
Anti-Excitotoxic Effects of N-Butylidenephthalide Revealed by Chemically Insulted Purkinje Progenitor Cells Derived from SCA3 iPSCs

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
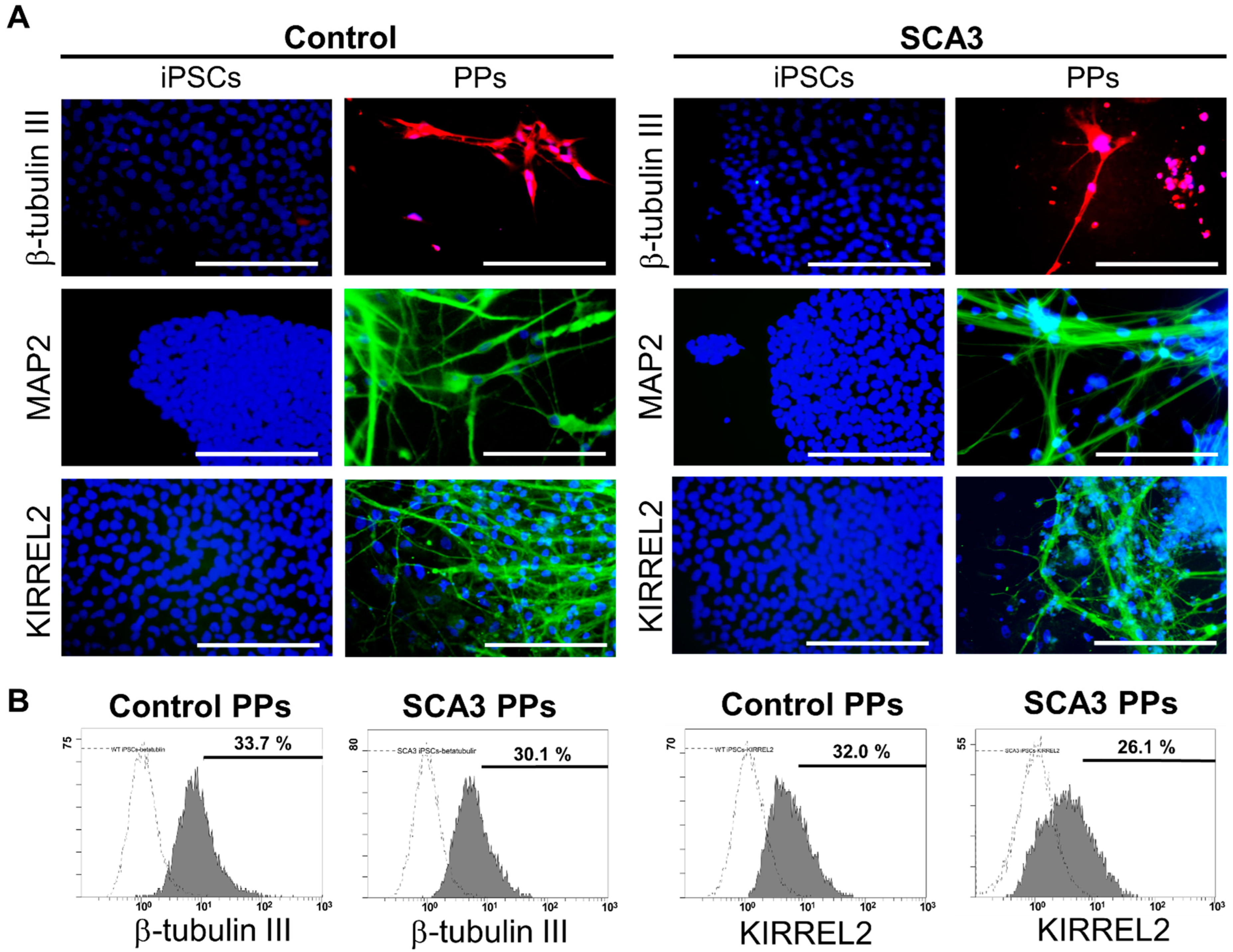
2.1. The Generation and Characterization of PPs
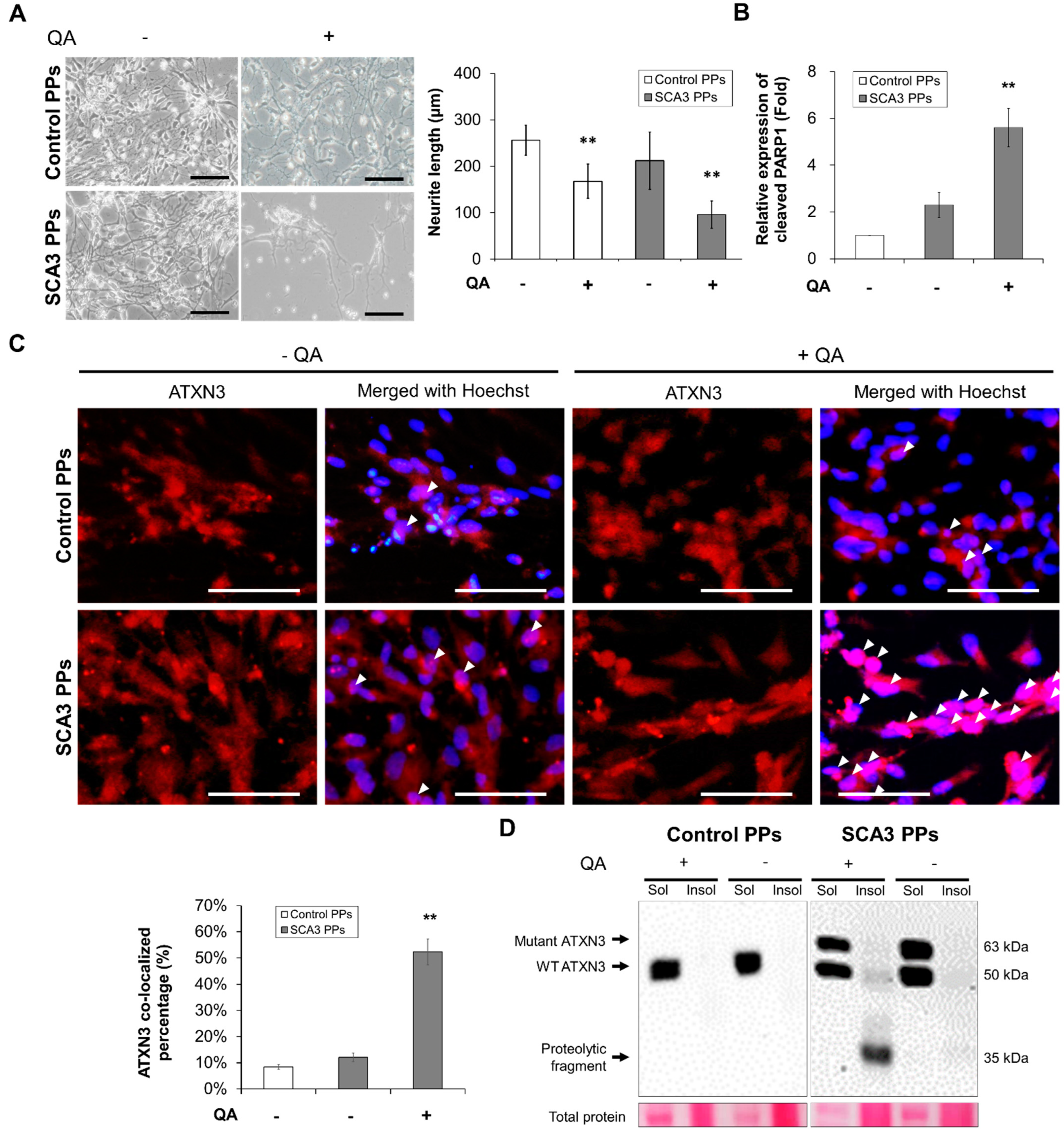
2.2. Chemical-Insulted SCA3 PPs Represent the Pathological Phenotype of SCA3
2.3. Chemical-Insulted SCA3 PPs Represent the Pathological Phenotype of SCA3
3. Discussion
4. Materials and Methods
4.1. Cell Culture of Control and SCA3 iPSCs
4.2. Differentiation of Control and SCA3 iPSCs into PPs
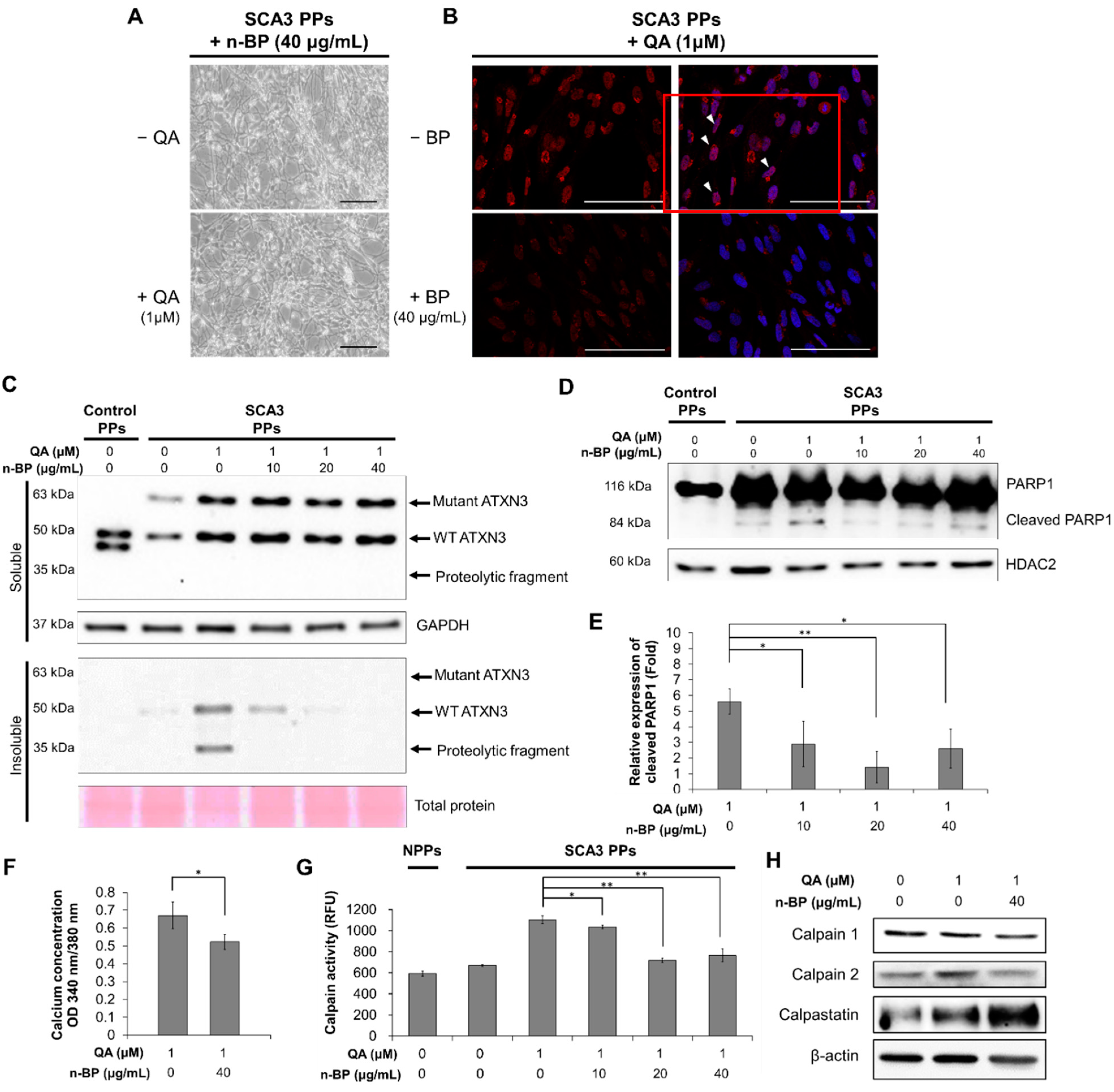
4.3. Induction of Excitotoxicity on the PPs and the Treatment of n-BP
4.4. Immunocytochemistry (ICC) Staining
4.5. Measurement of Neurite Length and Quantification of ATXN3 Co-Localization
4.6. Flow Cytometry
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Measurement of the Intracellular Calcium Concentration
4.9. Protein Extraction
4.10. Western Blot Analysis
4.11. Statistics
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Maschke, M.; Oehlert, G.; Xie, T.D.; Perlman, S.; Subramony, S.H.; Kumar, N.; Ptacek, L.J.; Gomez, C.M. Clinical feature profile of spinocerebellar ataxia type 1–8 predicts genetically defined subtypes. Mov. Disord. 2005, 20, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, R.A.M.; Toonen, L.J.A.; Gardiner, S.L.; van Roon-Mom, W.M.C. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Liang, X.; Long, Z.; Chen, Z.; Shi, Y.; Xia, K.; Meng, L.; Tang, B.; Qiu, R.; Jiang, H. Gene-Related Cerebellar Neurodegeneration in SCA3/MJD: A Case-Controlled Imaging-Genetic Study. Front. Neurol. 2019, 10, 1025. