
Determination of the Optimal Bacterial DNA Extraction Method to Explore the Urinary Microbiota



Abstract
:1. Introduction
2. Results
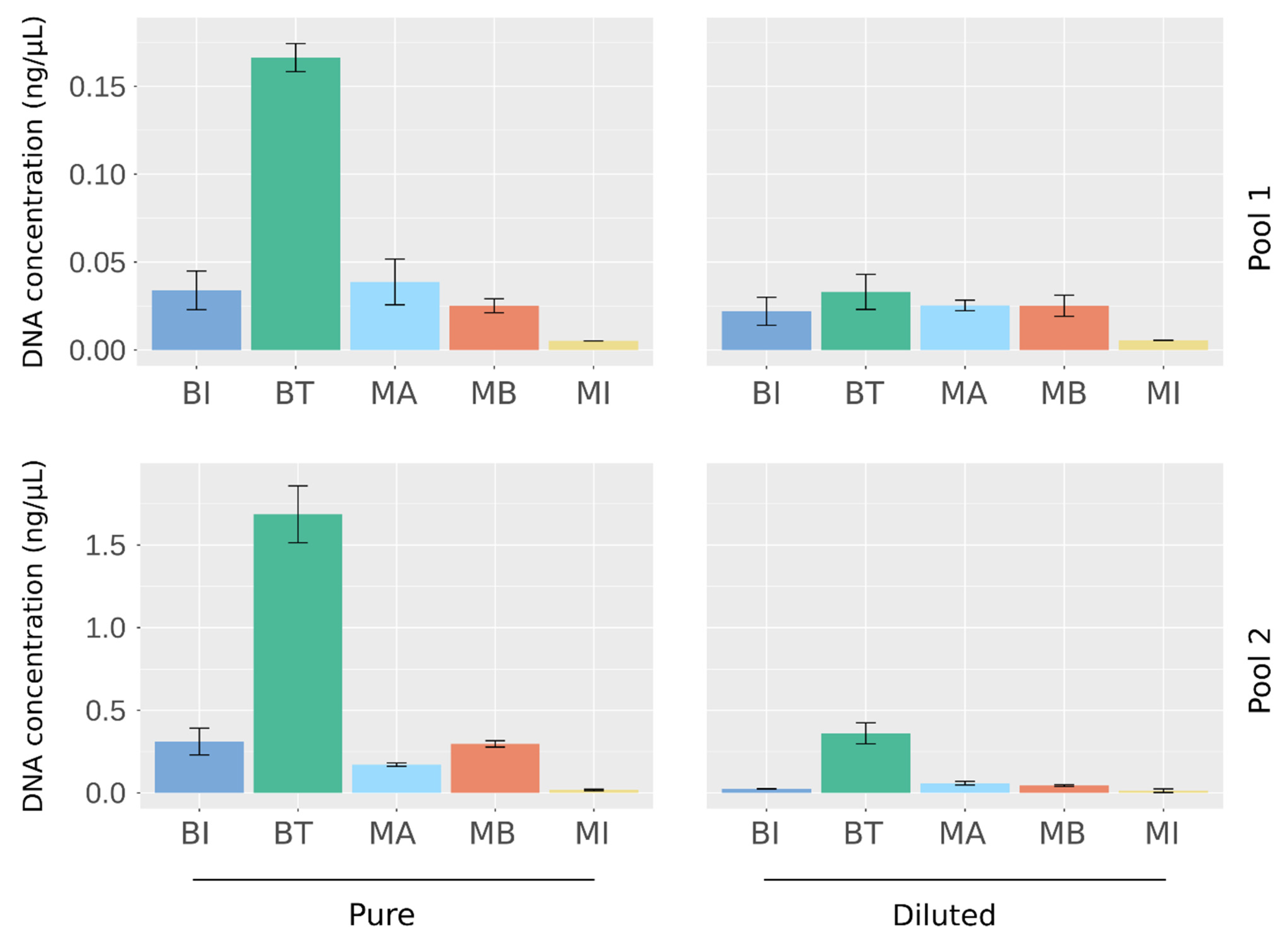
2.1. Quantification of DNA Extracted from Artificial Urine Samples
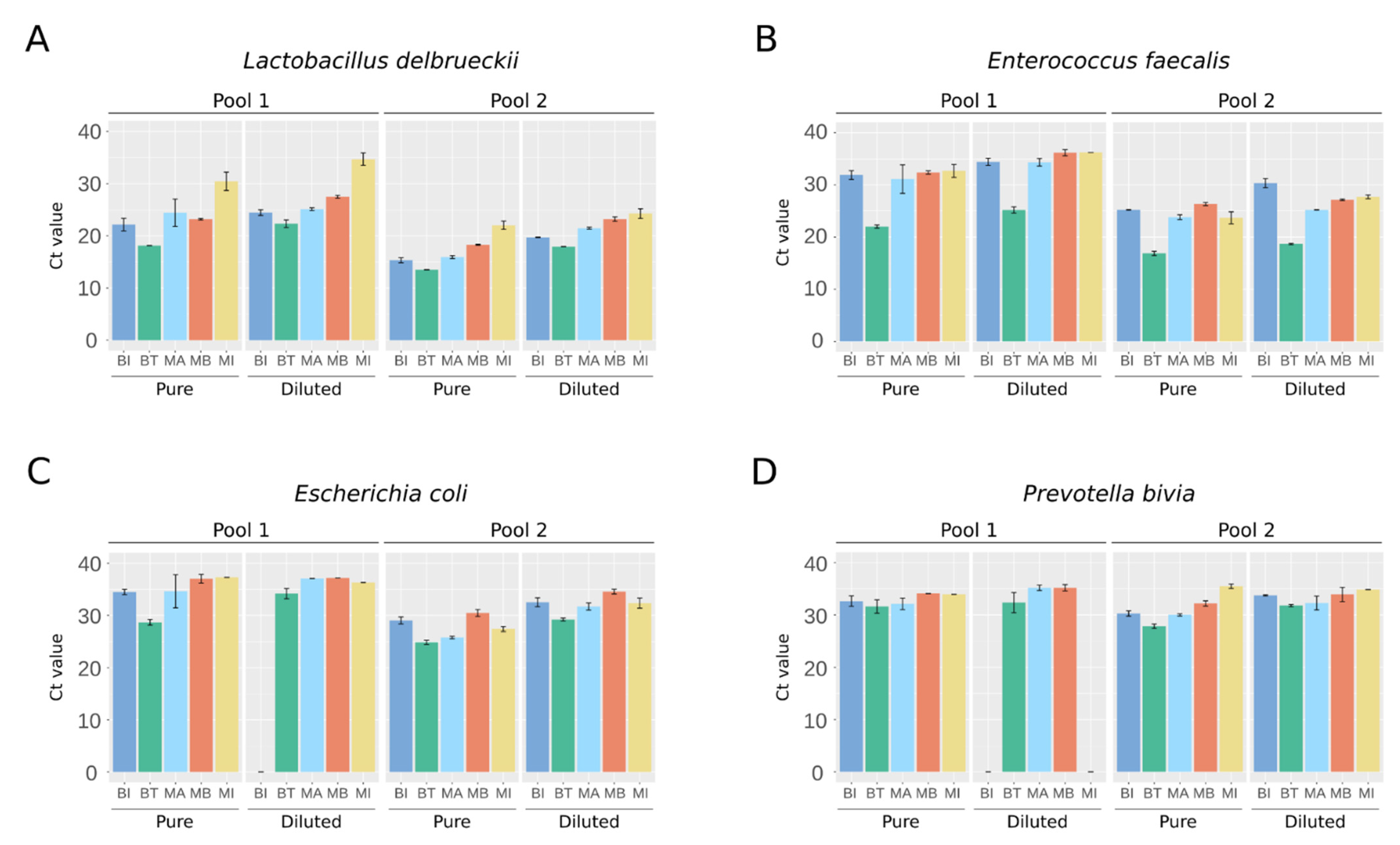
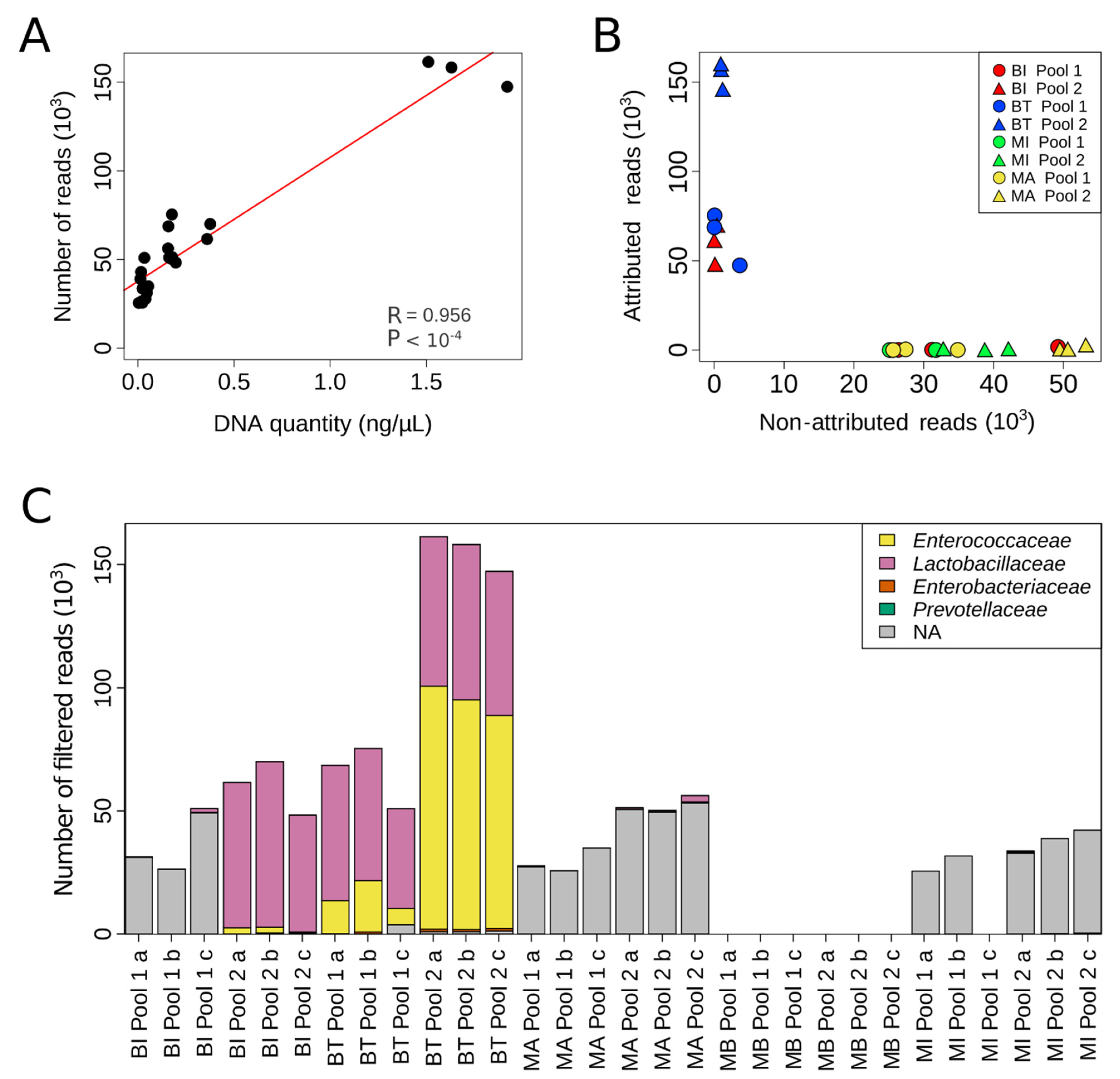
2.2. NGS Detection of Bacteria Present in Artificial Urine Samples
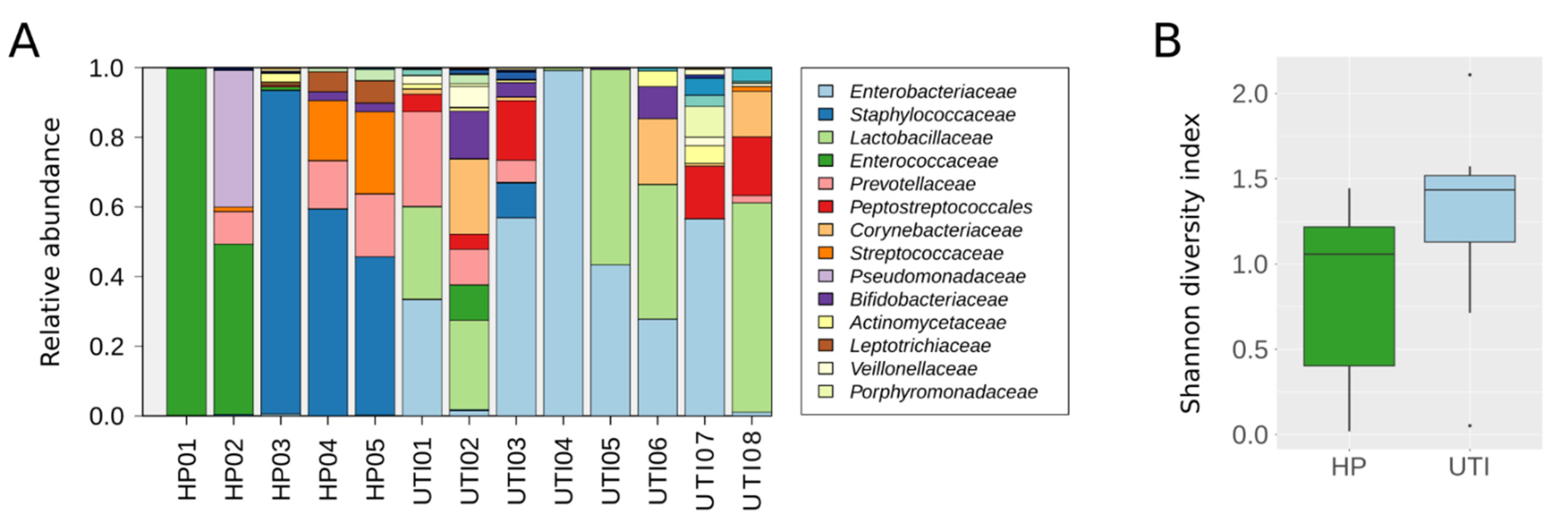
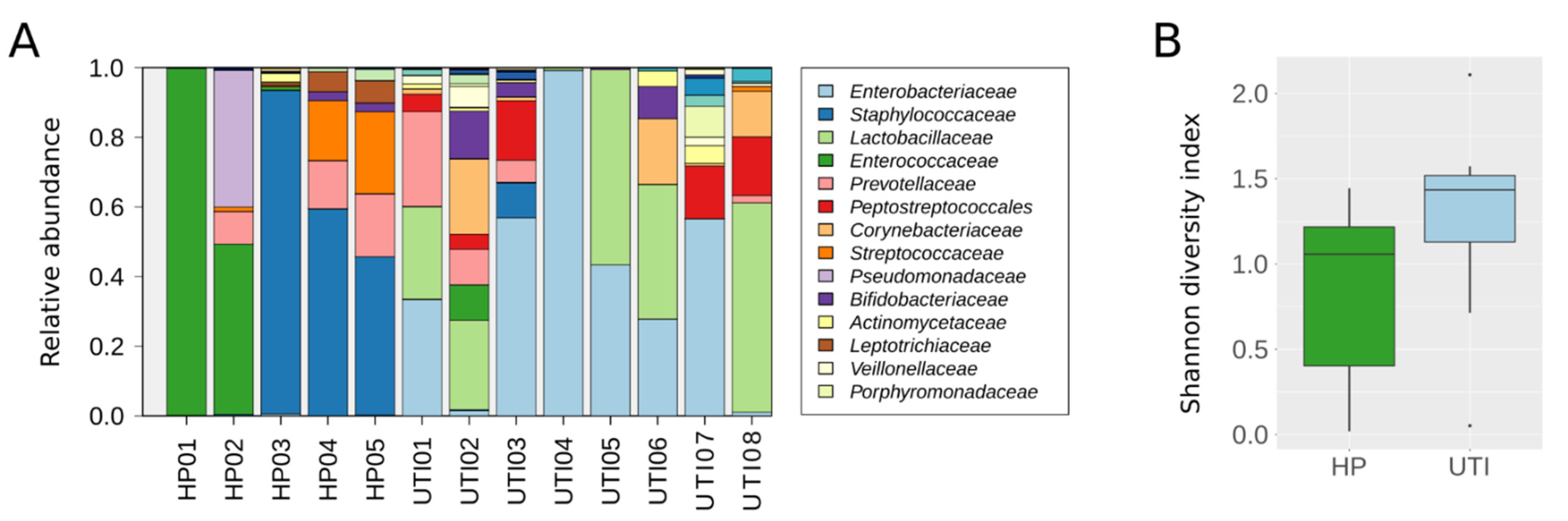
2.3. Evaluation of the BT Kit’s Performance on Human Clinical Samples
3. Discussion
4. Materials and Methods
4.1. Artificial Sample Preparation
4.2. DNA Extraction and Total Quantification
4.3. Human Sample Collection
4.4. Quantitative PCR
4.5. Next-Generation Sequencing Experiments
4.6. Bioinformatics Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thomas-White, K.; Brady, M.; Wolfe, A.J.; Mueller, E.R. The Bladder Is Not Sterile: History and Current Discoveries on the Urinary Microbiome. Curr. Bladder Dysfunct. Rep. 2016, 11, 18–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, S.A.; Razvi, H.; Dave, S.; Reid, G.; Burton, J. The microbiome of the urinary tract—A role beyond infection. Nat. Rev. Urol. 2015, 12, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.J.; Toh, E.; Shibata, N.; Rong, R.; Kenton, K.S.; Fitzgerald, M.; Mueller, E.R.; Schreckenberger, P.C.; Dong, Q.; Nelson, D.E.; et al. Evidence of Uncultivated Bacteria in the Adult Female Bladder. J. Clin. Microbiol. 