
From Marine Metabolites to the Drugs of the Future: Squalamine, Trodusquemine, Their Steroid and Triterpene Analogues †




Abstract
1. Introduction
2. Syntheses of Squalamine from Cholic Acids
2.1. Synthesis Based on 3β-Acetoxy-5-Cholic Acid
2.2. Synthesis of 24R- and 24S-Squalamine’s from Stigmasterol
2.3. Synthesis from 7α-(Benzyloxy)-3-Dioxolan-Cholestan-24R-Ol
2.4. Synthesis from Cholic Acid Sulfate
2.5. Synthesis from 3-Keto-23,24-Bisnorchol-4-En-22-Ol
2.6. Synthesis from Methyl 3-Keto-5α-Chenodeoxycholonate
2.7. Synthesis from Desmosterol
2.8. Synthesis from Methylhyodeoxycholonate
3. Syntheses of Squalamine Analogs
3.1. Synthesis of 3α-Episqualamine
3.2. Synthesis of Squalamine Analogs from Cholic Acids
3.3. Synthesis of Steroid Methylenepolyamines from Cholic, Deoxycholic, Chenodeoxycholic, Ursodeoxycholic and Lithocholic Acids
3.4. Synthesis from 22-Hydroxy-23,24-Dinorchol-4-En-3-One and Its Analogs
3.5. Synthesis of Steroid Methylenepolyamines from Cholestan, 4-Cholestene, 5-Cholesten-3-One, 6-Ketocholestanol and 3,7-Diketocholestene
3.6. Synthesis from 3-Keto-7-Hydroxycholestane
3.7. Synthesis of Squalamine Analogs from Cholesterol and Progesterone
3.8. Synthesis of Spermidino-7-Fluoro-3-Aminosteroids
3.9. Synthesis of Cholanic Acid Carboxamides with Alkane Polyamines
3.10. Synthesis of Steroid Carbamates
3.11. Synthesis of Aminopropoxysteroids
3.12. Synthesis of Squalamine Phosphate
3.13. Synthesis of Squalamine Analogues with Multiple Steroid Backbones
4. Synthesis of Trodusquemine and Its Analogs
5. Synthesis of Claramine and Its Analogues
6. Biological Properties of Squalamine, Trodusquemine and Their Analogues
6.1. Biological Activity of Squalamine and Trodusquemine
6.1.1. Antibacterial and Antiviral Activity
6.1.2. Neuroprotective Activity
6.1.3. Antiangiogenic and Antitumor Activity
6.2. Trodusquemine as a Unique PTP1B Inhibitor
6.2.1. Antiobesity and Weight Loss Activity
6.2.2. Anticancer Activity
6.2.3. Antiatherogenic Properties
6.2.4. Regenerative Properties
6.2.5. Anxiolytic Properties
6.3. Clinical Data
6.4. Synthetic Analogs of Squalamine and Trodusquemine and Structure-Activity Studies
6.5. Ceragenins as Antibiotics
7. Terpene- and Triterpene-Based Polyamine Derivatives
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | RAC-alpha serine/threonine-protein kinase |
| AMPK5′ | adenosine monophosphate-activated protein kinase |
| BOP | N-(benzenesulfonyl)-L-prolyl-L-O-(1-pyrrolidinylcarbonyl)-tyrosine sodium salt |
| BRK | BRICK1 subunit of SCAR/WAVE actin nucleating complex |
| DAT | dopamine active transporter |
| DAST | (diethylamino)sulfur trifluoride |
| (DHQD)2PHAL | hydroquinidine 1,4-phthalazinediyl diether |
| EEDQ | 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline |
| FAK | focal adhesion kinase |
| FRET | förster or fluorescence resonance energy transfer |
| GTPase | guanosine triphosphate hydrolase |
| HOBt | 1-hydroxybenzotriazole |
| HypF-N | N-terminal domain of the E. coli HypF carbamoyltransferase |
| IL13Rα2 | interleukin-13 receptor α2 |
| Jak2 | Janus kinase 2 |
| LDLR | low density lipoprotein receptor |
| LMO4 | LIM domain only 4 |
| MBC | minimum bactericidal concentration |
| MCP-1 | macrophage-1 chemoattractant protein |
| MDR | multidrug-resistant |
| mGluR5 | metabotropic glutamate receptor 5 |
| MHC | minimum hemolytic concentration |
| MNP | magnetic nanoparticles |
| mRNA | messenger RNA |
| MRSA | methicillin-resistant S. aureus |
| MECBS | 2-methyl-CBS-oxazaborolidine |
| NDM-1 | new Delhi metallo-beta-lactamase |
| NHE | sodium-hydrogen exchanger |
| PADS | petromyzonamine disulfate |
| PDMS | Polydimethylsiloxane |
| PI3K | phosphoinositide 3-kinase |
| PEI | Polyethyleneimines |
| PTP1B | protein tyrosine phosphatase 1B |
| PTSA | 4-toluenesulfonamide |
| shRNA | small hairpin RNA |
| SNP | silver nanoparticle |
| STAT | signal transducer and activator of transcription |
| TBHP | tert-butyl hydroperoxide solution |
| TEMPO | 2,2,6,6-tetramethylpiperidine 1-oxyl |
| Tyk2 | non-receptor tyrosine-protein kinase 2 |
| TMCS | trimethylsilyl chloride |
| VEGF | vascular endothelial growth factor |
References
- Casero, R.A.; Woster, P.M. Terminally alkylated polyamine analogues as chemotherapeutic agents. J. Med. Chem. 2001, 44, 1–26. [Google Scholar] [CrossRef]
- Karigiannis, G.; Papaioannou, D. Structure, biological activity and synthesis of polyamine analogues and conjugates. Eur. J. Org. Chem. 2000, 2000, 1841–1863. [Google Scholar] [CrossRef]
- Rogoza, L.N.; Salakhutdinov, N.F.; Tolstikov, G.A. Polymethyleneamine alkaloids of animal origin: I. Metabolites of marine and microbial organisms. Russ. J. Bioorg. Chem. 2005, 31, 507–520. [Google Scholar] [CrossRef]
- Rogoza, L.N.; Salakhutdinov, N.F.; Tolstikov, G.A. Polymethyleneamine alkaloids of animal origin: II. Polyamine neurotoxins. Russ. J. Bioorg. Chem. 2006, 32, 23–36. [Google Scholar] [CrossRef]
- Stone, R. Deja vu guides the way to new antimicrobial steroid. Science 1993, 259, 1125. [Google Scholar] [CrossRef]
- Moore, K.S.; Wehrli, S.; Roder, H.; Rogers, M.; Forrest, J.N., Jr.; McCrimmon, D.; Zasloff, M. Squalamine: An aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. USA 1993, 90, 1354–1358. [Google Scholar] [CrossRef]
- Rao, M.N.; Shinnar, A.E.; Noecker, L.A.; Chao, T.L.; Feibush, B.; Snyder, B.; Sharkansky, I.; Sarkahian, A.; Zhang, X.; Jones, S.R.; et al. Aminosterols from the Dogfish Shark Squalus acanthias. J. Nat. Prod. 2000, 63, 631–635. [Google Scholar] [CrossRef]
- Moriarty, R.M.; Tuladhar, S.M.; Guo, L.; Wehrli, S. Synthesis of squalamine. A steroidal antibiotic from the shark. Tetrahedron Lett. 1994, 35, 8103–8106. [Google Scholar] [CrossRef]
- Moriarty, R.M.; Enache, L.A.; Kinney, W.A.; Allen, C.S.; Canary, J.W.; Tuladhar, S.M.; Guo, L. Stereoselective synthesis of squalamine dessulfate. Tetrahedron Lett. 1995, 36, 5139–5142. [Google Scholar] [CrossRef]
- Yun, S.-S.; Li, W. Identification of squalamine in the plasma membrane of white blood cells in the sea lamprey, Petromyzon marinus. J. Lipid Res. 2007, 48, 2579–2586. [Google Scholar] [CrossRef]
- Brunel, J.M.; Letourneux, Y. Recent Advances in the Synthesis of Spermine and Spermidine Analogs of the Shark Aminosterol Squalamine. Eur. J. Org. Chem. 2003, 2003, 3897–3907. [Google Scholar] [CrossRef]
- Schmidt, E.J.; Boswell, J.S.; Walsh, J.P.; Schellenberg, M.M.; Winter, T.W.; Li, C.; Allman, G.W.; Savage, P.B. Activities of cholic acid-derived antimicrobial agents against multidrug-resistant bacteria. J. Antimicrob. Chemother. 2001, 47, 671–674. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cushnie, T.P.T.; Cushnie, B.; Lamb, A.J. Alkaloids: An overview of their antibacterial, antibiotic-enhancingand antivirulence activities. Int. J. Antimicrob. Agents 2014, 44, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Sills, A.K., Jr.; Williams, J.I.; Tyler, B.M.; Epstein, D.S.; Sipos, E.P.; Davis, J.D.; McLane, M.P.; Pitchford, S.; Cheshire, K.; Gannon, F.H.; et al. Squalamine inhibits angiogenesis and solid tumor growth in vivo and perturbsembryonic vasculature. Cancer Res. 1998, 58, 2784–2792. [Google Scholar]
- Westa, C.L.; Mao, Y.-K.; Delungahawatta, T.; Amin, J.Y.; Farhin, S.; McQuade, R.M.; Diwakarla, S.; Pustovit, R.; Stanisz, A.M.; Bienenstock, J.; et al. Squalamine restores the function of the enteric nervous system in mouse models of Parkinson’s disease. J. Parkinson’s Dis. 2020, 10, 1477–1491. [Google Scholar] [CrossRef]
- Hoye, T.R.; Dvornikovs, V.; Fine, J.M.; Anderson, K.R.; Jeffrey, C.S.; Muddiman, D.C.; Shao, F.; Sorensen, P.W.; Wang, J. Details of the structure determination of the sulfated steroids PSDS and PADS: New components of the Sea Lamprey (Petromyzon marinus) migratory pheromone. J. Org. Chem. 2007, 72, 7544–7550. [Google Scholar] [CrossRef]
- Fine, J.M.; Sorensen, P.W. Isolation and biological activity of the multi-component sea lamprey migratory pheromone. J. Chem. Ecol. 2008, 34, 1259–1267. [Google Scholar] [CrossRef]
- Seiler, N.; Knodgen, B.; Haegele, K. N-(3-Aminopropyl)pyrrolidin-2-one, a product of spermidine catabolism in vivo. Biochem. J. 1982, 208, 189–197. [Google Scholar] [CrossRef]
- Barbut, D.; Zasloff, M. Methods for Treating Blood Pressure Conditions Using Aminosterol Compositions. U.S. Patent 20200129528A1, 30 April 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Methods of Treating Alzheimer’s Disease Using Aminosterol Compositions. U.S. Patent 20200038412A, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Methods of Treating Parkinson’s Disease Using Aminosterol Compositions. U.S. Patent 20200038413A1, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Methods of Treating Constipation Using Aminosterol Compositions. U.S. Patent 20200038414A1, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Aminosterol Compositions and Methods of Using the Same for Treating Erectile Dysfunction. U.S. Patent 20200038415A1, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Methods of Treating Cardiac Conduction Defects Using Aminosterol Compositions. U.S. Patent 20200038416A1, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Methods and Compositions for Treating Cognitive Impairment. U.S. Patent 20200038417A1, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Methods of Treating Autism Spectrum Disorder Using Aminosterol Compositions. U.S. Patent 20200038418A1, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Methods of Treating Multiple System Atrophy Using Aminosterol Compositions. U.S. Patent 20200038419A1, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Aminosterol Compositions and Methods of Using the Same for Treating Depression. U.S. Patent 20200038420A1, 6 February 2020. [Google Scholar]