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.D.; Teixeira-Castro, A.; Maciel, P. From Pathogenesis to Novel Therapeutics for Spinocerebellar Ataxia Type 3: Evading Potholes on the Way to Translation. Neurotherapeutics 2019, 16, 1009–1031. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, H.S.; Moore, L.R.; Paulson, H.L. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol. Dis. 2020, 134, 104635. [Google Scholar] [CrossRef]
- Burnett, B.; Li, F.S.; Pittman, R.N. The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 2003, 12, 3195–3205. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Saha, S.; Chakraborty, A.; Silva-Fernandes, A.; Mandal, S.M.; Neves-Carvalho, A.; Liu, Y.; Pandita, R.K.; Hegde, M.L.; Hegde, P.M.; et al. The role of the mammalian DNA end-processing enzyme polynucleotide kinase 3’-phosphatase in spinocerebellar ataxia type 3 pathogenesis. PLoS Genet. 2015, 11, e1004749. [Google Scholar] [CrossRef] [Green Version]
- Kazachkova, N.; Raposo, M.; Montiel, R.; Cymbron, T.; Bettencourt, C.; Silva-Fernandes, A.; Silva, S.; Maciel, P.; Lima, M. Patterns of mitochondrial DNA damage in blood and brain tissues of a transgenic mouse model of Machado-Joseph disease. Neurodegen. Dis. 2013, 11, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Evert, B.O.; Vogt, I.R.; Kindermann, C.; Ozimek, L.; de Vos, R.A.; Brunt, E.R.; Schmitt, I.; Klockgether, T.; Wullner, U. Inflammatory genes are upregulated in expanded ataxin-3-expressing cell lines and spinocerebellar ataxia type 3 brains. J. Neurosci. 2001, 21, 5389–5396. [Google Scholar] [CrossRef] [Green Version]
- Rub, U.; de Vos, R.A.; Brunt, E.R.; Schultz, C.; Paulson, H.; Del Tredici, K.; Braak, H. Degeneration of the external cuneate nucleus in spinocerebellar ataxia type 3 (Machado-Joseph disease). Brain Res. 2002, 953, 126–134. [Google Scholar] [CrossRef]