2012, 50, 1376–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grine, G.; Lotte, R.; Chirio, D.; Chevalier, A.; Raoult, D.; Drancourt, M.; Ruimy, R. Co-culture of Methanobrevibacter smithii with enterobacteria during urinary infection. EBioMedicine 2019, 43, 333–337. [Google Scholar] [CrossRef] [Green Version]
- Pearce, M.M.; Hilt, E.E.; Rosenfeld, A.B.; Zilliox, M.J.; Thomas-White, K.; Fok, C.; Kliethermes, S.; Schreckenberger, P.C.; Brubaker, L.; Gai, X.; et al. The Female Urinary Microbiome: A Comparison of Women with and without Urgency Urinary Incontinence. mBio 2014, 5, e01283-14. [Google Scholar] [CrossRef] [Green Version]
- Komesu, Y.M.; Richter, H.E.; Carper, B.; Dinwiddie, D.L.; Lukacz, E.S.; Siddiqui, N.Y.; Sung, V.W.; Zyczynski, H.M.; Ridgeway, B.; Rogers, R.G.; et al. The urinary microbiome in women with mixed urinary incontinence compared to similarly aged controls. Int. Urogynecol. J. 2018, 29, 1785–1795. [Google Scholar] [CrossRef]
- Wu, J.F.; Muthusamy, A.; Al-Ghalith, G.A.; Knights, D.; Guo, B.; Wu, B.; Remmel, R.P.; Schladt, D.P.; Alegre, M.-L.; Oetting, W.S.; et al. Urinary microbiome associated with chronic allograft dysfunction in kidney transplant recipients. Clin. Transplant. 2018, 32, e13436. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, G.; Zhao, J.; Chen, J.; Chen, Y.; Huang, W.; Zhong, J.; Zeng, J. Profiling the Urinary Microbiota in Male Patients with Bladder Cancer in China. Front. Cell. Infect. Microbiol. 2018, 8, 167, Erratum in Front. Cell. Infect. Microbiol. 2018, 8, 429. [Google Scholar] [CrossRef]
- Popović, V.B.; Šitum, M.; Chow, C.-E.T.; Chan, L.S.; Roje, B.; Terzić, J. The urinary microbiome associated with bladder cancer. Sci. Rep. 2018, 8, 12157. [Google Scholar] [CrossRef] [Green Version]
- Bi, H.; Tian, Y.; Song, C.; Li, J.; Liu, T.; Chen, Z.; Chen, C.; Huang, Y.; Zhang, Y. Urinary microbiota—A potential biomarker and therapeutic target for bladder cancer. J. Med Microbiol. 2019, 68, 1471–1478. [Google Scholar] [CrossRef]
- Lewis, D.A.; Brown, R.; Williams, J.; White, P.; Jacobson, S.K.; Marchesi, J.R.; Drake, M.J. The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front. Cell. Infect. Microbiol. 2013, 3, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouts, D.E.; Szpakowski, S.; Purushe, J.; Torralba, M.; Waterman, R.C.; MacNeil, M.D.; Alexander, L.J.; Nelson, K.E. Next Generation Sequencing to Define Prokaryotic and Fungal Diversity in the Bovine Rumen. PLoS ONE 2012, 7, e48289. [Google Scholar] [CrossRef] [PubMed]
- Nienhouse, V.; Gao, X.; Dong, Q.; Nelson, D.E.; Toh, E.; McKinley, K.; Schreckenberger, P.; Shibata, N.; Fok, C.S.; Mueller, E.R.; et al. Interplay between Bladder Microbiota and Urinary Antimicrobial Peptides: Mechanisms for Human Urinary Tract Infection Risk and Symptom Severity. PLoS ONE 2014, 9, e114185. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.Y.; Song, E.-J.; Kim, S.H.; Lee, J.; Nam, Y.-D. Comparison of DNA extraction methods for human gut microbial community profiling. Syst. Appl. Microbiol. 2018, 41, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Fiedorová, K.; Radvanský, M.; Němcová, E.; Grombiříková, H.; Bosák, J.; Černochová, M.; Lexa, M.; Šmajs, D.; Freiberger, T. The Impact of DNA Extraction Methods on Stool Bacterial and Fungal Microbiota Community Recovery. Front. Microbiol. 2019, 10, 821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrand, J.; Patron, K.; Legrand-Frossi, C.; Frippiat, J.-P.; Merlin, C.; Alauzet, C.; Lozniewski, A. Comparison of seven methods for extraction of bacterial DNA from fecal and cecal samples of mice. J. Microbiol. Methods 2014, 105, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Gill, C.; Van De Wijgert, J.H.H.M.; Blow, F.; Darby, A.C. Evaluation of Lysis Methods for the Extraction of Bacterial DNA for Analysis of the Vaginal Microbiota. PLoS ONE 2016, 11, e0163148. [Google Scholar] [CrossRef] [Green Version]
- Mirsepasi, H.C.; Persson, S.; Struve, C.; Andersen, L.O.B.; Petersen, A.M.; Krogfelt, K.A. Microbial diversity in fecal samples depends on DNA extraction method: EasyMag DNA extraction compared to QIAamp DNA stool