- Barbut, D.; Zasloff, M. Aminosterol Compositions and Methods of Using the Same for Treating Schizophrenia. U.S. Patent 20200155574A1, 21 May 2020. [Google Scholar]
- Zasloff, M. Methods for Treating and Preventing Viral Infections. U.S. Patent 20200323883A1, 15 October 2020. [Google Scholar]
- Selinsky, B.S.; Zhou, Z.; Fojtik, K.G.; Jones, S.R.; Dollahon, N.R.; Shinnar, A.E. The aminosterol antibiotic squalamine permeabilizes large unilamellar phospholipid vesicles. Biochim. Biophys. Acta-Biomembr. 1998, 1370, 218–234. [Google Scholar] [CrossRef][Green Version]
- Williams, J.I. Squalamine: A new angiostatic steroid. Antiangiogenic Agents Cancer Ther. 1999, 1, 153–174. [Google Scholar] [CrossRef]
- Savage, P.B.; Li, C. Cholic acid derivatives: Novel antimicrobials. Expert Opin. Investig. Drugs 2000, 9, 263–272. [Google Scholar] [CrossRef]
- Shu, Y.; Jones, S.R.; Kinney, W.A.; Selinsky, B.S. The synthesis of spermine analogs of the shark aminosterol squalamine. Steroids 2002, 67, 291–304. [Google Scholar] [CrossRef]
- Savage, P.B. Cationic steroid antibiotics. Curr. Med. Chem.–Anti-Infect. Agents 2002, 1, 293–304. [Google Scholar] [CrossRef]
- Salunke, D.B.; Hazra, B.G.; Pore, V.S. Bile acide-polyamine conugates as synthetic ionophores. Archivoc 2003, 9, 115–125. [Google Scholar]
- Choucair, B.; Dherbomez, M.; Roussakis, C.; El Kihel, L. Synthesis of spermidinylcholestanol and spermidinylcholesterol, squalamine analogues. Tetrahedron 2004, 60, 11477–11486. [Google Scholar] [CrossRef]
- Alhanout, K.; M Rolain, J.; M Brunel, J. Squalamine as an Example of a New Potent Antimicrobial Agents Class: A Critical Review. Curr. Med. Chem. 2010, 17, 3909–3917. [Google Scholar] [CrossRef]
- Bourguet-Kondracki, M.-L.; Brunel, J.-M. Promises of the Unprecedented Aminosterol Squalamine. Outstanding Marine Molecules: Chemistry, Biology, Analysis, 1st ed.; Wiley-VCH Verlag GmbH&Co., KGaA: Weinheim, Germany, 2014; pp. 265–283, Print ISBN: 9783527334650, Online ISBN: 9783527681501. [Google Scholar] [CrossRef]
- Djouhri-Bouktab, L.; Rolain, J.M.; Brunel, J.M. Mini-Review: Polyamines Metabolism, Toxicity and Potent Therapeutical Use. Anti-Infect. Agents 2014, 12, 95–103. [Google Scholar] [CrossRef]
- Blanchet, M.; Brunel, J.M. Bile acid derivatives: From old molecules to a new potent therapeutic use: An overview. Curr. Med. Chem. 2018, 25, 3613–3636. [Google Scholar] [CrossRef] [PubMed]
- Négrel, S.; Brunel, J.-M. Synthesis and biological activities of naturally functionalized polyamines: An overview. Curr. Med. Chem. 2021, 28, 3406–3448. [Google Scholar] [CrossRef] [PubMed]
- Kinney, W.A.; Zhang, X.; Williams, J.I.; Johnston, S.; Michalak, R.S.; Deshpande, M.; Dostal, L.; Rosazza, J.P.N. A short formal synthesis of squalamine from a microbial metabolite. Org. Lett. 2000, 2, 2921–2922. [Google Scholar] [CrossRef]
- Weis, A.L.; Bakos, T.; Alferiev, I.; Zhang, X.; Shao, B.; Kinney, W.A. Synthesis of an azido spermidine equivalent. Tetrahedron Lett. 1999, 40, 4863–4864. [Google Scholar] [CrossRef]
- Wehrli, S.L.; Moore, K.S.; Roder, H.; Durell, S.; Zasloff, M. Structure of the novel steroidal antibiotic squalamine determined by two-dimensional NMR spectroscopy. Steroids 1993, 58, 370–378. [Google Scholar] [CrossRef]
- Pearson, A.J.; Chen, Y.S.; Han, G.R.; Hsu, S.Y.; Ray, T. A new method for the oxidation of alkenes to enones. An efficient synthesis of Δ5-7-oxo steroids. J. Chem. Soc. Perkin Trans. 1 1985, 267–273. [Google Scholar] [CrossRef]
- Tal, D.M.; Frisch, G.D.; Elliott, W.H. Bile acids LXIX. Selective K-selectride reduction of 3,7-diketo steroids. Tetrahedron 1984, 40, 851–854. [Google Scholar] [CrossRef]
- Arnostova, L.M.; Pouzar, V.; Drasar, P. Preparation of steroid hydroxy sulfates. Synth. Commun. 1990, 20, 1521–1529. [Google Scholar] [CrossRef]
- McGill, J.M.; LaBell, E.S.; Williams, M. Hydride reagents for stereoselective reductive amination. An improved preparation of 3-endo-tropanamine. Tetrahedron Lett. 1996, 37, 3977–3980. [Google Scholar] [CrossRef]
- Li, C.; Budge, L.P.; Driscoll, C.D.; Willardson, B.M.; Allman, G.W.; Savage, P.B. Incremental conversion of outer-membrane permeabilizers into potent antibiotics for gram-negative bacteria. J. Am. Chem. Soc. 1999, 121, 931–940. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, M.N.; Jones, S.R.; Shao, B.; Feibush, P.; McGuigan, M.; Tzodikov, N.; Feibush, B.; Sharkansky, I.; Snyder, B.; et al. Synthesis of squalamine utilizing a readily accessible spermidine equivalent. J. Org. Chem. 1998, 63, 8599–8603. [Google Scholar] [CrossRef]
- Liang, F.; Wan, S.; Li, Z.; Xiong, X.; Yang, L.; Zhou, X.; Wu, C. Medical applications of macrocyclic polyamines. Curr. Med. Chem. 2006, 13, 711–727. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.D.; Cai, F.; Zhou, W.S. A new highly stereoselective construction of the sidechain of squalamine through improved Sharpless catalytic asymmetric dihydroxylation. Tetrahedron Lett. 2001, 42, 2537–2539. [Google Scholar] [CrossRef]
- Zhou, X.-D.; Cai, F.; Zhou, W.-S. A stereoselective synthesis of squalamine. Tetrahedron 2002, 58, 10293–10299. [Google Scholar] [CrossRef]
- Zhang, D.-H.; Cai, F.; Zhou, X.-D.; Zhou, W.-S. A concise and stereoselective synthesis of squalamine. Org. Lett. 2003, 5, 3257–3259. [Google Scholar] [CrossRef]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic asymmetric dihidroxylation. Chem. Rev. 1994, 94, 2483–2549. [Google Scholar] [CrossRef]
- Iida, T.; Nishida, S.; Chang, F.C.; Niwa, T.; Goto, J.; Nambara, T. Potential bile acid metabolites. 19. The epimeric 3alpha,6,7beta-trihydroxy- and 3alpha,6,7beta,12alpha-tetrahydroxy-5alpha-cholanoic acids. Cherm. Pharm. Bull. 1993, 41, 763–765. [Google Scholar] [CrossRef][Green Version]
- Huang, L.F.; Zhou, W.S.; Sun, L.Q.; Pan, X.F. Studies on steroidal plant-growth regulators. Part 29. Osmium tetroxide-catalysed asymmetric dihydroxylation of the (22E,24R)- and the (22E,24S)-24-alkyl steroidal unsaturated side chain. J. Chem. Soc. Perkin Trans. 1 1993, 1683–1686. [Google Scholar] [CrossRef]
- Corey, E.J.; Grogan, M.J. Stereocontrolled syntheses of 24(S),25-epoxycholesterol and related oxysterols for studies on the activation of LXR receptors. Tetrahedron Lett. 1998, 39, 9351–9354. [Google Scholar] [CrossRef]
- Yadav, J.S.; Mysorekar, S.V. A facile conversion of tertiary alcohols to olefins. Synth. Commun. 1989, 19, 1057–1060. [Google Scholar] [CrossRef]
- Goodnow, R., Jr.; Konno, K.; Niwa, M.; Kallimopoulos, T.; Bukownic, R.; Lenares, D.; Nakanishi, K. Synthesis of glutamate receptor antagonist philanthotoxin-433 (PhTX-433) and its analogs. Tetrahedron 1990, 46, 3267–3286. [Google Scholar] [CrossRef]
- Okumura, K.; Nakamura, Y.; Takeuchi, S.; Kato, I.; Fujimoto, Y.; Ikekawa, N. Formal synthesis of squalamine from desmosterol. Chem. Pharm. Bull. 2003, 51, 1177–1182. [Google Scholar] [CrossRef]
- Jones, S.R.; Selinsky, B.S.; Rao, M.N.; Zhang, X.N.; Kinney, W.A.; Tham, F.S. Efficient route to 7α-(benzoyloxy)-3-dioxolane cholestan-24(R)-ol, a key intermediate in the synthesis of squalamine. J. Org. Chem. 1998, 63, 3786–3789. [Google Scholar] [CrossRef]
- Foricher, J.; Furbringer, C.; Pfoertner, K. Process for the Catalytic Oxidation of Isoprenoids Having Allylic Groups. U.S. Patent 5,030,739, 9 July 1991. [Google Scholar]
- Shen, J.M.; Zhou, X.D.; Zhou, W.S. Formal synthesis of squalamine from methylhyodeoxycholanate. Acta Chim. Sin. 2006, 64, 1513–1516. [Google Scholar]
- Zasloff, M. Use of Squalamine for the Manufacture of a Medicament for Inhibiting NHE. Europe Patent 0831837B1, 2 May 2003. [Google Scholar]
- Jones, S.R.; Kinney, W.A.; Zhang, X.; Jones, L.M.; Selinsky, B.S. The synthesis and characterization of analogs of the antimicrobial compound squalamine: 6β-hydroxy-3-aminosterols synthesized from hyodeoxycholic acid. Steroids 1996, 61, 565–571. [Google Scholar] [CrossRef]
- Aher, N.G.; Pore, V.S.; Mishra, N.N.; Shukla, P.K.; Gonnade, R.G. Design and synthesis of bile acid-based amino sterols as antimicrobial agents. Bioorg. Med. Chem. Lett. 2009, 19, 5411–5414. [Google Scholar] [CrossRef] [PubMed]
- Brunel, J.M.; Marc, J.P. Compounds That Are Analogs of Squalamine, Used as Antibacterial Agents. U.S. Patent 10729701B2, 4 August 2020. [Google Scholar]
- Kim, H.S.; Choi, B.S.; Kwon, K.S.; Lee, S.O.; Kwak, H.J.; Lee, C.H. Synthesis and antimicrobial activity of squalamine analogue. Bioorg. Med. Chem. 2000, 8, 2059–2065. [Google Scholar] [CrossRef]
- Choucair, B.; Dherbomez, M.; Roussakis, C.; El Kihel, L. Synthesis of 7α- and 7β-spermidinylcholesterol, squalamine analogues. Bioorg. Med. Chem. Lett. 2004, 14, 4213–4216. [Google Scholar] [CrossRef]
- Kim, H.-S.; Khan, S.N.; Jadhav, J.R.; Jeong, J.-W.; Jung, K.; Kwak, J.-H. A concise synthesis and antimicrobial activities of 3-polyamino-23,24-bisnorcholanes as steroid–polyamine conjugates. Bioorg. Med. Chem. Lett. 2011, 21, 3861–3865. [Google Scholar] [CrossRef]
- Salmi, C.; Loncle, C.; Vidal, N.; Letourneux, Y.; Brunel, J.M. New stereoselective titanium reductive amination synthesis of 3-amino and polyaminosterol derivatives possessing antimicrobial activities. Eur. J. Med. Chem. 2008, 43, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Salmi, C.; Loncle, C.; Vidal, N.; Laget, M.; Letourneux, Y.; Brunel, J. New 3-aminosteroid derivatives as a new family of topical antibacterial agents active against methicillin-resistant staphylococcus aureus (MRSA). Lett. Drug Des. Discov. 2008, 5, 169–172. [Google Scholar] [CrossRef]