- Rub, U.; Brunt, E.R.; Deller, T. New insights into the pathoanatomy of spinocerebellar ataxia type 3 (Machado-Joseph disease). Curr. Opin. Neurol. 2008, 21, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Sibon, O.C.M.; Dijkers, P.F. Inhibition of NF-κB in astrocytes is sufficient to delay neurodegeneration induced by proteotoxicity in neurons. J. Neuroinflamm. 2018, 15, 261. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Hong, Z.X.; Lin, S.Z.; Harn, H.J. Identifying Therapeutic Targets for Spinocerebellar Ataxia Type 3/Machado-Joseph Disease through Integration of Pathological Biomarkers and Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 3063. [Google Scholar] [CrossRef] [PubMed]
- Rub, U.; Schols, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.W.; Deller, T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.A.; Mantele, S.; Mollenkopf, P.; Boy, J.; Kehlenbach, R.H.; Riess, O.; Schmidt, T. Identification and functional dissection of localization signals within ataxin-3. Neurobiol. Dis. 2009, 36, 280–292. [Google Scholar] [CrossRef]
- Reina, C.P.; Zhong, X.; Pittman, R.N. Proteotoxic stress increases nuclear localization of ataxin-3. Hum. Mol. Genet. 2010, 19, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Bichelmeier, U.; Schmidt, T.; Hubener, J.; Boy, J.; Ruttiger, L.; Habig, K.; Poths, S.; Bonin, M.; Knipper, M.; Schmidt, W.J.; et al. Nuclear localization of ataxin-3 is required for the manifestation of symptoms in SCA3: In vivo evidence. J. Neurosci. 2007, 27, 7418–7428. [Google Scholar] [CrossRef] [Green Version]