mini kit extraction. BMC Res. Notes 2014, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Selway, C.A.; Eisenhofer, R.; Weyrich, L.S. Microbiome applications for pathology: Challenges of low microbial biomass samples during diagnostic testing. J. Pathol. Clin. Res. 2020, 6, 97–106. [Google Scholar] [CrossRef]
- Karstens, L.; Siddiqui, N.Y.; Zaza, T.; Barstad, A.; Amundsen, C.L.; Sysoeva, T.A. Benchmarking DNA isolation kits used in analyses of the urinary microbiome. Sci. Rep. 2021, 11, 6186. [Google Scholar] [CrossRef]
- Ackerman, A.L.; Anger, J.T.; Khalique, M.U.; Ackerman, J.E.; Tang, J.; Kim, J.; Underhill, D.M.; Freeman, M.R. The NIH Multidisciplinary Approach to the Study of Chronic Pelvic Pain (MAPP) Optimization of DNA extraction from human urinary samples for mycobiome community profiling. PLoS ONE 2019, 14, e0210306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munch, M.M.; Chambers, L.C.; Manhart, L.E.; Domogala, D.; Lopez, A.; Fredricks, D.N.; Srinivasan, S. Optimizing bacterial DNA extraction in urine. PLoS ONE 2019, 14, e0222962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, S.; Cohen, D.B.; Ravel, J.; Abdo, Z.; Forney, L.J. Evaluation of Methods for the Extraction and Purification of DNA from the Human Microbiome. PLoS ONE 2012, 7, e33865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrofchak, R.; Madden, C.; Evans, M.V.; Hale, V.L. Evaluating extraction methods to study canine urine microbiota. PLoS ONE 2021, 16, e0253989. [Google Scholar] [CrossRef] [PubMed]