- Salmi, C.; Loncle, C.; Vidal, N.; Laget, M.; Letourneux, Y.; Brunel, J.M. Antimicrobial activities of 3-amino- and polyaminosterol analogues of squalamine and trodusquemine. J. Inzyme Inhib. Med. Chem. 2008, 23, 860–865. [Google Scholar] [CrossRef]
- Boes, A.; Brunel, J.M.; Derouaux, A.; Kerff, F.; Bouhss, A.; Touze, T.; Breukink, E.; Terrak, M. Squalamine and aminosterol mimics inhibit the peptidoglycan glycosyltransferase activity of PBP1B. Antibiotics 2020, 9, 373. [Google Scholar] [CrossRef] [PubMed]
- Loncle, C.; Salmi, C.; Letourneux, Y.; Brunel, J.M. Synthesis of new 7-aminosterol squalamine analogues with high antimicrobial activities through a stereoselective titanium reductive amination reaction. Tetrahedron 2007, 63, 12968–12974. [Google Scholar] [CrossRef]
- Alhanout, K.; Brunel, J.M.; Raoult, D.; Rolain, J.M. In vitro antibacterial activity of aminosterols against multidrug-resistant bacteria from patients with cystic fibrosis. J. Antimicrob. Chemother. 2009, 64, 810–814. [Google Scholar] [CrossRef]
- Alhanout, K.; Brunel, J.M.; Ranque, S.; Rolain, J.M. In vitro antifungal activity of aminosterols against moulds isolated from cystic fibrosis patients. J. Antimicrob. Chemother. 2010, 65, 1307–1309. [Google Scholar] [CrossRef]
- Blanchet, M.; Borselli, D.; Brunel, J.M. Polyamine derivatives: A revival of an old neglected scaffold to fight resistant Gram-negative bacteria? Future Med. Chem. 2016, 8, 963–973. [Google Scholar] [CrossRef]
- Djouhri, L.; Vidal, N.; Rolain, J.M.; Brunel, J.M. Synthesis of new 3,20-bispolyaminosteroid squalamine analogues and evaluation of their antimicrobial activities. J. Med. Chem. 2011, 54, 7417–7421. [Google Scholar] [CrossRef]
- Sakr, A.; Laurent, F.; Brunel, J.-M.; Dagher, T.N.; Blin, O.; Rolain, J.-M. Polyaminosteroid Analogues as Potent Antibacterial Agents Against Mupirocin- Resistant Staphylococcus aureus Strains. Anti-Infect. Agents 2020, 18, 239–244. [Google Scholar] [CrossRef]
- Khan, S.N.; Kim, B.J.; Kim, H.-S. Synthesis and antimicrobial activity of 7-fluoro-3-aminosteroids. Bioorg. Med. Chem. Lett. 2007, 17, 5139–5142. [Google Scholar] [CrossRef]
- Khabnadideh, S.; Tan, C.L.; Croft, S.L.; Kendrick, H.; Yardley, V.; Gilbert, I.H. Squalamine analogues as potential anti-trypanosomal and anti-leishmanial compounds. Bioorg. Med. Chem. Lett. 2000, 10, 1237–1239. [Google Scholar] [CrossRef]
- Dvorken, L.V.; Smyth, R.B.; Mislow, K. Stereochemistry of the 1,2,3,4-dibenz-1,3-cyclooctadiene system. J. Am. Chem. Soc. 1958, 80, 486–492. [Google Scholar] [CrossRef]
- Blagbrough, I.S.; Al-Hadithi, D.; Geall, A.J. DNA condensation by bile acid conjugates of thermine and spermine. Pharm. Pharmacol. Commun. 1999, 5, 139–144. [Google Scholar] [CrossRef]
- Vida, N.; Svobodová, H.; Rárová, L.; Drašar, P.; Šaman, D.; Cvačka, J.; Wimmer, Z. Polyamine conjugates of stigmasterol. Steroids 2012, 77, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, R.A.S.; Bucki, R.; Janmey, P.A.; Diamond, S.L. A series of cationic sterol lipids with gene transfer and bactericidal activity. Bioorg. Med. Chem. 2009, 17, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Blagbrough, I.S.; Geall, A.J.; Neal, A.P. Polyamines and novel polyamine conjugates interact with DNA in ways that can be exploited in non-viral gene therapy. Biochem. Soc. Trans. 2003, 31, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Geall, A.J.; Taylor, R.J.; Earll, M.E.; Eaton, M.A.W.; Blagbrough, I.S. Synthesis of cholesteryl polyamine carbamates: pKa studies and condensation of calf thymus DNA. Bioconjug. Chem. 2000, 11, 314–326. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kwon, K.-C.; Kim, K.S.; Lee, C.H. Synthesis and antimicrobial activity of new 3α-Hydroxy-23,24-bisnorcholane polyamine carbamates. Bioorg. Med. Chem. Lett. 2001, 11, 3065–3068. [Google Scholar] [CrossRef]
- Savage, P.B. Design, synthesis and characterization of cationic peptide and steroid antibiotics. Eur. J. Org. Chem. 2002, 2002, 759–768. [Google Scholar] [CrossRef]
- Lai, X.Z.; Feng, Y.; Pollard, J.; Chin, J.N.; Rybak, M.J.; Bucki, R.; Epand, R.F.; Epand, R.M.; Savage, P.B. Ceragenins: Cholic acidbased mimics of antimicrobial peptides. Acc. Chem. Res. 2008, 41, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Epand, R.F.; Savage, P.B. Ceragenins (cationic steroid compounds), a novel class of antimicrobial agents. Drug News Perspect. 2008, 21, 307–311. [Google Scholar] [CrossRef]
- Leszczyńska, K.; Namiot, A.; Fein, D.E.; Wen, Q.; Namiot, Z.; Savage, P.B.; Diamond, S.; Janmey, P.A.; Bucki, R. Bactericidal activities of the cationic steroid CSA-13 and the cathelicidin peptide LL-37 against Helicobacter pylori in simulated gastric juice. BMC Microbiol. 2009, 9, 187. [Google Scholar] [CrossRef]
- Bucki, R.; Sostarecz, A.G.; Byfield, F.J.; Savage, P.B.; Janmey, P.A. Resistance of the antibacterial agent ceragenin CSA-13 to inactivation by DNA or F-actin and its activity in cystic fibrosis sputum. J. Antimicrob. Chemother. 2007, 60, 535–545. [Google Scholar] [CrossRef]
- Bucki, R.; Namiot, D.B.; Namiot, Z.; Savage, P.B.; Janmey, P.A. Salivary mucins inhibit antibacterial activity of the cathelicidin-derived LL-37 peptide but not the cationic steroid CSA-13. J. Antimicrob. Chemother. 2008, 62, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Leszczyńska, K.; Namiot, A.; Cruz, K.; Byfield, F.J.; Won, E.; Mendez, G.; Sokołowski, W.; Savage, P.B.; Bucki, R.; Janmey, P.A. Potential of ceragenin CSA-13 and its mixture with pluronic F-127 as treatment of topical bacterial infections. J. Appl. Microbiol. 2011, 110, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Hoppenes, M.A.; Sylvester, C.B.; Qureshi, A.T.; Scherr, T.; Czapski, D.R.; Duran, R.S.; Savage, P.B.; Hayes, D. Ceragenin mediated selectivity of antimicrobial silver nanoparticles. ACS Appl. Mater. Interfaces 2014, 6, 13900–13908. [Google Scholar] [CrossRef] [PubMed]
- Niemirowicz, K.; Surel, U.; Wilczewska, A.Z.; Mystkowska, J.; Piktel, E.; Gu, X.; Namiot, Z.; Kułakowska, A.; Savage, P.B.; Bucki, R. Bactericidal activity and biocompatibility of ceragenin-coated magnetic nanoparticles. J. Nanobiotechnol. 2015, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Bernard, E.M.; Sadownik, A.; Regen, S.L.; Armstrong, D. Antimicrobial activities of squalamine mimics. Antimicrob. Agents Chemother. 1997, 41, 1433–1438. [Google Scholar] [CrossRef]
- Saha, S.; Savage, P.B.; Bal, M. Enhancement of the efficacy of erythromycin in multiple antibiotic-resistant gram-negative bacterial pathogens. J. Appl. Microbiol. 2008, 105, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, J.-P.; Brunel, J.-M.; Chevalier, J.; Pages, J.-M. Squalamine, an original chemosensitizer to combat antibiotic-resistant Gram-negative bacteria. J. Antimicrob. Chemother. 2010, 65, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Yin, N.; Liu, Y.; Cardenas-Garcia, J.; Evanson, R.; Orsak, T.; Fan, M.; Turin, G.; Savage, P.B. Origins of cell selectivity of cationic steroid antibiotics. J. Am. Chem. Soc. 2004, 126, 13642–13648. [Google Scholar] [CrossRef]
- Zasloff, M. Formulations Comprising Aminosterols. U.S. Patent 8623416B2, 7 January 2014. [Google Scholar]
- Vicens, M.; Medarde, M.; Macias, R.I.R.; Larena, M.G.; Villafaina, A.; Serrano, M.A.; Marin, J.J.G. Novel cationic and neutral glycocholic acid and polyamine conjugates able to inhibit transporters involved in hepatic and intestinal bile acid uptake. Bioorg. Med. Chem. 2007, 15, 2359–2367. [Google Scholar] [CrossRef]
- Blagbrough, I.S.; Geall, A.J. Practical synthesis of unsymmetrical polyamine amides. Tetrahedron Lett. 1998, 39, 439–442. [Google Scholar] [CrossRef]
- Chen, W.-H.; Shao, X.-B.; Moellering, R.; Wennersten, C.; Regen, S.L. A bioconjugate approach toward squalamine mimics: Insight into the mechanism of biological action. Bioconjug. Chem. 2006, 17, 1582–1591. [Google Scholar] [CrossRef]
- Chen, W.-H.; Wennersten, C.; Moellering, R.C.; Regen, S.L. Towards squalamine mimics: Synthesis and antibacterial activities of head-to-tail dimeric sterol-polyamine conjugates. Chem. Biodivers. 2013, 10, 385–393. [Google Scholar] [CrossRef]
- Xiao, Q.; Sun, J.; Ju, Y.; Zhao, Y.; Cui, Y. Facile synthesis of 3β-cholesterol H-phosphonates. Chem. Lett. 2003, 32, 522–523. [Google Scholar] [CrossRef]
- Xiao, Q.; Sun, J.; Sun, Q.; Ju, Y.; Zhao, Y.F.; Cui, Y.X. Synthesis of AZT 5′-O-hydrogen phospholipids and their derivatives. Synthesis 2003, 2003, 107–112. [Google Scholar] [CrossRef]
- Ji, S.H.; Xiao, Q.; Ju, Y.; Zhao, Y. Synthesis of novel dimeric steroidal-nucleoside phosphoramidates. Chem. Lett. 2005, 34, 944–945. [Google Scholar] [CrossRef]
- Wu, D.; Ji, S.; Wu, Y.; Ju, Y.; Zhao, Y. Design, synthesis, and antitumor activity of bile acid-polyamine-nucleoside conjugates. Bioorg. Med. Chem. Lett. 2007, 17, 2983–2986. [Google Scholar] [CrossRef] [PubMed]
- Shawakfeh, K.Q.; Al-Said, N.H.; Abboushi, E.K. Synthesis of new di- and triamine diosgenin dimers. Tetrahedron 2010, 66, 1420–1423. [Google Scholar] [CrossRef]
- Bajaj, A.; Kondaiah, P.; Bhattacharya, S. Synthesis and gene transfection efficacies of PEI-cholesterol-based lipopolymers. Bioconjug. Chem. 2008, 19, 1640–1651. [Google Scholar] [CrossRef]
- Mishra, R.; Mishra, S. Updates in bile acid-bioactive molecule conjugates and their applications. Steroids 2020, 159, 108639–108654. [Google Scholar] [CrossRef]
- Errico, S.; Lucchesi, G.; Odino, D.; Muscat, S.; Capitini, C.; Bugelli, C.; Canale, C.; Ferrando, R.; Grasso, G.; Barbut, D.; et al. Making biological membrane resistant to the toxicity of misfolded protein oligomers: A lesson from trodusquemine. Nanoscale 2020, 12, 22596–22614. [Google Scholar] [CrossRef]