- Hübener, J.; Weber, J.J.; Richter, C.; Honold, L.; Weiss, A.; Murad, F.; Breuer, P.; Wüllner, U.; Bellstedt, P.; Paquet-Durand, F.; et al. Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3). Hum. Mol. Genet. 2013, 22, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Jacquard, C.; Trioulier, Y.; Cosker, F.; Escartin, C.; Bizat, N.; Hantraye, P.; Cancela, J.M.; Bonvento, G.; Brouillet, E. Brain mitochondrial defects amplify intracellular [Ca2+] rise and neurodegeneration but not Ca2+ entry during NMDA receptor activation. FASEB J. 2006, 20, 1021–1023. [Google Scholar] [CrossRef] [Green Version]
- Rajamani, K.; Liu, J.W.; Wu, C.H.; Chiang, I.T.; You, D.H.; Lin, S.Y.; Hsieh, D.K.; Lin, S.Z.; Harn, H.J.; Chiou, T.W. n-Butylidenephthalide exhibits protection against neurotoxicity through regulation of tryptophan 2, 3 dioxygenase in spinocerebellar ataxia type 3. Neuropharmacology 2017, 117, 434–446. [Google Scholar] [CrossRef]
- Haacke, A.; Hartl, F.U.; Breuer, P. Calpain Inhibition Is Sufficient to Suppress Aggregation of Polyglutamine-expanded Ataxin-3. J. Biol. Chem. 2007, 282, 18851–18856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, A.T.; Gonçalves, N.; Koeppen, A.; Déglon, N.; Kügler, S.; Duarte, C.B.; Pereira de Almeida, L. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain 2012, 135, 2428–2439. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lin, S.Y.; Liu, J.W.; Lin, S.Z.; Harn, H.J.; Chiou, T.W. n-Butylidenephthalide Modulates Autophagy to Ameliorate Neuropathological Progress of Spinocerebellar Ataxia Type 3 through mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 6339. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, D.E.; Carroll, T.S.; Kocabas, A.; Zhu, X.; Behesti, H.; Faust, P.L.; Stalbow, L.; Fang, Y.; Hatten, M.E. Novel genetic features of human and mouse Purkinje cell differentiation defined by comparative transcriptomics. Proc. Natl. Acad. Sci. USA 2020, 117, 15085–15095. [Google Scholar] [CrossRef]