- Hrbacek, J.; Morais, D.; Cermak, P.; Hanacek, V.; Zachoval, R. Alpha-diversity and microbial community structure of the male urinary microbiota depend on urine sampling method. Sci. Rep. 2021, 11, 23758. [Google Scholar] [CrossRef] [PubMed]
- Pohl, H.G.; Groah, S.L.; Pérez-Losada, M.; Ljungberg, I.; Sprague, B.M.; Chandal, N.; Caldovic, L.; Hsieh, M. The Urine Microbiome of Healthy Men and Women Differs by Urine Collection Method. Int. Neurourol. J. 2020, 24, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Gottschick, C.; Deng, Z.-L.; Vital, M.; Masur, C.; Abels, C.; Pieper, D.H.; Wagner-Döbler, I. The urinary microbiota of men and women and its changes in women during bacterial vaginosis and antibiotic treatment. Microbiome 2017, 5, 99. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kit | Full Name | Supplier | Lysis Method | Extraction Method | DNA Binding Principle | |||
|---|---|---|---|---|---|---|---|---|
| Chemical | Enzymatic | Mecanical | Heat | |||||
| BI | QIAmp BiOstic Bacteremia | Qiagen | Yes | PK | Yes | 70 °C | Manual | Silica spin-column |
| BT | DNeasy Blood and Tissue | Qiagen | Yes | PK and Ly | No | No | Manual | Silica spin-column |
| MA | MagNA Pure Compact | Roche | Yes | PK and Ly | No | 95 °C | Automated | Magnetic beads |