- Govers, R.M.T.; Brunel, J.M. Anti-Diabetic Aminosteroid. Derivatives. Patent WO2013057422A1, 25 April 2013. [Google Scholar]
- Pandey, Z.N.R.; Zhou, X.; Zaman, T.; Cruz, S.A.; Qin, Z.; Lu, M.; Keyhanian, K.; Brunel, J.M.; Stewart, A.F.R.; Chen, H.H. LMO4 is required to maintain hypothalamic insulin signaling. Biochem. Biophys. Res. Commun. 2014, 450, 666–672. [Google Scholar] [CrossRef]
- Blanchet, M.; Borselli, D.; Rodallec, A.; Peiretti, F.; Vidal, N.; Bolla, J.-M.M.; Digiorgio, C.; Morrison, K.R.; Wuest, W.M.; Brunel, J.M. Claramines: A New Class of Broad-Spectrum Antimicrobial Agents with Bimodal Activity. ChemMedChem 2018, 13, 1018–1027. [Google Scholar] [CrossRef]
- Mancini, I.; Defant, A.; Guella, G. Recent synthesis of marine natural products with antibacterial activities. Anti-Infect. Agents Med. Chem. 2007, 6, 17–48. [Google Scholar] [CrossRef]
- Pietras, R.J.; Weinberg, O.K. Antiangiogenic Steroids in Human Cancer Therapy. Evid. Based Complement. Alternat. Med. 2005, 2, 49–57. [Google Scholar] [CrossRef]
- Wnorowska, U.; Fiedoruk, K.; Piktel, E.; Prasad, S.V.; Sulik, M.; Janion, M.; Daniluk, T.; Savage, P.B.; Bucki, R. Nanoantibiotics containing membrane-active human cathelicidin LL-37 or synthetic ceragenins attached to the surface of magnetic nanoparticles as novel and innovative therapeutic tools: Current status and potential future applications. J. Nanobiotechnol. 2020, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Rauter, A.P. Membrane-targeting antibiotics: Recent developments outside the peptide space. Future Med. Chem. 2019, 11, 211–228. [Google Scholar] [CrossRef]
- Butler, M.W.; Stahlschmidt, Z.R.; Ardia, D.R.; Davies, S.; Davis, J.; Guillette, L.J.; Johnson, N.; McCormick, S.D.; McGraw, K.J.; DeNardo, D.F. Thermal sensitivity of immune function: Evidence against a generalist-specialist trade-off among endothermic and ectothermic vertebrates. Am. Nat. 2013, 181, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.B.; Li, C.; Taotafa, U.; Ding, B.; Guan, Q. Antibacterial properties of cationic steroid antibiotics. FEMS Microbiol. Lett. 2002, 217, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Alhanout, K.; Giorgio, C.D.; Meo, M.D.; Brunel, J.M. Non-Genotoxic Assessment of a Natural Antimicrobial Agent: Squalamine. Anti-Infect. Agents 2014, 12, 75–79. [Google Scholar] [CrossRef]
- Salmi, C.; Loncle, C.; Vidal, N.; Letourneux, Y.; Fantini, J.; Maresca, M.; Taïeb, N.; Pagès, J.M.; Brunel, J.M.; Page, J. Squalamine: An Appropriate Strategy against the Emergence of Multidrug Resistant Gram-Negative Bacteria? PLoS ONE 2008, 3, e2765. [Google Scholar] [CrossRef]
- Alhanout, K.; Djouhri, L.; Vidal, N.; Brunel, J.M.; Piarroux, R.; Ranque, S. In vitro activity of aminosterols against yeasts involved in blood stream infections. Med. Mycol. 2011, 49, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Khelaifia, S.; Brunel, J.M.; Michel, J.B.; Drancourt, M. In-vitro archaeacidal activity of biocides against human-associated archaea. PLoS ONE 2013, 8, e62738. [Google Scholar] [CrossRef]
- Khelaifia, S.; Drancourt, M. Susceptibility of archaea to antimicrobial agents: Applications to clinical microbiology. Clin. Microbiol. Infect. 2012, 18, 841–848. [Google Scholar] [CrossRef]
- Nicol, M.; Mlouka, M.A.B.; Berthe, T.; Di Martino, P.; Jouenne, T.; Brunel, J.M.; Dé, E. Anti-persister activity of squalamine against Acinetobacter baumannii. Int. J. Antimicrob. Agents 2019, 53, 337–342. [Google Scholar] [CrossRef]
- Coulibaly, O.; Alhanout, K.; L’ollivier, C.; Brunel, J.M.; Thera, M.A.; Djimdé, A.A.; Doumbo, O.K.; Piarroux, R.; Ranque, S. In vitro activity of aminosterols against dermatophytes. Med. Mycol. 2013, 51, 309–312. [Google Scholar] [CrossRef][Green Version]
- Kwon, D.H.; Lu, C.D. Polyamines increase antibiotic susceptibility in pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Lu, C.D. Polyamine effects on antibiotic susceptibility in bacteria. Antimicrob. Agents Chemother. 2007, 51, 2070–2077. [Google Scholar] [CrossRef]
- Alhanout, K.; Malesinki, S.; Vidal, N.; Peyrot, V.; Rolain, J.M.; Brunel, J.M. New insights into the antibacterial mechanism of action of squalamine. J. Antimicrob. Chemother. 2010, 65, 1688–1693. [Google Scholar] [CrossRef]
- Di Pasquale, E.; Salmi-Smail, C.; Brunel, J.M.; Sanchez, P.; Fantini, J.; Maresca, M. Biophysical studies of the interaction of squalamine and other cationic amphiphilic molecules with bacterial and eukaryotic membranes: Importance of the distribution coefficient in membrane selectivity. Chem. Phys. Lipids 2010, 163, 131–140. [Google Scholar] [CrossRef]
- Selinsky, B.S.; Smith, R.; Frangiosi, A.; Vonbaur, B.; Pedersen, L. Squalamine is not a proton ionophore. Biochim. Biophys. Acta-Biomembr. 2000, 1464, 135–141. [Google Scholar] [CrossRef][Green Version]
- Russo, T.A.; Mylotte, D. Expression of the K54 and O4 specific antigen has opposite effects on the bactericidal activity of squalamine against an extraintestinal isolate of Escherichia coli. FEMS Microbiol. Lett. 1998, 162, 311–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghodbane, R.; Ameen, S.M.; Drancourt, M.; Brunel, J.M. In vitro antimicrobial activity of squalamine derivatives against mycobacteria. Tuberculosis 2013, 93, 565–566. [Google Scholar] [CrossRef]
- Asmar, S.; Drancourt, M. Chlorhexidine decontamination of sputum for culturing Mycobacterium tuberculosis. BMC Microbiol. 2015, 15, 155. [Google Scholar] [CrossRef]
- Djouhri-Bouktab, L.; Alhanout, K.; Andrieu, V.; Raoult, D.; Rolain, J.M.; Brunel, J.M. Squalamine ointment for Staphylococcus aureus skin decolonization in a mouse model. J. Antimicrob. Chemother. 2011, 66, 1306–1310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Djouhri-Bouktab, L.; Alhanout, K.; Andrieu, V.; Stremler, N.; Dubus, J.C.; Raoult, D.; Rolain, J.M.; Brunel, J.M. Soluble squalamine tablets for the rapid disinfection of home nebulizers of cystic fibrosis patients. J. Cyst. Fibros. 2012, 11, 555–559. [Google Scholar] [CrossRef][Green Version]
- Hraiech, S.; Brégeon, F.; Brunel, J.M.; Rolain, J.M.; Lepidi, H.; Andrieu, V.; Raoult, D.; Papazian, L.; Roch, A. Antibacterial efficacy of inhaled squalamine in a rat model of chronic Pseudomonas aeruginosa pneumonia. J. Antimicrob. Chemother. 2012, 67, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, O.; Thera, M.A.; Koné, A.K.; Siaka, G.; Traoré, P.; Djimdé, A.A.; Brunel, J.M.; Gaudart, J.; Piarroux, R.; Doumbo, O.K.; et al. A double-blind randomized placebo-controlled clinical trial of squalamine ointment for tinea capitis treatment. Mycopathologia 2015, 179, 187–193. [Google Scholar] [CrossRef][Green Version]
- Zasloff, M.; Adams, A.P.; Beckerman, B.; Campbell, A.; Han, Z.; Luijten, E.; Meza, I.; Julander, J.; Mishra, A.; Qu, W.; et al. Squalamine as a broad-spectrum systemic antiviral agent with therapeutic potential. Proc. Natl. Acad. Sci. USA 2011, 108, 15978–15983. [Google Scholar] [CrossRef]
- Ozaslan, M. Squalamine: May be an effective viral control. Int. J. Virol. 2012, 8, 285–287. [Google Scholar] [CrossRef][Green Version]
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Müller, M.B.D.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.A.; Cascella, R.; et al. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017. [Google Scholar] [CrossRef]
- Limbocker, R.; Staats, R.; Chia, S.; Ruggeri, F.S.; Mannini, B.; Xu, C.K.; Perni, M.; Cascella, R.; Bigi, A.; Sasser, L.R.; et al. Squalamine and Its Derivatives Modulate the Aggregation of Amyloid-β and α-Synuclein and Suppress the Toxicity of Their Oligomers. Front. Neurosci. 2021, 15, 541–558. [Google Scholar] [CrossRef] [PubMed]
- Perni, M.; Flagmeier, P.; Limbocker, R.; Cascella, R.; Aprile, F.A.; Galvagnion, C.; Heller, G.T.; Meisl, G.; Chen, S.W.; Kumita, J.R.; et al. Multistep inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem. Biol. 2018, 13, 2308–2319. [Google Scholar] [CrossRef]
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 2020, 3, 435. [Google Scholar] [CrossRef] [PubMed]
- Errico, S.; Ramshini, H.; Capitini, C.; Canale, C.; Spaziano, M.; Barbut, D.; Calamai, M.; Zasloff, M.; Oropesa-Nuñez, R.; Vendruscolo, M.; et al. Quantitative Measurement of the Affinity of Toxic and Nontoxic Misfolded Protein Oligomers for Lipid Bilayers and of Its Modulation by Lipid Composition and Trodusquemine. ACS Chem. Neurosci. 2021, 12, 3189–3202. [Google Scholar] [CrossRef]
- Perni, M.; van der Goot, A.; Limbocker, R.; van Ham, T.J.; Aprile, F.A.; Xu, C.K.; Flagmeier, P.; Thijssen, K.; Sormanni, P.; Fusco, G.; et al. Comparative Studies in the A30P and A53T α-Synuclein C. Elegans Strains to Investigate the Molecular Origins of Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 492–502. [Google Scholar] [CrossRef]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine enhances Aβ42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 2019, 10, 225. [Google Scholar] [CrossRef] [PubMed]
- Odino, D.; Errico, S.; Canale, C.; Ferrando, R.; Chiti, F.; Relini, A. Interaction between Biomimetic Lipid Membranes and Trodusquemine: An Atomic Force Microscopy Study. Biophys. J. 2021, 120, 325a. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, Z.; Sharmin, F.; Lin, W.; Ricke, K.M.; Zasloff, M.A.; Stewart, A.F.R.; Chen, H.-H. Tyrosine Phosphatase PTP1B Impairs Presynaptic NMDA Receptor-Mediated Plasticity in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2021, 156, 105402. [Google Scholar] [CrossRef]