- Watson, L.M.; Wong, M.M.K.; Vowles, J.; Cowley, S.A.; Becker, E.B.E. A Simplified Method for Generating Purkinje Cells from Human-Induced Pluripotent Stem Cells. Cerebellum 2018, 17, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, B.; Pan, N.; Fu, L.; Wang, C.; Song, G.; An, J.; Liu, Z.; Zhu, W.; Guan, Y.; et al. Differentiation of human induced pluripotent stem cells to mature functional Purkinje neurons. Sci. Rep. 2015, 5, 9232. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P. Excitotoxic and excitoprotective mechanisms: Abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromol. Med. 2003, 3, 65–94. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, R.J. Neuroprotection, excitotoxicity and NMDA antagonists. Arq. Neuro-Psiquiatr. 2000, 58, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, T.S.; Tu, H.; Nelson, O.; Pook, M.; Hammer, R.; Nukina, N.; Bezprozvanny, I. Deranged calcium signaling and neurodegeneration in spinocerebellar ataxia type 3. J. Neurosci. 2008, 28, 12713–12724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento-Ferreira, I.; Santos-Ferreira, T.; Sousa-Ferreira, L.; Auregan, G.; Onofre, I.; Alves, S.; Dufour, N.; Colomer Gould, V.F.; Koeppen, A.; Deglon, N.; et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2011, 134, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Onofre, I.; Mendonca, N.; Lopes, S.; Nobre, R.; de Melo, J.B.; Carreira, I.M.; Januario, C.; Goncalves, A.F.; de Almeida, L.P. Fibroblasts of Machado Joseph Disease patients reveal autophagy impairment. Sci. Rep. 2016, 6, 28220. [Google Scholar] [CrossRef] [PubMed]