| MB | AMPure XP Beads | Beckman-Coulter | Yes | PK and Ly | No | 95 °C | Manual | Magnetic beads |
| MI | QIAmp DNA Microbiome | Qiagen | Yes | PK | Yes | No | Manual | Silica spin-column |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vendrell, J.A.; Henry, S.; Cabello-Aguilar, S.; Heckendorn, E.; Godreuil, S.; Solassol, J. Determination of the Optimal Bacterial DNA Extraction Method to Explore the Urinary Microbiota. Int. J. Mol. Sci. 2022, 23, 1336. https://doi.org/10.3390/ijms23031336
Vendrell JA, Henry S, Cabello-Aguilar S, Heckendorn E, Godreuil S, Solassol J. Determination of the Optimal Bacterial DNA Extraction Method to Explore the Urinary Microbiota. International Journal of Molecular Sciences. 2022; 23(3):1336. https://doi.org/10.3390/ijms23031336
Chicago/Turabian StyleVendrell, Julie A., Steven Henry, Simon Cabello-Aguilar, Elise Heckendorn, Sylvain Godreuil, and Jérôme Solassol. 2022. "Determination of the Optimal Bacterial DNA Extraction Method to Explore the Urinary Microbiota" International Journal of Molecular Sciences 23, no. 3: 1336. https://doi.org/10.3390/ijms23031336
APA StyleVendrell, J. A., Henry, S., Cabello-Aguilar, S., Heckendorn, E., Godreuil, S., & Solassol, J. (2022). Determination of the Optimal Bacterial DNA Extraction Method to Explore the Urinary Microbiota. International Journal of Molecular Sciences, 23(3), 1336. https://doi.org/10.3390/ijms23031336