- Ricke, K.M.; Cruz, S.A.; Qin, Z.; Farrokhi, K.; Sharmin, F.; Zhang, L.; Zasloff, M.A.; Stewart, A.F.R.; Chen, H.-H. Neuronal Protein Tyrosine Phosphatase 1B Hastens Amyloid β-Associated Alzheimer’s Disease in Mice. J. Neurosci. 2020, 40, 1581–1593. [Google Scholar] [CrossRef]
- Limbocker, R.; Errico, S.; Barbut, D.; Knowles, T.P.J.; Vendruscolo, M.; Chiti, F.; Zasloff, M. Squalamine and Trodusquemine: Two natural products for neurodegenerative diseases, from physical chemistry to the clinic. Nat. Prod. Rep. 2021, 38. [Google Scholar] [CrossRef]
- Li, D.; Williams, J.I.; Pietras, R.J. Squalamine and cisplatin block angiogenesis and growth of human ovarian cancer cells with or without HER-2 gene overexpression. Oncogene 2002, 21, 2805–2814. [Google Scholar] [CrossRef]
- Akhter, S.; Nath, S.K.; Tse, G.M.; Williams, J.; Zasloff, M.; Donowitz, M. Squalamine, a novel cationic steroid, specifically inhibits the brush- border NA+/H+exchanger isoform NHE3. Am. J. Physiol.-Cell Physiol. 1999, 276, 136–144. [Google Scholar] [CrossRef]
- Williams, J.I.; Weitman, S.; Gonzalez, C.M.; Jundt, C.H.; Marty, J.; Stringer, S.D.; Holroyd, K.J.; Mclane, M.P.; Chen, Q.; Zasloff, M.; et al. Squalamine treatment of human tumors in nu/nu mice enhances platinum-based chemotherapies. Clin. Cancer Res. 2001, 7, 724–733. [Google Scholar]
- Teicher, B.A.; Williams, J.I.; Takeuchi, H.; Ara, G.; Herbst, R.S.; Buxton, D. Potential of the aminosterol, squalamine in combination therapy in the rat 13,762 mammary carcinoma and the murine Lewis lung carcinoma. Anticancer Res. 1998, 18, 2567–2573. [Google Scholar]
- Márquez-Garbán, D.C.; Gorrín-Rivas, M.; Chen, H.W.; Sterling, C.; Elashoff, D.; Hamilton, N.; Pietras, R.J. Squalamine blocks tumor-associated angiogenesis and growth of human breast cancer cells with or without HER-2/neu overexpression. Cancer Lett. 2019, 449, 66–75. [Google Scholar] [CrossRef]
- Higgins, R.D.; Yan, Y.; Geng, Y.; Zasloff, M.; Williams, J.I. Regression of retinopathy by squalamine in a mouse model. Pediatr. Res. 2004, 56, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Ciulla, T.A.; Criswell, M.H.; Danis, R.P.; Williams, J.I.; McLane, M.P.; Holroyd, K.J. Squalamine lactate reduces choroidal neovascularization in a laser-injury model in the rat. Retina 2003, 23, 808–814. [Google Scholar] [CrossRef]
- Genaidy, M.; Kazi, A.A.; Peyman, G.A.; Passos-Machado, E.; Farahat, H.G.; Williams, J.I.; Holroyd, K.J.; Blake, D.A. Effect of squalamine on iris neovascularization in monkeys. Retina 2002, 22, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.; Hardy, E.; Kinney, W.J.; Chao, T.; Zasloff, M.; Fauci, A. MSI-1436, a novel aminosterol, inhibits HIV replication in vitro and in vivo infected mononuclear cells. In Proceedings of the 4th Conference on Retroviruses and Opportunistic Infections, Washington, DC, USA, 22–26 January 1997; p. 231. [Google Scholar]
- Alper, S.L.; Chernova, M.N.; Williams, J.; Zasloff, M.; Law, F.Y.; Knauf, P.A. Differential inhibition of AE1 and AE2 anion exchangers by oxonol dyes and by novel polyaminosterol analogs of the shark antibiotic squalamine. Biochem. Cell Biol. 1998, 76, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Chernova, M.N.; Vandorpe, D.H.; Clark, J.S.; Williams, J.I.; Zasloff, M.A.; Jiang, L.; Alper, S.L. Apparent receptor-mediated activation of Ca2+-dependent conductive Cl− transport by shark-derived polyaminosterols. Am. J. Physiol. Integr. Comp. Physiol. 2005, 289, R1644–R1658. [Google Scholar] [CrossRef]
- Zasloff, M.; Williams, J.; Kinney, W.; Anderson, M.; McLane, M. Therapeutic Uses for an Aminosterol. Compound. Patent WO9744044, 27 November 1997. [Google Scholar]
- Zasloff, M.; Williams, J.; Chen, Q.; Anderson, M.; Maeder, T.; Holroyd, K.; Jones, S.; Kinney, W.; Cheshire, K.; McLane, M. A spermine-coupled cholesterol metabolite from the shark with potent appetite suppressant and antidiabetic properties. Int. J. Obes. 2001, 25, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Patel, H.R.; Takahashi, N.; Qi, Y.; Hileman, S.M.; Zasloff, M.A. Appetite suppression and weight reduction by a centrally active aminosterol. Diabetes 2002, 51, 2099–2104. [Google Scholar] [CrossRef]
- Roitman, M.F.; Wescott, S.; Cone, J.J.; McLane, M.P.; Wolfe, H.R. MSI-1436 reduces acute food intake without affecting dopamine transporter activity. Pharmacol. Biochem. Behav. 2010, 97, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Cho, H. Protein tyrosine phosphatase 1B (PTP1B) and obesity. Vitam. Horm. 2013, 91, 405–424. [Google Scholar] [CrossRef]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Haj, F.G.; Zabolotny, J.M.; Kim, Y.B.; Kahn, B.B.; Neel, B.G. Liver-specific Protein-tyrosine Phosphatase 1B (PTP1B) Re-expression Alters Glucose Homeostasis of PTP1B–/–Mice. J. Biol. Chem. 2005, 280, 15038–15046. [Google Scholar] [CrossRef]
- Delibegovic, M.; Zimmer, D.; Kauffman, C.; Rak, K.; Hong, E.G.; Cho, Y.R.; Kim, J.K.; Kahn, B.B.; Neel, B.G.; Bence, K.K. Liver-specific deletion of Protein-Tyrosine Phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 2009, 58, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J. Protein-tyrosine phosphatase 1B (PTP1B): A novel therapeutic target for type 2 diabetes mellitus, obesity and related states of insulin resistance. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2001, 1, 265–275. [Google Scholar] [CrossRef]
- Koren, S.; Fantus, I.G. Inhibition of the protein tyrosine phosphatase PTP1B: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef]
- Taylor, S. Inhibitors of Protein Tyrosine Phosphatase 1B (PTP1B). Curr. Top. Med. Chem. 2003, 3, 759–782. [Google Scholar] [CrossRef]
- Lantz, K.A.; Hart, S.G.E.E.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; Mclane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity 2010, 18, 1516–1523. [Google Scholar] [CrossRef]
- Pandey, N.R.; Zhou, X.; Qin, Z.; Zaman, T.; Gomez-Smith, M.; Keyhanian, K.; Anisman, H.; Brunel, J.M.; Stewart, A.F.R.; Chen, H.-H. The LIM domain only 4 protein is a metabolic responsive inhibitor of protein tyrosine phosphatase 1B that controls hypothalamic leptin signaling. J. Neurosci. 2013, 33, 12647–12655. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.-M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases—From housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, Y.; Zhu, S.; Liang, W.; Wang, Z.; Wang, Y.; Lv, T.; Yao, Y.; Yuan, D.; Song, Y. PTP1B promotes cell proliferation and metastasis through activating src and ERK1/2 in non-small cell lung cancer. Cancer Lett. 2015, 359, 218–225. [Google Scholar] [CrossRef]
- Lessard, L.; Labbé, D.P.; Deblois, G.; Beǵin, L.R.; Hardy, S.; Mes-Masson, A.M.; Saad, F.; Trotman, L.C.; Giguére, V.; Tremblay, M.L. PTP1B is an androgen receptor-regulated phosphatase that promotes the progression of prostate cancer. Cancer Res. 2012, 72, 1529–1537. [Google Scholar] [CrossRef]
- Wang, J.; Liu, B.; Chen, X.; Su, L.; Wu, P.; Wu, J.; Zhu, Z. PTP1B expression contributes to gastric cancer progression. Med. Oncol. 2012, 29, 948–956. [Google Scholar] [CrossRef]
- Zhu, S.; Bjorge, J.D.; Fujita, D.J. PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Res. 2007, 67, 10129–10137. [Google Scholar] [CrossRef]
- Stuible, M.; Doody, K.M.; Tremblay, M.L. PTP1B and TC-PTP: Regulators of transformation and tumorigenesis. Cancer Metastasis Rev. 2008, 27, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dubé, N.; Read, M.; Penney, J.; Paquet, M.; Han, Y.; Kennedy, B.P.; Muller, W.J.; Tremblay, M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007, 39, 338–346. [Google Scholar] [CrossRef]
- Easty, D.; Gallagher, W.; Bennett, D.C. Protein tyrosine phosphatases, new targets for cancer therapy. Curr. Cancer Drug Targets 2006, 6, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Lin, G.; Lucito, R.; Tonks, N.K. Protein-tyrosine Phosphatase 1B antagonized signaling by insulin-like growth factor-1 receptor and kinase BRK/PTK6 in ovarian cancer cells. J. Biol. Chem. 2013, 288, 24923–24934. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.P. Diabetes and cardiovascular disease: The “common soil” hypothesis. Diabetes 1995, 44, 369–374. [Google Scholar] [CrossRef]
- Halcox, J.; Misra, A. Type 2 diabetes mellitus, metabolic syndrome, and mixed dyslipidemia: How similar, how different, and how to treat? Metab. Syndr. Relat. Disord. 2015, 13, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.J.; Watts, G.F. Endothelial dysfunction in diabetes: Pathogenesis, significance, and treatment. Rev. Diabet. Stud. 2013, 10, 133–156. [Google Scholar] [CrossRef]
- Bornfeldt, K.E.; Tabas, I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011, 14, 575–585. [Google Scholar] [CrossRef]
- Pansuria, M.; Xi, H.; Li, L.; Yang, X.F.; Wang, H. Insulin resistance, metabolic stress, and atherosclerosis. Front. Biosci. (Sch. Ed.) 2012, 4, 916–931. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; Li, Q.; Freund, B.; Feather, D.; Abramov, R.; Wu, I.H.; Chen, K.; Yamamoto-Hiraoka, J.; Goldenbogen, J.; Sotiropoulos, K.B.; et al. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein e null mice. Cell Metab. 2010, 11, 379–389. [Google Scholar] [CrossRef]
- Thompson, D.; Morrice, N.; Grant, L.; Le Sommer, S.; Lees, E.K.; Mody, N.; Wilson, H.M.; Delibegovic, M. Pharmacological inhibition of protein tyrosine phosphatase 1B protects against atherosclerotic plaque formation in the LDLR−/− mouse model of atherosclerosis. Clin. Sci. 2017, 131, 2489–2501. [Google Scholar] [CrossRef]
- Lu, B.; Atala, A. Small molecules and small molecule drugs in regenerative medicine. Drug Discov. Today 2014, 19, 801–808. [Google Scholar] [CrossRef]