- Watchon, M.; Yuan, K.C.; Mackovski, N.; Svahn, A.J.; Cole, N.J.; Goldsbury, C.; Rinkwitz, S.; Becker, T.S.; Nicholson, G.A.; Laird, A.S. Calpain Inhibition Is Protective in Machado-Joseph Disease Zebrafish Due to Induction of Autophagy. J. Neurosci. 2017, 37, 7782–7794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soong, B.W.; Syu, S.H.; Wen, C.H.; Ko, H.W.; Wu, M.L.; Hsieh, P.C.; Hwang, S.M.; Lu, H.E. Generation of induced pluripotent stem cells from a patient with spinocerebellar ataxia type 3. Stem Cell Res. 2017, 18, 29–32. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.-H.; Chiang, I.-T.; Liu, J.-W.; Hsieh, J.; Lee, J.-H.; Lu, H.-E.; Tso, H.-S.; Deng, Y.-C.; Kao, J.-C.; Wu, J.-R.; et al. Anti-Excitotoxic Effects of N-Butylidenephthalide Revealed by Chemically Insulted Purkinje Progenitor Cells Derived from SCA3 iPSCs. Int. J. Mol. Sci. 2022, 23, 1391. https://doi.org/10.3390/ijms23031391
Yang H-H, Chiang I-T, Liu J-W, Hsieh J, Lee J-H, Lu H-E, Tso H-S, Deng Y-C, Kao J-C, Wu J-R, et al. Anti-Excitotoxic Effects of N-Butylidenephthalide Revealed by Chemically Insulted Purkinje Progenitor Cells Derived from SCA3 iPSCs. International Journal of Molecular Sciences. 2022; 23(3):1391. https://doi.org/10.3390/ijms23031391
Chicago/Turabian StyleYang, Hsin-Han, I-Tsang Chiang, Jen-Wei Liu, Jeanne Hsieh, Jui-Hao Lee, Huai-En Lu, Hwa-Sung Tso, Yu-Chen Deng, Jo-Chi Kao, Jhen-Rong Wu, and et al. 2022. "Anti-Excitotoxic Effects of N-Butylidenephthalide Revealed by Chemically Insulted Purkinje Progenitor Cells Derived from SCA3 iPSCs" International Journal of Molecular Sciences 23, no. 3: 1391. https://doi.org/10.3390/ijms23031391
APA StyleYang, H.-H., Chiang, I.-T., Liu, J.-W., Hsieh, J., Lee, J.-H., Lu, H.-E., Tso, H.-S., Deng, Y.-C., Kao, J.-C., Wu, J.-R., Harn, H.-J., & Chiou, T.-W. (2022). Anti-Excitotoxic Effects of N-Butylidenephthalide Revealed by Chemically Insulted Purkinje Progenitor Cells Derived from SCA3 iPSCs. International Journal of Molecular Sciences, 23(3), 1391. https://doi.org/10.3390/ijms23031391