemic Anaplastic Large Cell Lymphoma. Ther. Adv. Hematol. 2012, 3, 209–225. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor Efficacy of a Combination of CMC-544 (Inotuzumab Ozogamicin), a CD22-Targeted Cytotoxic Immunoconjugate of Calicheamicin, and Rituximab against Non-Hodgkin’s B-Cell Lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [CrossRef]
- Sawas, A.; Savage, K.J.; Perez, R.P.; Advani, R.H.; Melhem-Bertrandt, A.; Lackey, J.; Trave, F.; Anand, B.; Huang, Y.; Vincent, M.; et al. A First in Human Experience of the Anti-CD37 Antibody-Drug Conjugate AGS67E in Lymphoid Malignancies. J. Clin. Oncol. 2016, 34, 7549. [Google Scholar] [CrossRef]
- Advani, R.H.; Lebovic, D.; Chen, A.; Brunvand, M.; Goy, A.; Chang, J.E.; Hochberg, E.; Yalamanchili, S.; Kahn, R.; Lu, D.; et al. Phase I Study of the Anti-CD22 Antibody–Drug Conjugate Pinatuzumab Vedotin with/without Rituximab in Patients with Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Clin. Cancer Res. 2017, 23, 1167–1176. [Google Scholar] [CrossRef]
- Stathis, A.; Freedman, A.S.; Flinn, I.W.; Maddocks, K.J.; Weitman, S.; Berdeja, J.G.; Mejia, A.V.; Zucca, E.; Green, R.; Romanelli, A.; et al. A Phase I Study of IMGN529, an Antibody-Drug Conjugate (ADC) Targeting CD37, in Adult Patients with Relapsed or Refractory B-Cell Non-Hodgkin’s Lymphoma (NHL). Blood 2014, 124, 1760. [Google Scholar] [CrossRef]
- Fathi, A.T.; Borate, U.; DeAngelo, D.J.; O’Brien, M.M.; Trippett, T.; Shah, B.D.; Hale, G.A.; Foran, J.M.; Silverman, L.B.; Tibes, R.; et al. A Phase 1 Study of Denintuzumab Mafodotin (SGN-CD19A) in Adults with Relapsed or Refractory B-Lineage Acute Leukemia (B-ALL) and Highly Aggressive Lymphoma. Blood 2015, 126, 1328. [Google Scholar] [CrossRef]
- Trnĕný, M.; Verhoef, G.; Dyer, M.J.; Ben Yehuda, D.; Patti, C.; Canales, M.; Lopez, A.; Awan, F.T.; Montgomery, P.G.; Janikova, A.; et al. A Phase II Multicenter Study of the Anti-CD19 Antibody Drug Conjugate Coltuximab Ravtansine (SAR3419) in Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma Previously Treated with Rituximab-Based Immunotherapy. Haematologica 2018, 103, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Zhylko, A.; Winiarska, M.; Graczyk-Jarzynka, A. The Great War of Today: Modifications of CAR-T Cells to Effectively Combat Malignancies. Cancers 2020, 12, 2030. [Google Scholar] [CrossRef] [PubMed]
- Miazek-Zapala, N.; Slusarczyk, A.; Kusowska, A.; Zapala, P.; Kubacz, M.; Winiarska, M.; Bobrowicz, M. The “Magic Bullet” Is Here? Cell-Based Immunotherapies for Hematological Malignancies in the Twilight of the Chemotherapy Era. Cells 2021, 10, 1511. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V. Immunotherapy: Tisagenlecleucel-the First Approved CAR-T-Cell Therapy: Implications for Payers and Policy Makers. Nat. Rev. Clin. Oncol. 2018, 15, 11–12. [Google Scholar] [CrossRef]
- Al-Mansour, M.; Al-Foheidi, M.; Ibrahim, E. Efficacy and Safety of Second-Generation CAR T-Cell Therapy in Diffuse Large B-Cell Lymphoma: A Meta-Analysis. Mol. Clin. Oncol. 2020, 13, 33. [Google Scholar] [CrossRef]
- Maloney, D.G. Anti-CD19 CAR T Cell Therapy for Lymphoma-off to the Races! Nat. Rev. Clin. Oncol. 2019, 16, 279–280. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of Resistance to CAR T Cell Therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Laurent, C.; Syrykh, C.; Hamon, M.; Adélaïde, J.; Guille, A.; Escudié, F.; Jalowicki, G.; Fina, F.; Bardet, A.; Mescam, L.; et al. Resistance of B-Cell Lymphomas to CAR T-Cell Therapy Is Associated With Genomic Tumor Changes Which Can Result in Transdifferentiation. Am. J. Surg. Pathol. 2021. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-Cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Ruella, M.; Maus, M.V. Catch Me If You Can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput. Struct. Biotechnol. J. 2016, 14, 357–362. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-Negative Myeloid Phenotype Allows Immune Escape of MLL-Rearranged B-ALL from CD19 CAR-T-Cell Therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Ledererova, A.; Dostalova, L.; Kozlova, V.; Peschelova, H.; Ladungova, A.; Culen, M.; Loja, T.; Verner, J.; Pospisilova, S.; Smida, M.; et al. Hypermethylation of CD19 Promoter Enables Antigen-Negative Escape to CART-19 in Vivo and in Vitro. J. Immunother. Cancer 2021, 9, e002352. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T Cell Trogocytosis and Cooperative Killing Regulate Tumour Antigen Escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.D.; Ziccheddu, B.; Coughlin, C.A.; Faramand, R.; Griswold, A.J.; Reid, K.M.; Landgren, O.; Locke, F.L.; Maura, F.; Davila, M.L.; et al. Genomic Drivers of Large B-Cell Lymphoma Resistance to CD19 CAR-T Therapy. Blood 2021, 138, 42. [Google Scholar] [CrossRef]