- Smith, A.M.; Maguire-Nguyen, K.K.; Rando, T.A.; Zasloff, M.A.; Strange, K.B.; Yin, V.P. The protein tyrosine phosphatase 1B inhibitor MSI-1436 stimulates regeneration of heart and multiple other tissues. NPJ Regen. Med. 2017, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Khoury, Z.H.; Salameh, F. Trodusquemine: Potential Utility in Wound Regeneration. Regen. Eng. Transl. Med. 2021, 7, 118–119. [Google Scholar] [CrossRef]
- Griebel, G.; Holmes, A. 50 years of hurdles and hope in anxiolytic drug discovery. Nat. Rev. Drug Discov. 2013, 12, 667–687. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Zhou, X.; Pandey, N.R.; Vecchiarelli, H.A.; Stewart, C.A.; Zhang, X.; Lagace, D.C.; Brunel, J.M.; Béïque, J.C.; Stewart, A.F.R.; et al. Chronic stress induces anxiety via an amygdalar intracellular cascade that impairs endocannabinoid signaling. Neuron 2015, 85, 1319–1331. [Google Scholar] [CrossRef]
- Krishnan, N.; Tonks, N.K. Anxious moments for the protein tyrosine phosphatase PTP1B. Trends Neurosci. 2015, 38, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Zhang, L.; Cruz, S.A.; Stewart, A.F.R.; Chen, H.-H. Activation of Tyrosine Phosphatase PTP1B in Pyramidal Neurons Impairs Endocannabinoid Signaling by Tyrosine Receptor Kinase TrkB and Causes Schizophrenia-like Behaviors in Mice. Neuropsychopharmacology 2020, 45, 1884–1895. [Google Scholar] [CrossRef]
- Bhargava, P.; Marshall, J.L.; Dahut, W.; Rizvi, N.; Trocky, N.; Williams, J.I.; Hait, H.; Song, S.; Holroyd, K.J.; Hawkins, M.J. A phase I and pharmacokinetic study of squalamine, a novel antiangiogenic agent, in patients with advanced cancers. Clin. Cancer Res. 2001, 7, 3912–3919. [Google Scholar] [PubMed]
- Hao, D.; Hammond, L.A.; Eckhardt, S.G.; Patnaik, A.; Takimoto, C.H.; Schwartz, G.H.; Goetz, A.D.; Tolcher, A.W.; McCreery, H.A.; Mamun, K.; et al. A Phase I and pharmacokinetic study of squalamine, an aminosterol angiogenesis inhibitor. Clin. Cancer Res. 2003, 9, 2465–2471. [Google Scholar] [PubMed]
- Herbst, R.S.; Hammond, L.A.; Carbone, D.P.; Tran, H.T.; Holroyd, K.J.; Desai, A.; Williams, J.I.; Bekele, B.N.; Hait, H.; Allgood, V.; et al. A phase I/IIA trial of continuous five-day infusion of squalamine lactate (MSI-1256F) plus carboplatin and paclitaxel in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2003, 9, 4108–4115. [Google Scholar]
- Ciulla, T.; Oliver, A.; Gast, M.J.; Evizon, S. Squalamine lactate for the treatment of age-related macular degeneration. Expert Rev. Ophthalmol. 2007, 2, 165–175. [Google Scholar] [CrossRef]
- Wroblewski, J.J.; Hu, A.Y. Topical squalamine 0.2% and intravitreal ranibizumab 0.5 mg as combination therapy for macular edema due to branch and central retinal vein occlusion: An open-label, randomized study. Ophthalmic Surg. Lasers Imaging Retin. 2016, 47, 914–923. [Google Scholar] [CrossRef]
- Zeitz, O.; Joussen, A.M. Eye drops instead of intravitreal injections? The dream of treating macular diseases by topically administered drugs. Klin. Monbl. Augenheilkd. 2017, 234, 1088–1093. [Google Scholar] [CrossRef]
- Rosenfeld, P.J.; Feuer, W.J. Lessons from recent phase III trial failures: Don’t design phase III trials based on retrospective subgroup analyses from phase II trials. Ophthalmology 2018, 125, 1488–1491. [Google Scholar] [CrossRef]
- Kesen, M.R.; Cousins, S.W. Encyclopedia of the Eye, 1st ed.; Academic Press: New York, NY, USA, 2010; pp. 257–265. ISBN 9780123742032. [Google Scholar] [CrossRef]
- Deng, G.; Dewa, T.; Regen, S.L. A synthetic ionophore that recognizes negatively charged phospholipid membranes. J. Am. Chem. Soc. 1996, 118, 8975–8976. [Google Scholar] [CrossRef]
- Tessema, T.D.; Gassler, F.; Shu, Y.; Jones, S.; Selinsky, B.S. Structure-activity relationships in aminosterol antibiotics: The effect of stereochemistry at the 7-OH group. Bioorg. Med. Chem. Lett. 2013, 23, 3377–3381. [Google Scholar] [CrossRef]
- Brunel, J.M.; Blanchet, M.; Marc, J.P. Amide Derivatives of Squalamine for the Treatment of Infections. U.S. Patent 0780101B2, 22 September 2020. [Google Scholar]
- Qin, Z.; Pandey, N.R.; Zhou, X.; Stewart, C.A.; Hari, A.; Huang, H.; Stewart, A.F.R.; Brunel, J.M.; Chen, H.-H. Functional properties of Claramine: A novel PTP1B inhibitor and insulin-mimetic compound. Biochem. Biophys. Res. Commun. 2015, 458, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, R.A.; Martín-Regalado, Á.; Jaén, M.; Zannikou, M.; Zhang, P.; de los Ríos, V.; Balyasnikova, I.V.; Casal, J.I. Protein Tyrosine Phosphatase-1B inhibition disrupts IL13Rα2-promoted invasion and metastasis in cancer cells. Cancers 2020, 12, 500. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Konidaris, K.F.; Gasser, G.; Tonks, N.K. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J. Biol. Chem. 2018, 293, 1517–1525. [Google Scholar] [CrossRef]
- Lou, Y.; Schapman, D.; Mercier, D.; Alexandre, S.; Dé, E.; Brunel, J.-M.; Kébir, N.; Thébault, P. Modification of poly(dimethyl siloxane) surfaces with an antibacterial claramine-derivative through click-chemistry grafting. React. Funct. Polym. 2022, 170, 105102. [Google Scholar] [CrossRef]
- Birteksoz-Tan, A.S.; Zeybek, Z.; Hacioglu, M.; Savage, P.B.; Bozkurt-Guzel, C. In vitro activities of antimicrobial peptides and ceragenins against Legionella pneumophila. J. Antibiot. 2019, 72, 291–297. [Google Scholar] [CrossRef]
- Hashemi, M.M.; Holden, B.S.; Durnas, B.; Buck, R.; Savage, P.B. Ceragenins as mimics of endogenous antimicrobial peptides. J. Antimicrob. Agents 2017, 3, 1–11. [Google Scholar] [CrossRef]
- Howell, M.D.; Streib, J.E.; Kim, B.E.; Lesley, L.J.; Dunlap, A.P.; Geng, D.; Feng, Y.; Savage, P.B.; Leung, D.Y.M. Ceragenins: A class of antiviral compounds to treat orthopox infections. J. Investig. Dermatol. 2009, 129, 2668–2675. [Google Scholar] [CrossRef]
- Moscoso, M.; Esteban-Torres, M.; Menéndez, M.; García, E. In vitro bactericidal and bacteriolytic activity of ceragenin CSA-13 against planktonic cultures and biofilms of Streptococcus pneumoniae and other pathogenic Streptococci. PLoS ONE 2014, 9, e101037. [Google Scholar] [CrossRef]
- Li, C.; Peters, A.S.; Meredith, E.L.; Allman, G.W.; Savage, P.B. Design and synthesis of potent sensitizers of gram-negative bacteria based on a cholic acid scaffolding. J. Am. Chem. Soc. 1998, 120, 2961–2962. [Google Scholar] [CrossRef]
- Oyardi, O.; Savage, P.B.; Akcali, A.; Erturan, Z.; Bozkurt-Guzel, C. Ceragenins exhibiting promising antimicrobial activity against various multidrug resistant Gram negative bacteria. Istanbul J. Pharm. 2019, 48, 68–72. [Google Scholar] [CrossRef]
- Dao, A.; Mills, R.J.; Kamble, S.; Savage, P.B.; Little, D.G.; Schindeler, A. The application of ceragenins to orthopedic surgery and medicine. J. Orthop. Res. 2020, 38, 1883–1894. [Google Scholar] [CrossRef]
- Mills, R.J.; Boyling, A.; Cheng, T.L.; Peacock, L.; Savage, P.B.; Tägil, M.; Little, D.G.; Schindeler, A. CSA-90 reduces periprosthetic joint infection in a novel rat model challenged with local and systemic Staphylococcus aureus. J. Orthop. Res. 2020, 38, 2065–2073. [Google Scholar] [CrossRef]
- Williams, D.L.; Sinclair, K.D.; Jeyapalina, S.; Bloebaum, R.D. Characterization of a novel active release coating to prevent biofilm implant-related infections. J. Biomed. Mater. Res. Part B Appl. Biomater. 2013, 101, 1078–1089. [Google Scholar] [CrossRef]
- Sinclair, K.D.; Pham, T.X.; Williams, D.L.; Farnsworth, R.W.; Loc-Carrillo, C.M.; Bloebaum, R.D. Model development for determining the efficacy of a combination coating for the prevention of perioperative device related infections: A pilot study. J. Biomed. Mater. Res. Part B Appl. Biomater. 2013, 101, 1143–1153. [Google Scholar] [CrossRef]
- Sinclair, K.D.; Pham, T.X.; Farnsworth, R.W.; Williams, D.L.; Loc-Carrillo, C.; Horne, L.A.; Ingebretsen, S.H.; Bloebaum, R.D. Development of a broad spectrum polymer-released antimicrobial coating for the prevention of resistant strain bacterial infections. J. Biomed. Mater. Res. Part A 2012, 100, 2732–2738. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Jennings, J.D.; Snarr, J.; Chaudhary, V.; Pollard, J.E.; Savage, P.B. Optimization of ceragenins for prevention of bacterial colonization of hydrogel contact lenses. Investig. Opthalmol. Vis. Sci. 2013, 54, 6217–6223. [Google Scholar] [CrossRef][Green Version]
- Jueati, R.; Jittikoon, J.; Vajragupta, O.; Pimthon, J. Molecular dynamics and experimental investigations of membrane perturbation by ceragenin CSA-13. Mahidol Univ. J. Pharm. Sci. 2012, 39, 25–32. [Google Scholar]
- Bozkurt-Guzel, C.; Savage, P.B.; Gerceker, A.A. In vitro activities of the novel ceragenin CSA-13, alone or in combination with colistin, tobramycin, and ciprofloxacin, against Pseudomonas aeruginosa strains isolated from cystic fibrosis patients. Chemotherapy 2011, 57, 505–510. [Google Scholar] [CrossRef]
- Mitchell, G.; Silvis, M.R.; Talkington, K.C.; Budzik, J.M.; Dodd, C.E.; PbBuba, J.; Oki, E.A.; Trotta, K.L.; Licht, D.; Jimenez-Morales, D.; et al. Ceragenins and antimicrobial peptides kill bacteria through distinct mechanisms. bioRxiv 2020. [Google Scholar] [CrossRef]
- Damar-Çelik, D.; Mataracı-Kara, E.; Savage, P.B.; Özbek-Çelik, B. Antibacterial and antibiofilm activities of ceragenins against Achromobacter species isolated from cystic fibrosis patients. J. Chemother. 2021, 33, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Mmuoegbulam, A.; Holden, B.; Coburn, J.; Wilson, J.; Taylor, M.; Reiley, J.; Baradaran, D.; Stenquist, T.; Deng, S.; et al. Susceptibility of Multidrug-Resistant Bacteria, isolated from water and plants in Nigeria, to Ceragenins. Int. J. Environ. Res. Public Health 2018, 15, 2758. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.M.; Holden, B.S.; Coburn, J.; Taylor, M.F.; Weber, S.; Hilton, B.; Zaugg, A.L.; McEwan, C.; Carson, R.; Andersen, J.L.; et al. Proteomic analysis of resistance of gram-negative bacteria to chlorhexidine and impacts on susceptibility to colistin, antimicrobial peptides, and ceragenins. Front. Microbiol. 2019, 10, 210. [Google Scholar] [CrossRef]
- Hacioglu, M.; Oyardi, O.; Bozkurt-Guzel, C.; Savage, P.B. Antibiofilm activities of ceragenins and antimicrobial peptides against fungal-bacterial mono and multispecies biofilms. J. Antibiot. 2020, 73, 455–462. [Google Scholar] [CrossRef]
- Hacioglu, M.; Haciosmanoglu, E.; Birteksoz-Tan, A.S.; Bozkurt-Guzel, C.; Savage, P.B. Effects of ceragenins and conventional antimicrobials on Candida albicans and Staphylococcus aureus mono and multispecies biofilms. Diagn. Microbiol. Infect. Dis. 2019, 95, 114863. [Google Scholar] [CrossRef] [PubMed]
- Olekson, M.A.; You, T.; Savage, P.B.; Leung, K.P. Antimicrobial ceragenins inhibit biofilms and affect mammalian cell viability and migration in vitro. FEBS Open Bio 2017, 7, 953–967. [Google Scholar] [CrossRef]
- Savage, P.B. 846 Effects of Ceragenins on Pseudomonas Aeruginosa biofilm formation in burn wounds in a porcine model. J. Burn Care Res. 2020, 41, S262–S263. [Google Scholar] [CrossRef]
- Piktel, E.; Pogoda, K.; Roman, M.; Niemirowicz, K.; Tokajuk, G.; Wróblewska, M.; Szynaka, B.; Kwiatek, W.M.; Savage, P.B.; Bucki, R. Sporicidal activity of ceragenin CSA-13 against Bacillus subtilis. Sci. Rep. 2017, 7, 44452. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Joseph, G.; Korza, G.; He, L.; Yuan, J.H.; Dong, W.; Setlow, B.; Li, Y.Q.; Savage, P.B.; Setlow, P. Effects of the microbicide ceragenin CSA-13 on and properties of Bacillus subtilis spores prepared on two very different media. J. Appl. Microbiol. 2019, 127, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, S.J.; Skłodowski, K.; Piktel, E.; Suprewicz, Ł.; Fiedoruk, K.; Daniluk, T.; Wolak, P.; Savage, P.B.; Bucki, R. NDM-1 carbapenemase-producing Enterobacteriaceae are highly susceptible to ceragenins CSA-13, CSA-44, and CSA-131. Infect. Drug Resist. 2020, 13, 3277–3294. [Google Scholar] [CrossRef]
- Bozkurt-Guzel, C.; Inci, G.; Oyardi, O.; Savage, P.B. Synergistic activity of ceragenins against carbapenem-resistant Acinetobacter baumannii strains in both checkerboard and dynamic time-kill assays. Curr. Microbiol. 2020, 77, 1419–1428. [Google Scholar] [CrossRef]
- Wnorowska, U.; Piktel, E.; Durnaś, B.; Fiedoruk, K.; Savage, P.B.; Bucki, R.; Durna, B.; Fiedoruk, K.; Savage, P.B.; Bucki, R. Use of ceragenins as a potential treatment for urinary tract infections. BMC Infect. Dis. 2019, 19, 369. [Google Scholar] [CrossRef]
- Ozbek-Celik, B.; Damar-Celik, D.; Mataraci-Kara, E.; Bozkurt-Guzel, C.; Savage, P.B. Comparative in vitro activities of first and second-generation ceragenins alone and in combination with antibiotics against Multidrug-Resistant Klebsiella pneumoniae strains. Antibiotics 2019, 8, 130. [Google Scholar] [CrossRef]
- Durnaś, B.; Wnorowska, U.; Pogoda, K.; Deptuła, P.; Wątek, M.; Piktel, E.; Głuszek, S.; Gu, X.; Savage, P.B.; Niemirowicz, K.; et al. Candidacidal activity of selected ceragenins and human cathelicidin LL-37 in experimental settings mimicking infection sites. PLoS ONE 2016, 11, e0157242. [Google Scholar] [CrossRef]
- Hashemi, M.M.; Rovig, J.; Holden, B.S.; Taylor, M.F.; Weber, S.; Wilson, J.; Hilton, B.; Zaugg, A.L.; Ellis, S.W.; Yost, C.D.; et al. Ceragenins are active against drug-resistant Candida auris clinical isolates in planktonic and biofilm forms. J. Antimicrob. Chemother. 2018, 73, 1537–1545. [Google Scholar] [CrossRef]
- Hacioglu, M.; Guzel, C.B.; Savage, P.B.; Tan, A.S.B. Antifungal susceptibilities, in vitro production of virulence factors and activities of ceragenins against Candida spp. isolated from vulvovaginal candidiasis. Med. Mycol. 2019, 57, 291–299. [Google Scholar] [CrossRef]
- Niemirowicz, K.; Piktel, E.; Wilczewska, A.; Markiewicz, K.; Durnaś, B.; Wątek, M.; Puszkarz, I.; Wróblewska, M.; Niklińska, W.; Savage, P.B.; et al. Core–shell magnetic nanoparticles display synergistic antibacterial effects against Pseudomonas aeruginosa and Staphylococcus aureus when combined with cathelicidin LL-37 or selected ceragenins. Int. J. Nanomed. 2016, 11, 5443–5455. [Google Scholar] [CrossRef]
- Durnaś, B.; Piktel, E.; Wątek, M.; Wollny, T.; Góźdź, S.; Smok-Kalwat, J.; Niemirowicz, K.; Savage, P.B.; Bucki, R. Anaerobic bacteria growth in the presence of cathelicidin LL-37 and selected ceragenins delivered as magnetic nanoparticles cargo. BMC Microbiol. 2017, 17, 167. [Google Scholar] [CrossRef]
- Niemirowicz, K.; Durnaś, B.; Tokajuk, G.; Piktel, E.; Michalak, G.; Gu, X.; Kułakowska, A.; Savage, P.B.; Bucki, R. Formulation and candidacidal activity of magnetic nanoparticles coated with cathelicidin LL-37 and ceragenin CSA-13. Sci. Rep. 2017, 7, 4610. [Google Scholar] [CrossRef]
- Hashemi, M.; Holden, B.; Taylor, M.; Wilson, J.; Coburn, J.; Hilton, B.; Nance, T.; Gubler, S.; Genberg, C.; Deng, S.; et al. Antibacterial and antifungal activities of poloxamer Mmicelles containing ceragenin CSA-131 on ciliated tissues. Molecules 2018, 23, 596. [Google Scholar] [CrossRef] [PubMed]
- Piktel, E.; Bucki, R. Therapeutic potential of ceragenins and nanosystems containing membrane active compounds in the anti-cancer therapies. Postępy Pol. Med. Farm. 2019, 6, 21–32. [Google Scholar] [CrossRef]
- Kuroda, K.; Fukuda, T.; Okumura, K.; Yoneyama, H.; Isogai, H.; Savage, P.B.; Isogai, E. Ceragenin CSA-13 induces cell cycle arrest and antiproliferative effects in wild-type and p53 null mutant HCT116 colon cancer cells. Anticancer Drugs 2013, 24, 826–834. [Google Scholar] [CrossRef]
- Piktel, E.; Prokop, I.; Wnorowska, U.; Król, G.; Cieśluk, M.; Niemirowicz, K.; Savage, P.B.; Bucki, R. Ceragenin CSA-13 as free molecules and attached to magnetic nanoparticle surfaces induce caspase-dependent apoptosis in human breast cancer cells via disruption of cell oxidative balance. Oncotarget 2018, 9, 21904–21920. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Niemirowicz, K.; Prokop, I.; Wilczewska, A.; Wnorowska, U.; Piktel, E.; Wątek, M.; Savage, P.; Bucki, R. Magnetic nanoparticles enhance the anticancer activity of cathelicidin LL-37 peptide against colon cancer cells. Int. J. Nanomed. 2015, 10, 3843–3853. [Google Scholar] [CrossRef] [PubMed]
- Piktel, E.; Markiewicz, K.H.; Wilczewska, A.Z.; Daniluk, T.; Chmielewska, S.; Niemirowicz-Laskowska, K.; Mystkowska, J.; Paprocka, P.; Savage, P.B.; Bucki, R. Quantification of synergistic effects of ceragenin CSA-131 combined with iron oxide magnetic nanoparticles against cancer cells. Int. J. Nanomed. 2020, 15, 4573–4589. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Medvedeva, N.I.; Tolstikov, G.A. Synthesis of a triterpene-spermidine conjugate. Russ. J. Org. Chem. 2012, 48, 1366–1369. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Tolstikov, G.A.; Baikova, I.P.; Zaprutko, L.; Apryshko, G.N. Synthesis and antitumor activity of aminopropoxy derivatives of betulin, erythrodiol, and uvaol. Russ. J. Bioorg. Chem. 2011, 37, 369–379. [Google Scholar] [CrossRef]
- Giniyatullina, G.V.; Smirnova, I.E.; Kazakova, O.B.; Yavorskaya, N.P.; Golubeva, I.S.; Zhukova, O.S.; Pugacheva, R.B.; Apryshko, G.N.; Poroikov, V.V. Synthesis and anticancer activity of aminopropoxytriterpenoids. Med. Chem. Res. 2015, 24, 3423–3436. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Mustafin, A.G.; Babkov, D.A.; Sokolova, E.V.; Spasov, A.A. Evaluation of cytotoxicity and α-glucosidase inhibitory activity of amide and polyamino-derivatives of lupane triterpenoids. Molecules 2020, 25, 4833–4856. [Google Scholar] [CrossRef]
- Giniyatullina, G.V.; Flekhter, O.B.; Tolstikov, G.A. Synthesis of squalamine analogues on the basis of lupane triterpenoids. Mendeleev Commun. 2009, 19, 32–33. [Google Scholar] [CrossRef]
- Giniyatullina, G.V.; Kazakova, O.B. Synthesis of O- and N-aminopropyltriterpenoids based on messagenin. Chem. Nat. Compd. 2018, 54, 913–916. [Google Scholar] [CrossRef]
- Giniyatullina, G.V.; Kazakova, O.B. Synthesis and cytotoxicity of lupane mono- and bis-piperazinylamides. Chem. Nat. Compd. 2021, 57, 698–705. [Google Scholar] [CrossRef]
- Giniyatullina, G.V.; Kazakova, O.B.; Sorokina, I.V.; Zhukova, N.A.; Tolstikova, T.G.; Tolstikov, G.A. Synthesis of aminopropylamino derivatives of betulinic and oleanolic acids. Russ. J. Bioorg. Chem. 2013, 39, 329–337. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Giniyatullina, G.V.; Tolstikov, G.A. Synthesis of A-secomethylenamino- and substituted amidoximotriterpenoids. Russ. J. Bioorg. Chem. 2011, 37, 619–625. [Google Scholar] [CrossRef]
- Borselli, D.; Lieutaud, A.; Thefenne, H.; Garnotel, J.; Pagès, E.M.; Brunel, J.M.; Bolla, J.M. Polyamino-isoprenic derivatives block intrinsic resistance of P. aeruginosa to doxycycline and chloramphenicol in vitro. PLoS ONE 2016, 11, e0154490. [Google Scholar] [CrossRef]
- Berti, L.; Bolla, J.M.; Brunel, J.M.; Casanova, J.P.F.; Lorenzi, V. Use of Polyaminoisoprenyl Derivatives in Antibiotic or Antiseptic. Treatment. Patent WO2012113891Al, 30 August 2012. [Google Scholar]