- Fousek, K.; Watanabe, J.; Joseph, S.K.; George, A.; An, X.; Byrd, T.T.; Morris, J.S.; Luong, A.; Martínez-Paniagua, M.A.; Sanber, K.; et al. CAR T-Cells That Target Acute B-Lineage Leukemia Irrespective of CD19 Expression. Leukemia 2021, 35, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Shalabi, H.; Kraft, I.L.; Wang, H.-W.; Yuan, C.M.; Yates, B.; Delbrook, C.; Zimbelman, J.D.; Giller, R.; Stetler-Stevenson, M.; Jaffe, E.S.; et al. Sequential Loss of Tumor Surface Antigens Following Chimeric Antigen Receptor T-Cell Therapies in Diffuse Large B-Cell Lymphoma. Haematologica 2018, 103, e215–e218. [Google Scholar] [CrossRef]
- Majzner, R.G.; Frank, J.M.; Mount, C.; Tousley, A.; Kurtz, D.M.; Sworder, B.; Murphy, K.A.; Manousopoulou, A.; Kohler, K.; Rotiroti, M.C.; et al. CD58 Aberrations Limit Durable Responses to CD19 CAR in Large B Cell Lymphoma Patients Treated with Axicabtagene Ciloleucel but Can Be Overcome through Novel CAR Engineering. Blood 2020, 136, 53. [Google Scholar] [CrossRef]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-Cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef]
- Cai, Q.; Zhang, M.; Li, Z. Potential Strategies against Resistance to CAR T-Cell Therapy in Haematological Malignancies. Ther. Adv. Med. Oncol. 2020, 12, 1758835920962963. [Google Scholar] [CrossRef]
- Liu, Y.; Barta, S.K. Diffuse Large B-Cell Lymphoma: 2019 Update on Diagnosis, Risk Stratification, and Treatment. Am. J. Hematol. 2019, 94, 604–616. [Google Scholar] [CrossRef] [PubMed]


| Name | Target | Type | Mode of Action | Efficacy in Clinical and Preclinical Studies |
|---|---|---|---|---|
| Blinatumomab | CD3/CD19 | bispecific antibody (BiTE) | CD3-positive T-cell recruitment |
|
| Epcoritamab (DuoBody-CD3xCD20) | CD3/CD20 | bispecific antibody (BiTE) | CD3-positive T-cell recruitment |
|
| [(CD20)2xCD16] | CD20/CD16 | bispecific tribody | CD16-positive effector cell recruitment |
|
| NI-1701 | CD19/CD47 | bispecific antibody | ADCP enhancement via blocking immune checkpoint receptor |
|
| CD20-HLA-DR DVD-Ig | CD20/HLA-DR | bispecific antibody | Increase in selectivity against NHL cells versus healthy cells |
|
| Bs20×22 | CD20/CD22 | bispecific antibody (RTX/HB22.7 platform) | Induction of apoptosis |
|
| Bs20x22 | CD20/CD22 | bispecific antibody (hA20 (veltuzumab)/hLL2 (epratuzumab) platform) | ADCC enhancement via antigen crosslinking |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kusowska, A.; Kubacz, M.; Krawczyk, M.; Slusarczyk, A.; Winiarska, M.; Bobrowicz, M. Molecular Aspects of Resistance to Immunotherapies—Advances in Understanding and Management of Diffuse Large B-Cell Lymphoma. Int. J. Mol. Sci. 2022, 23, 1501. https://doi.org/10.3390/ijms23031501
Kusowska A, Kubacz M, Krawczyk M, Slusarczyk A, Winiarska M, Bobrowicz M. Molecular Aspects of Resistance to Immunotherapies—Advances in Understanding and Management of Diffuse Large B-Cell Lymphoma. International Journal of Molecular Sciences. 2022; 23(3):1501. https://doi.org/10.3390/ijms23031501
Chicago/Turabian StyleKusowska, Aleksandra, Matylda Kubacz, Marta Krawczyk, Aleksander Slusarczyk, Magdalena Winiarska, and Malgorzata Bobrowicz. 2022. "Molecular Aspects of Resistance to Immunotherapies—Advances in Understanding and Management of Diffuse Large B-Cell Lymphoma" International Journal of Molecular Sciences 23, no. 3: 1501. https://doi.org/10.3390/ijms23031501
APA StyleKusowska, A., Kubacz, M., Krawczyk, M., Slusarczyk, A., Winiarska, M., & Bobrowicz, M. (2022). Molecular Aspects of Resistance to Immunotherapies—Advances in Understanding and Management of Diffuse Large B-Cell Lymphoma. International Journal of Molecular Sciences, 23(3), 1501. https://doi.org/10.3390/ijms23031501