- Lieutaud, A.; Pieri, C.; Bolla, J.M.; Brunel, J.M. New polyaminoisoprenyl antibiotics enhancers against two multidrug-resistant gram-negative bacteria from Enterobacter and Salmonella species. J. Med. Chem. 2020, 63, 10496–10508. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.; Knorrscheidt, A.; Flemming, F.; Wiemann, J.; Sommerwerk, S.; Pavel, I.Z.; Al-Harrasi, A.; Csuk, R. Synthesis and proapoptotic activity of oleanolic acid derived amides. Bioorg. Chem. 2016, 68, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Giniyatullina, G.V.; Kazakova, O.B.; Salimova, E.V.; Tolstikov, G.A. Synthesis of new betulonic and oleanonic acid amides. Chem. Nat. Compd. 2011, 47, 68–72. [Google Scholar] [CrossRef]
- Emmerich, D.; Vanchanagiri, K.; Baratto, L.C.; Schmidt, H.; Paschke, R. Synthesis and studies of anticancer properties of lupane-type triterpenoid derivatives containing a cisplatin fragment. Eur. J. Med. Chem. 2014, 2014, 460–466. [Google Scholar] [CrossRef]
- Csuk, R.; Schwarz, S.; Kluge, R.; Ströhl, D. Synthesis and biological activity of some antitumor active derivatives from glycyrrhetinic acid. Eur. J. Med. Chem. 2010, 45, 5718–5723. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, O.B.; Giniyatullina, G.V.; Medvedeva, N.I.; Tolstikov, G.A.; Apryshko, G.N. Synthesis and cytotoxicity of triterpene seven-membered cyclic amines. Russ. J. Bioorg. Chem. 2014, 40, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, N.I.; Kazakova, O.B.; Lopatina, T.V.; Smirnova, I.E.; Giniyatullina, G.V.; Baikova, I.P.; Kataev, V.E. Synthesis and antimycobacterial activity of triterpenic A-ring azepanes. Eur. J. Med. Chem. 2018, 143, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Lia, T.; Fana, P.; Ye, Y.; Luo, Q.; Lou, H. Ring A-modified derivatives from the natural triterpene 3-O-acetyl-11-keto-β-boswellic acid and their cytotoxic activity. Anti-Cancer Agents Med. Chem. 2017, 17, 1153–1167. [Google Scholar] [CrossRef]
- Antimonova, A.N.; Uzenkova, N.V.; Petrenko, N.I.; Shakirov, M.M.; Shul’ts, E.E.; Tolstikov, G.A. Synthesis of betulonic acid amides. Chem. Nat. Compd. 2008, 44, 327–333. [Google Scholar] [CrossRef]
- Bai, K.K.; Yu, Z.; Chen, F.L.; Li, F.; Li, W.Y.; Guo, Y.H. Synthesis and evaluation of ursolic acid derivatives as potent cytotoxic agents. Bioorg. Med. Chem. Lett. 2012, 22, 2488–2493. [Google Scholar] [CrossRef]
- Ma, C.M.; Cai, S.Q.; Cui, J.R.; Wang, R.Q.; Tu, P.F.; Hattori, M.; Daneshtalab, M. The cytotoxic activity of ursolic acid derivatives. Eur. J. Med. Chem. 2005, 40, 582–589. [Google Scholar] [CrossRef]
- Schwarz, S.; Lucas, S.D.; Sommerwerk, S.; Csuk, R. Amino derivatives of glycyrrhetinic acid as potential inhibitors of cholinesterases. Bioorg. Med. Chem. 2014, 22, 3370–3378. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y.; Wang, C.M.; Cheng, H.L.; Chen, H.J.; Hsu, Y.M.; Lin, Y.C.; Chou, C.-H. Biological activity of oleanane triterpene derivatives obtained by chemical derivatization. Molecules 2013, 18, 13003–13019. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Hao, Y.; Zhang, G.; Wang, S.F.; Miao, T.T.; Zhang, K.P. Synthesis, in vitro antimicrobial and cytotoxic activities of new carbazole derivatives of ursolic acid. Bioorg. Med. Chem. Lett. 2015, 25, 554–557. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, L.; Li, L.; Liu, J.; Li, H.; Zhang, L.; Chen, L.; Cheng, K.; Zheng, M.; Wen, X.; et al. Identification of pentacyclic triterpenes derivatives as potent inhibitors against glycogen phosphorylase based on 3D-QSAR studies. Eur. J. Med. Chem. 2011, 46, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Wang, J.; Zhang, D.M.; Shu, C.; Cao, H.H.; Sun, P.H.; Wu, X.M.; Ye, W.C.; Chen, W.M. Synthesis and antiproliferative evaluation of 23-hydroxybetulinic acid derivatives. Eur. J. Med. Chem. 2011, 46, 2490–2502. [Google Scholar] [CrossRef]
- Tian, T.; Liu, X.; Lee, E.S.; Sun, J.; Feng, Z.; Zhao, L.; Zhao, C. Synthesis of novel oleanolic acid and ursolic acid in C-28 position derivatives as potential anticancer agents. Arch. Pharm. Res. 2017, 40, 458–468. [Google Scholar] [CrossRef]
- Urano, E.; Ablan, S.D.; Mandt, R.; Pauly, G.T.; Sigano, D.M.; Schneider, J.P.; Martin, D.E.; Nitz, T.J.; Wild, C.T.; Freed, E.O. Alkyl amine bevirimat derivatives are potent and broadly active HIV-1 maturation inhibitors. Antimicrob. Agents Chemother. 2016, 60, 190–197. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Leal, A.S.; Valdeira, A.S.; Gonçalves, B.M.F.; Alho, D.P.S.; Figueiredo, S.A.C.; Silvestre, S.M.; Mendes, V.I.S. Oleanane-, ursane-, and quinone methide friedelane-type triterpenoid derivatives: Recent advances in cancer treatment. Eur. J. Med. Chem. 2017, 142, 95–130. [Google Scholar] [CrossRef]
- Luo, S.; Li, P.; Li, S.; Du, Z.; Hu, X.; Fu, Y.; Zhang, Z. N, N-Dimethyl tertiary amino group mediated dual pancreas- and lung-targeting therapy against acute pancreatitis. Mol. Pharm. 2017, 14, 1771–1781. [Google Scholar] [CrossRef]
- Rodŕigues-Hernández, D.; Demuner, A.J.; Barbosa, L.C.A.; Csuk, R.; Heller, L. Hederagenin as a triterpene template for the development of new antitumor compounds. Eur. J. Med. Chem. 2015, 105, 57–62. [Google Scholar] [CrossRef]
- Ortiz, A.; Soumeillant, M.; Savage, S.A.; Strotman, N.A.; Haley, M.; Benkovics, T.; Nye, J.; Xu, Z.; Tan, Y.; Ayers, S.; et al. Synthesis of HIV-maturation inhibitor BMS-955176 from betulin by an enabling oxidation strategy. J. Org. Chem. 2017, 82, 4958–4963. [Google Scholar] [CrossRef]
- Spivak, A.; Khalitova, R.; Nedopekina, D.; Dzhemileva, L.; Yunusbaeva, M.; Odinokov, V.; D’yakonov, V.; Dzhemilev, U. Synthesis and evaluation of anticancer activities of novel C-28 guanidine-functionalized triterpene acid derivatives. Molecules 2018, 23, 3000. [Google Scholar] [CrossRef] [PubMed]
- Kahnt, M.; Hoenke, S.; Fischer, L.; Al-Harrasi, A.; Csuk, R. Synthesis and cytotoxicity evaluation of DOTA-conjugates of ursolic acid. Molecules 2019, 24, 2254–2273. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Gao, Y.; Zhan, M.; Qiu, N.; Piao, Y.; Zhou, Z.; Shen, Y. Glycyrrhizin acid and glycyrrhetinic acid modified polyethyleneimine for targeted DNA delivery to hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 5074–5098. [Google Scholar] [CrossRef]
- Bildziukevich, U.; Malík, M.; Özdemir, Z.; Rárová, L.; Janovská, L.; Šlouf, M.; Šaman, D.; Šarek, J.; Wimmer, Z. Spermine amides of selected triterpenoid acids: Dynamic supramolecular systems formation influences cytotoxicity of the drugs. J. Mater. Chem. B 2020, 8, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.Y.; Khalitova, R.R.; Nedopekina, D.A.; Gubaidullin, R.R. Antimicrobial properties of amine- and guanidine-functionalized derivatives of betulinic, ursolic and oleanolic acids: Synthesis and structure/activity evaluation. Steroids 2020, 154, 108530–108542. [Google Scholar] [CrossRef]
- Bildziukevich, U.; Vida, N.; Rárová, L.; Kolár, M.; Šaman, D.; Havlíček, L.; Drašar, P.; Wimmer, Z. Polyamine derivatives of betulinic acid and β-sitosterol: A comparative investigation. Steroids 2015, 100, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Bildziukevich, U.; Kaletová, E.; Šaman, D.; Sievänen, E.; Kolehmainen, E.T.; Šlouf, M.; Wimmer, Z. Spectral and microscopic study of self-assembly of novel cationic spermine amides of betulinic acid. Steroids 2017, 117, 90–96. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Brunel, J.M.; Khusnutdinova, E.F.; Negrel, S.; Giniyatullina, G.V.; Lopatina, T.V.; Petrova, A.V. A-ring modified triterpenoids and their spermidine-aldimines with strong antibacterial activity. Molbank 2019, 2019, M1078–M1086. [Google Scholar] [CrossRef]
- Qian, K.; Kuo, R.; Chen, C.; Huang, L.; Morris-Natschke, S.L.; Lee, K. Anti-AIDS agents 81. Design, synthesis, and structure-activity relationship study of betulinic acid and moronic acid derivatives as potent HIV maturation inhibitors. J. Med. Chem. 2010, 53, 3133–3141. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, D.; Barbosa, L.C.A.; Demuner, A.J.; Martins, J.P.A.; Fischer (nee Heller), L.; Csuk, R. Hederagenin amide derivatives as potential antiproliferative agents. Eur. J. Med. Chem. 2019, 168, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, Z.; Saman, D.; Bertula, K.; Lahtinen, M.; Bednarova, L.; Pazderkova, M.; Rarova, L.; Nonappa; Wimmer, Z. Rapid self-healing and thixotropic organogelation of amphiphilic oleanolic acid−spermine conjugates. Langmuir 2021, 37, 2693–2706. [Google Scholar] [CrossRef] [PubMed]
- Kazakova, O.; Rubanik, L.; Smirnova, I.; Poleschuk, N.; Petrova, A.; Kapustsina, Y.; Baikova, I.; Tret’yakova, E.; Khusnutdinova, E. Synthesis and in vitro activity of oleanolic acid derivatives against Chlamydia trachomatis and Staphylococcus aureus. Med. Chem. Res. 2021, 30, 1408–1418. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Sinou, V.; Babkov, D.A.; Kazakova, O.B.; Brunel, J.M. Development of new antimicrobial oleanonic acid polyamine conjugates. Antibiotics 2022, 11, 94. [Google Scholar] [CrossRef]
























































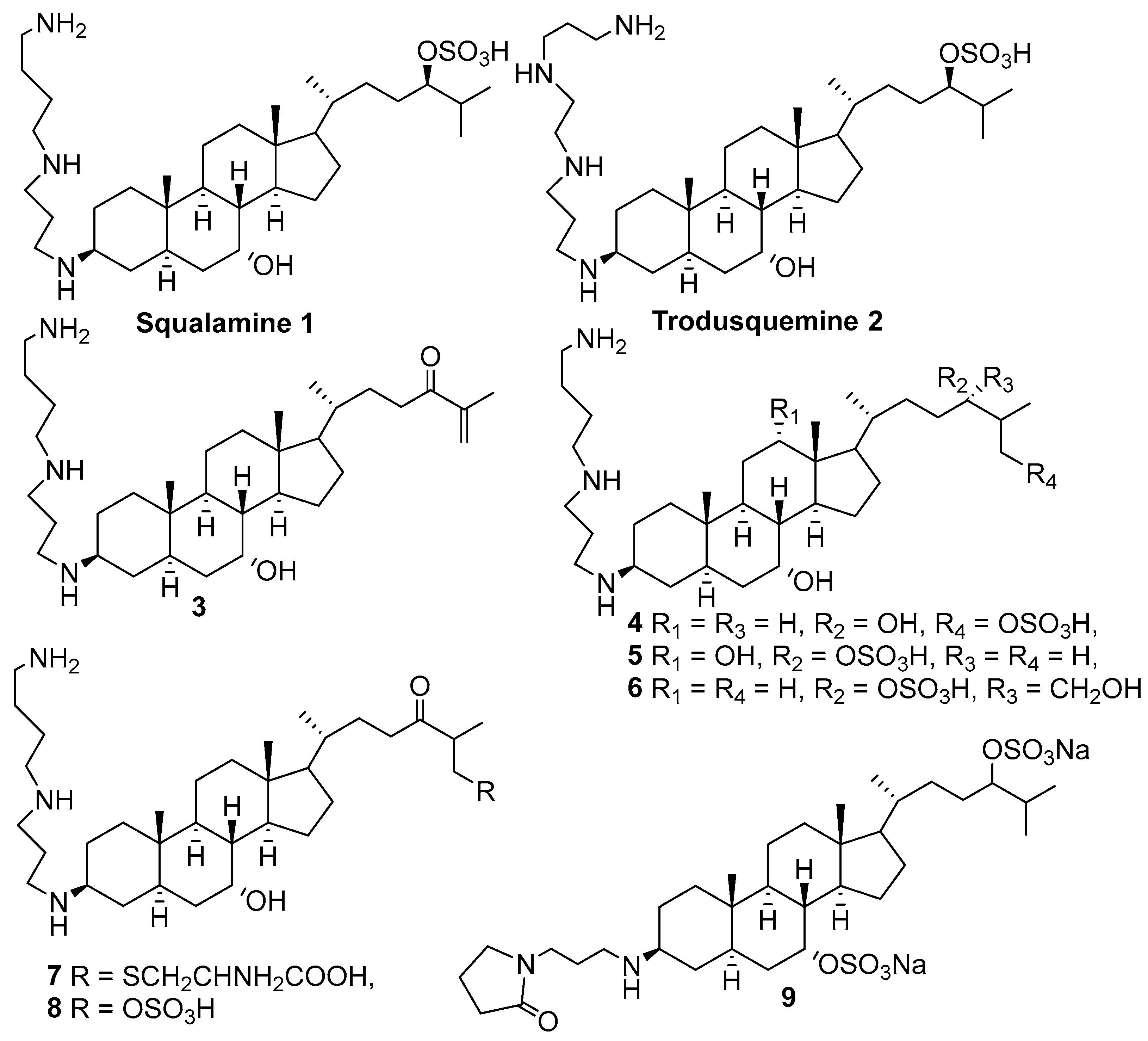{kind=link}
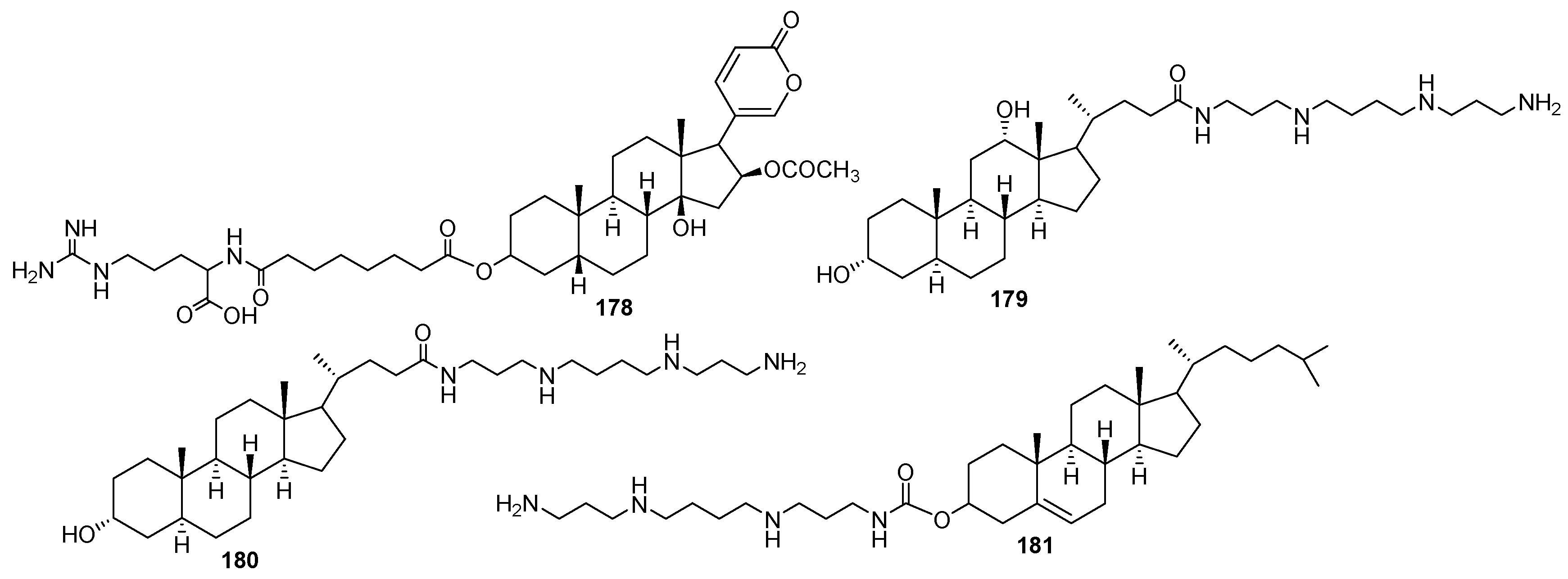{kind=link}
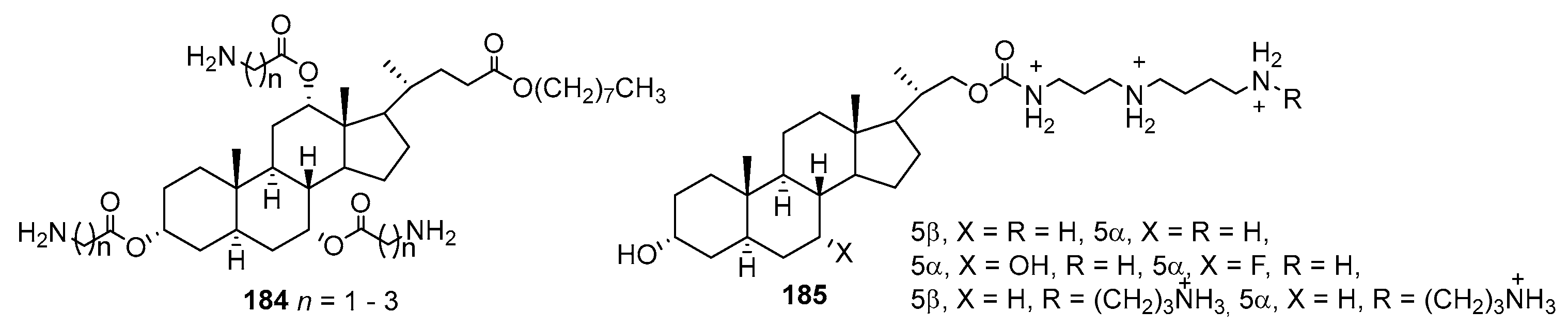{kind=link}
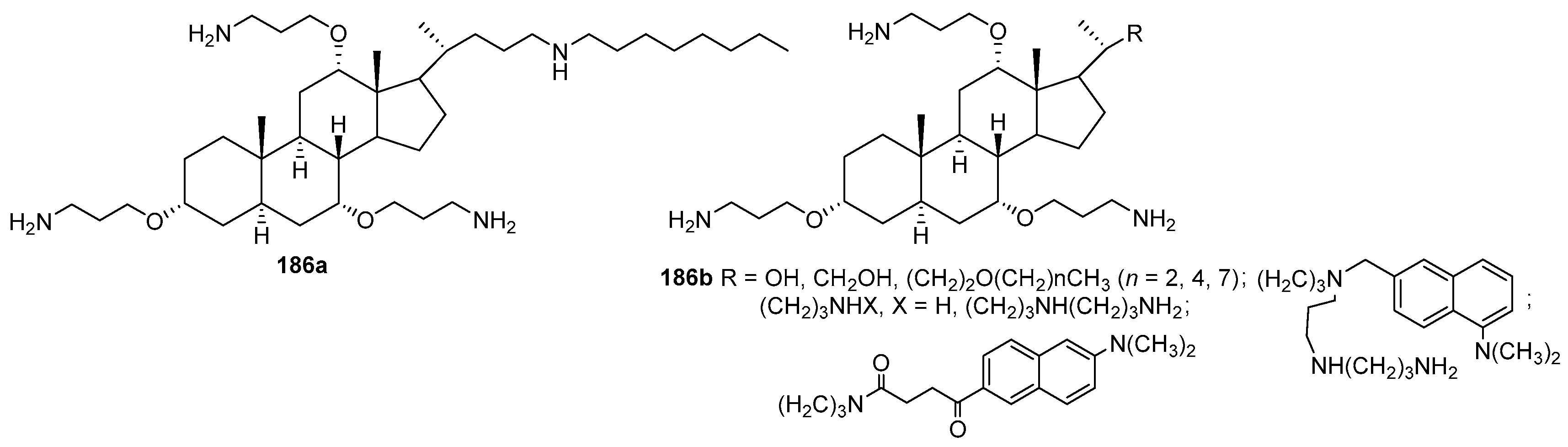{kind=link}
{kind=link}
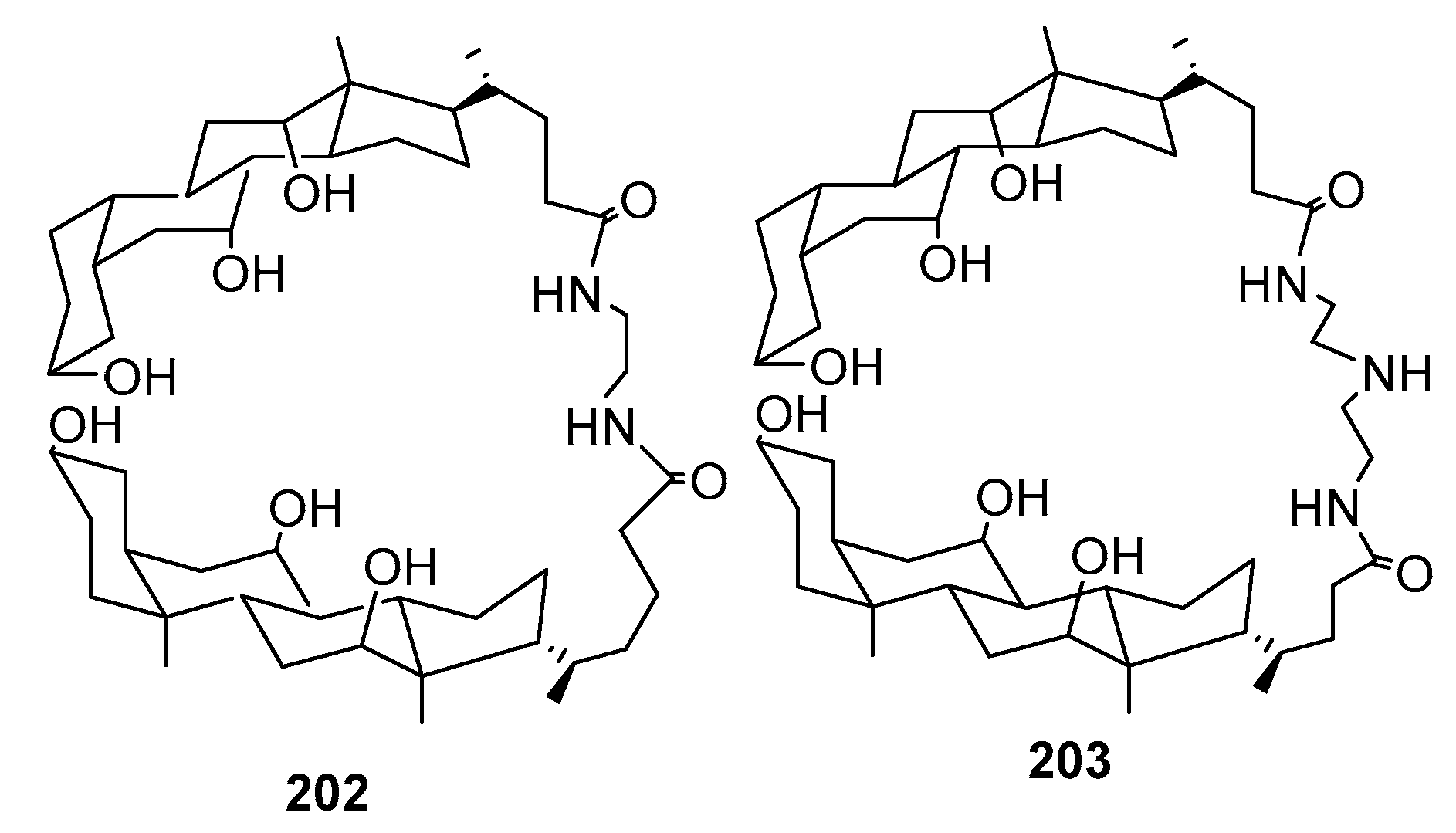{kind=link}
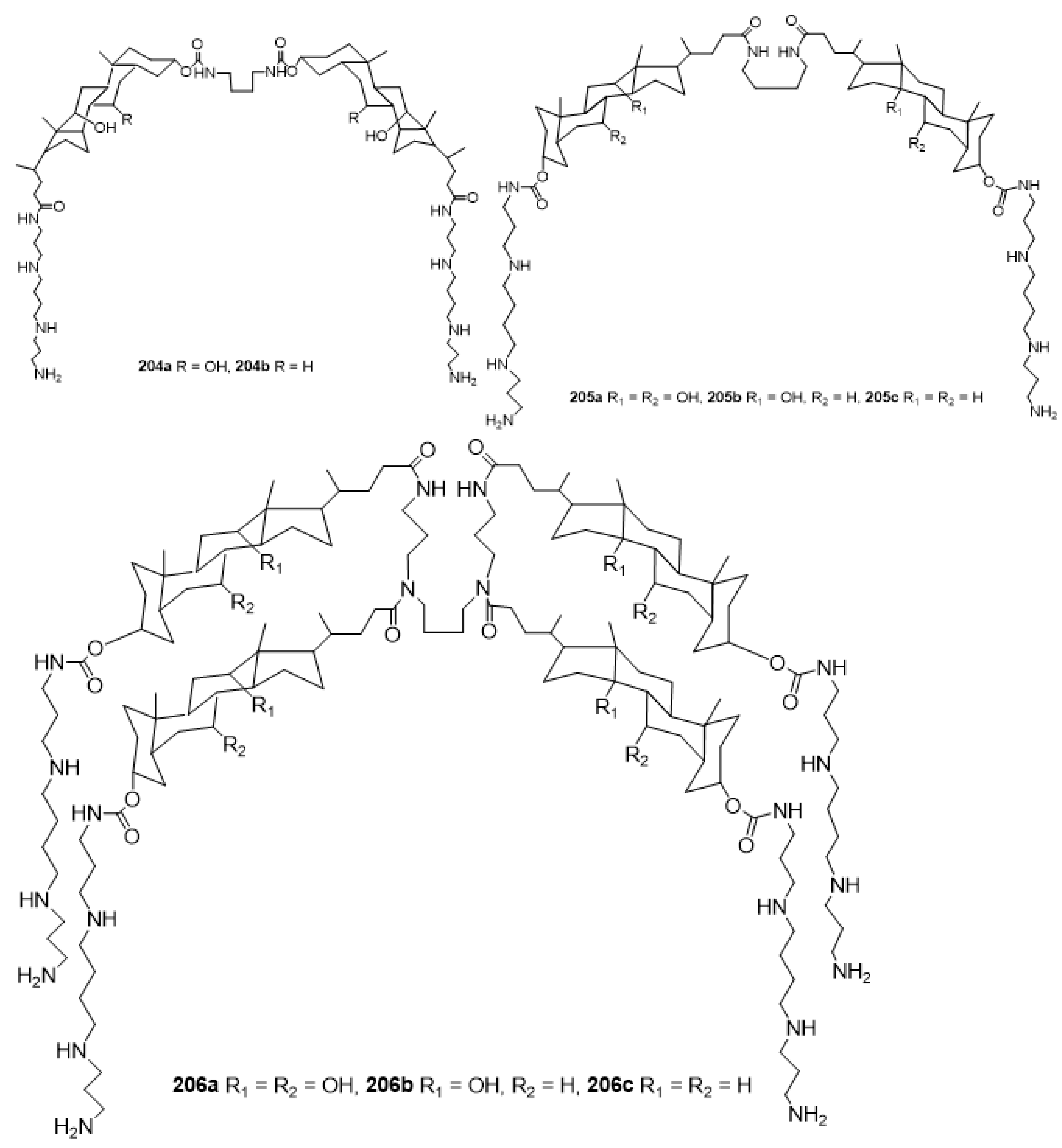{kind=link}
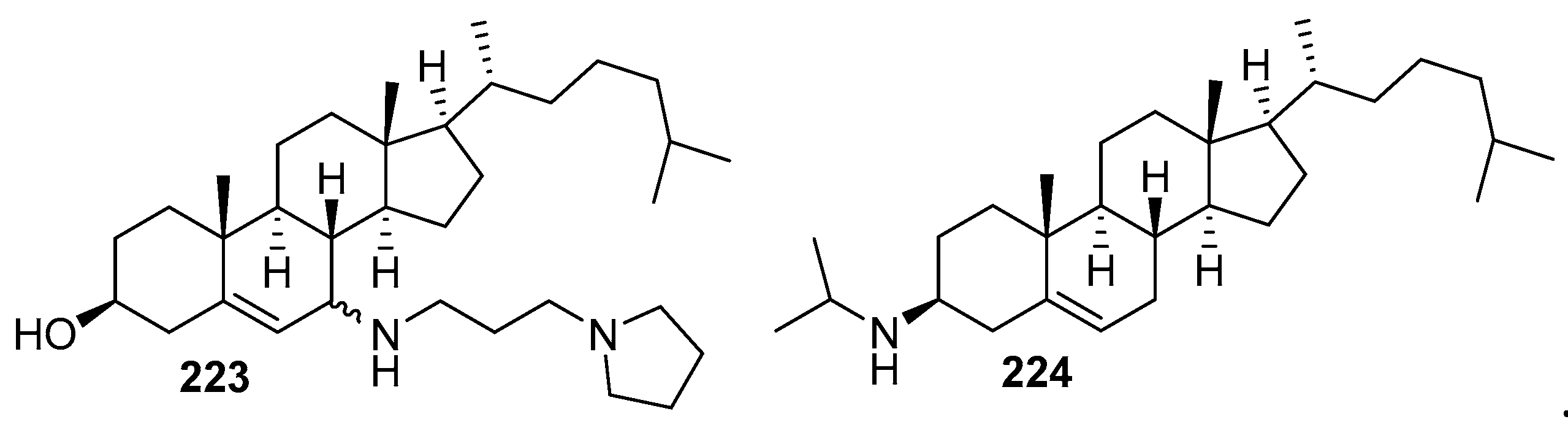{kind=link}
{kind=link}
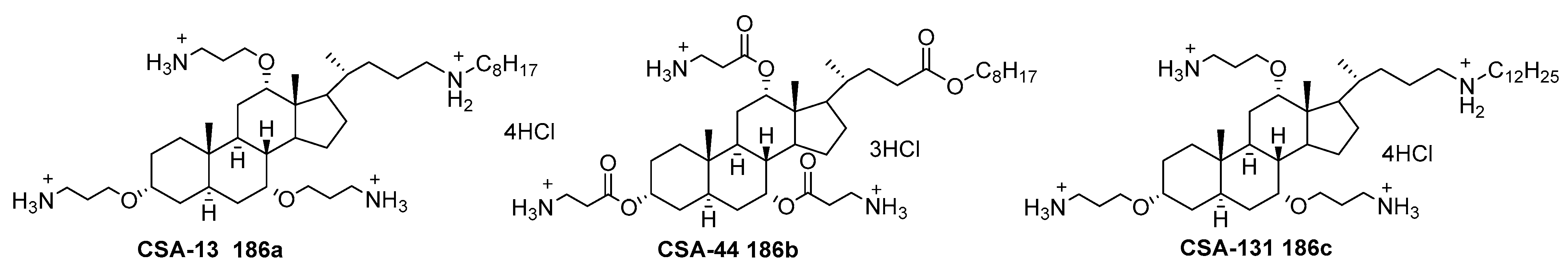{kind=link}
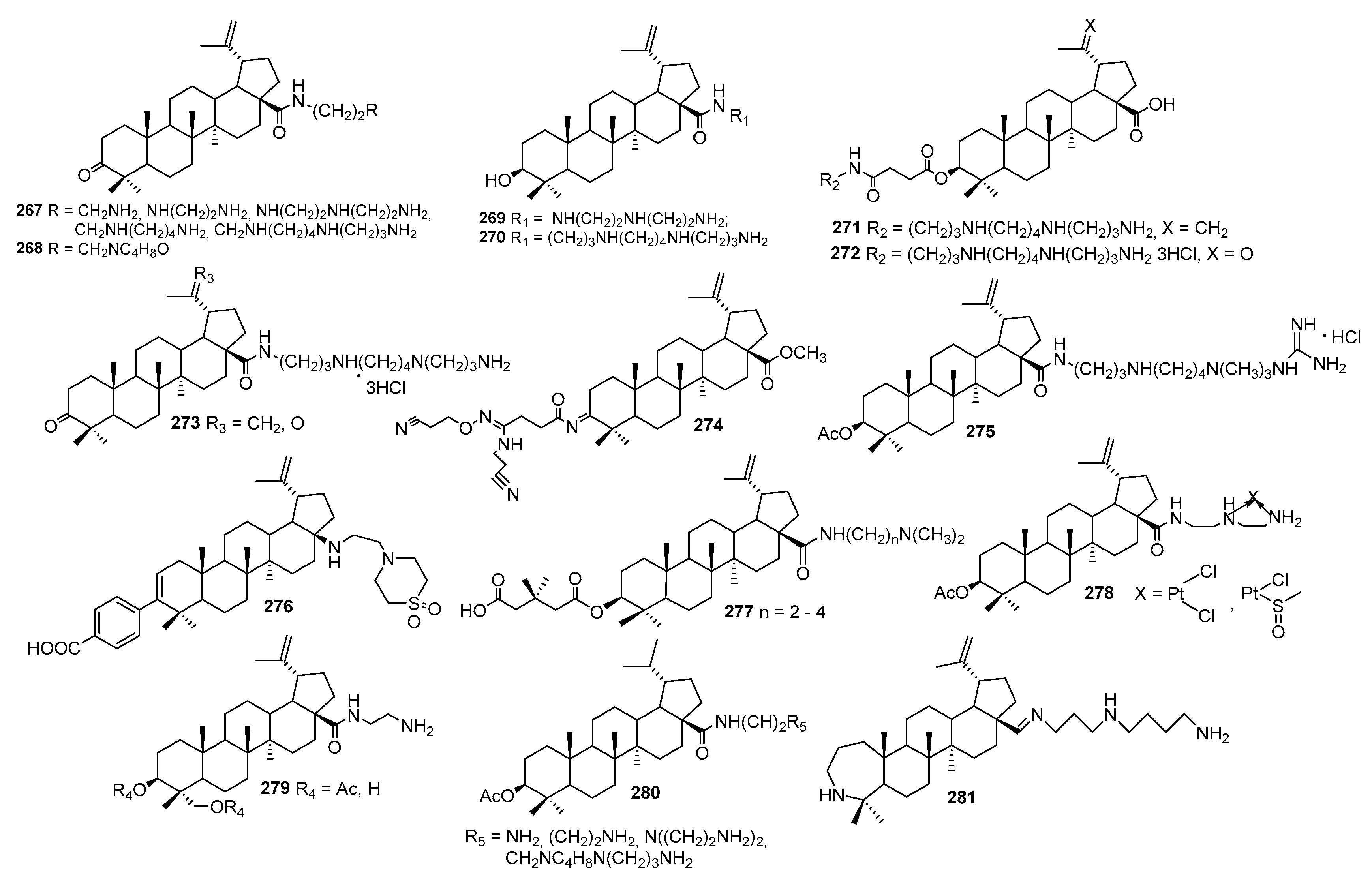{kind=link}
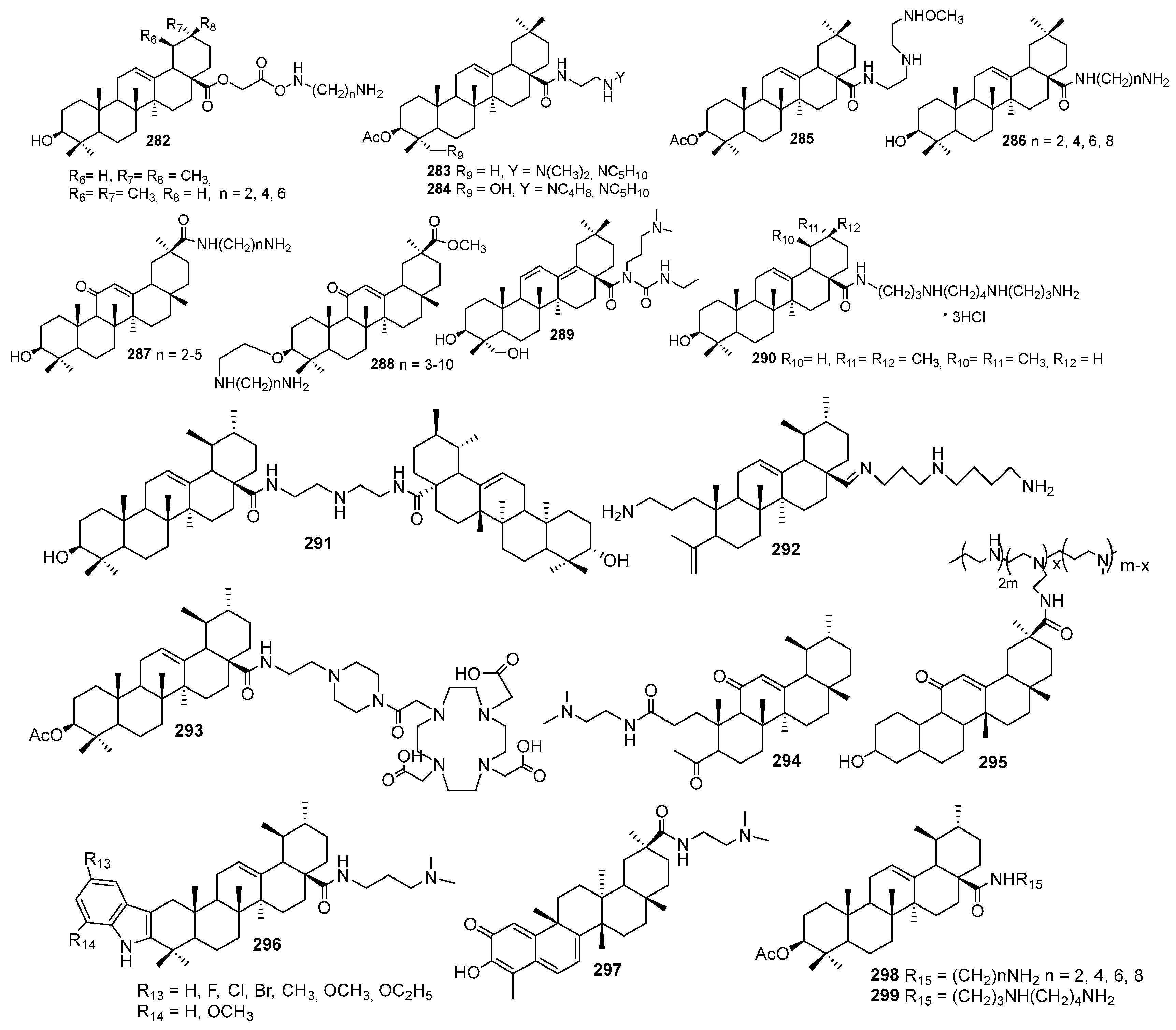{kind=link}
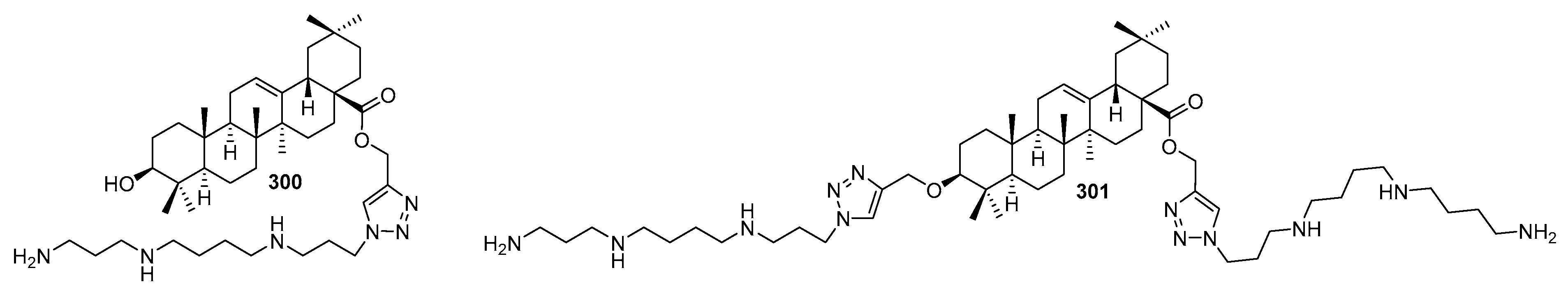{kind=link}
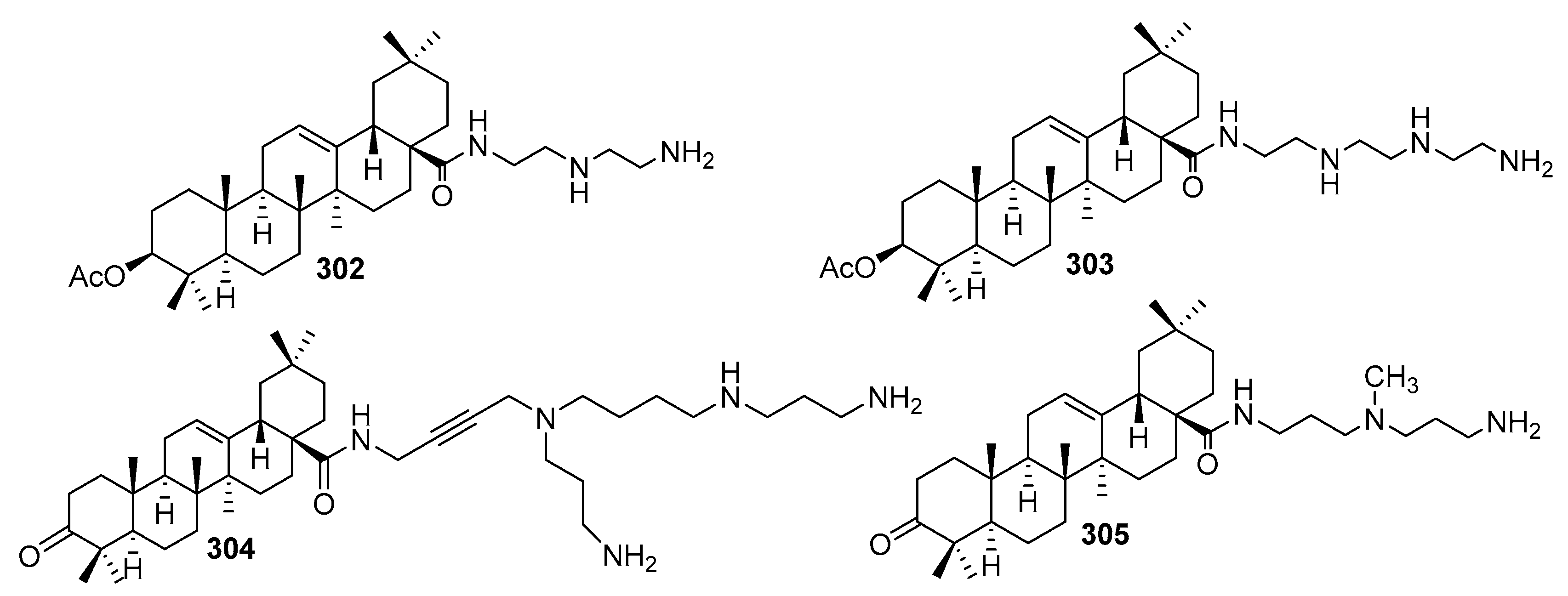{kind=link}
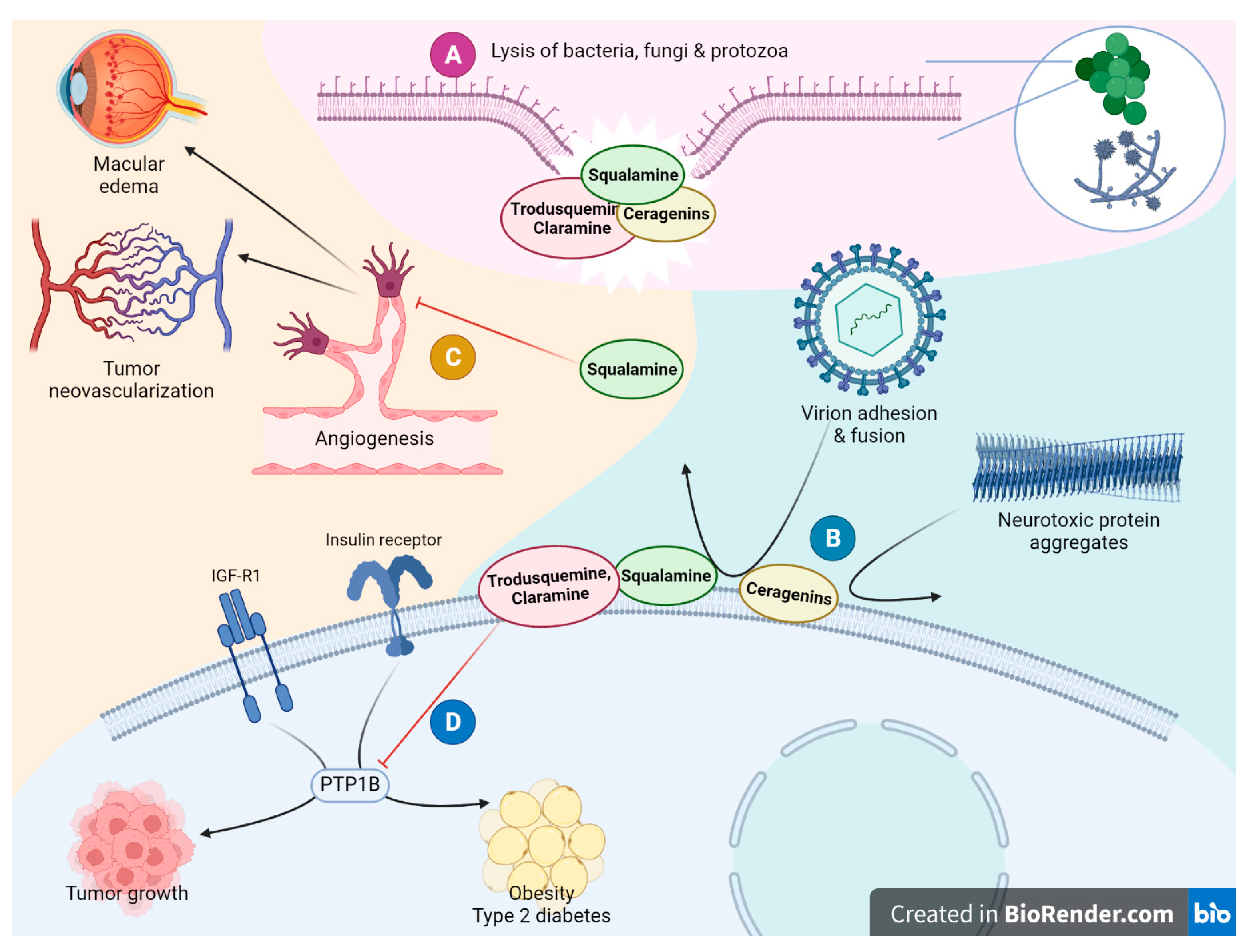{kind=link}
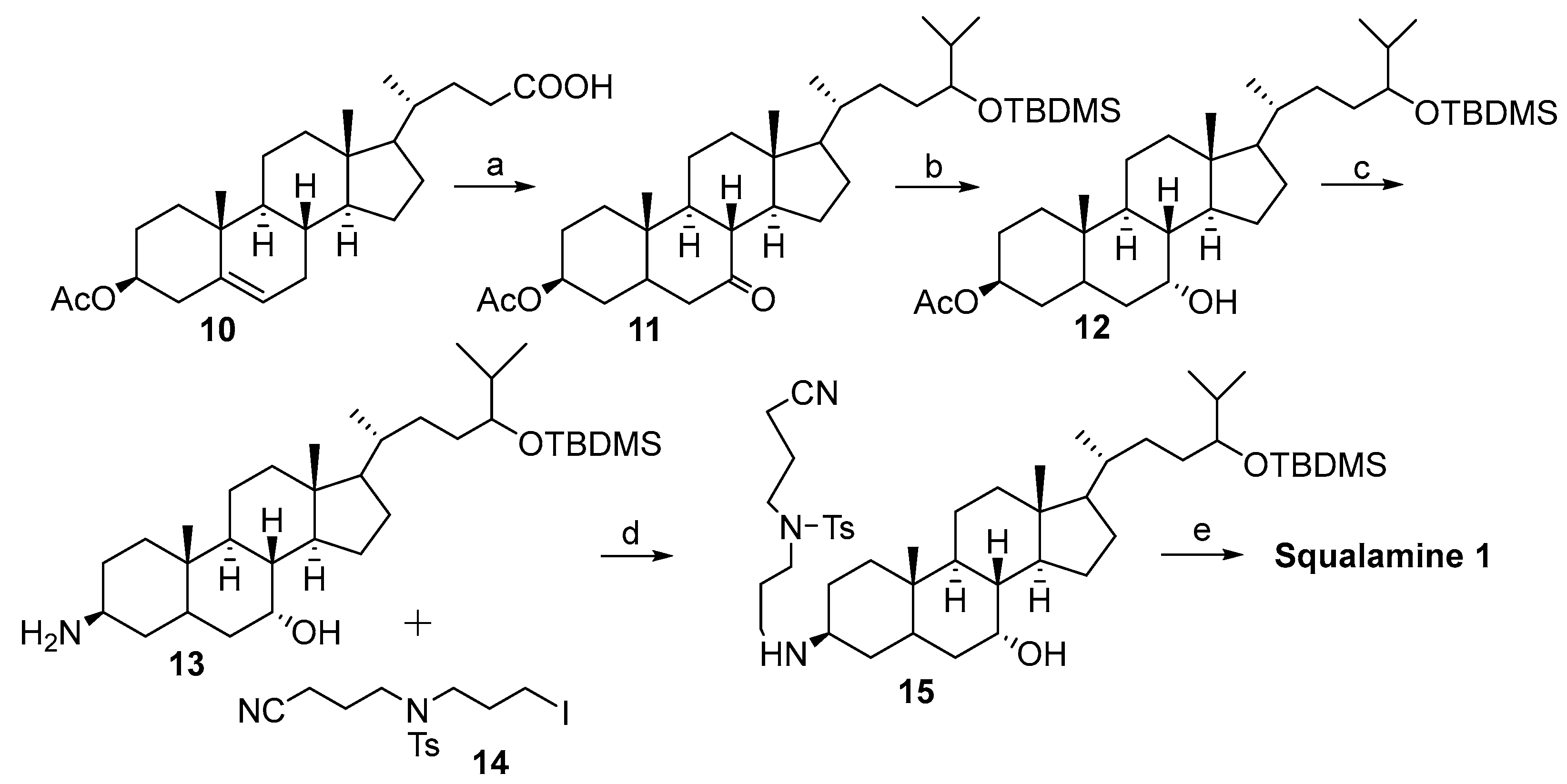{kind=link}
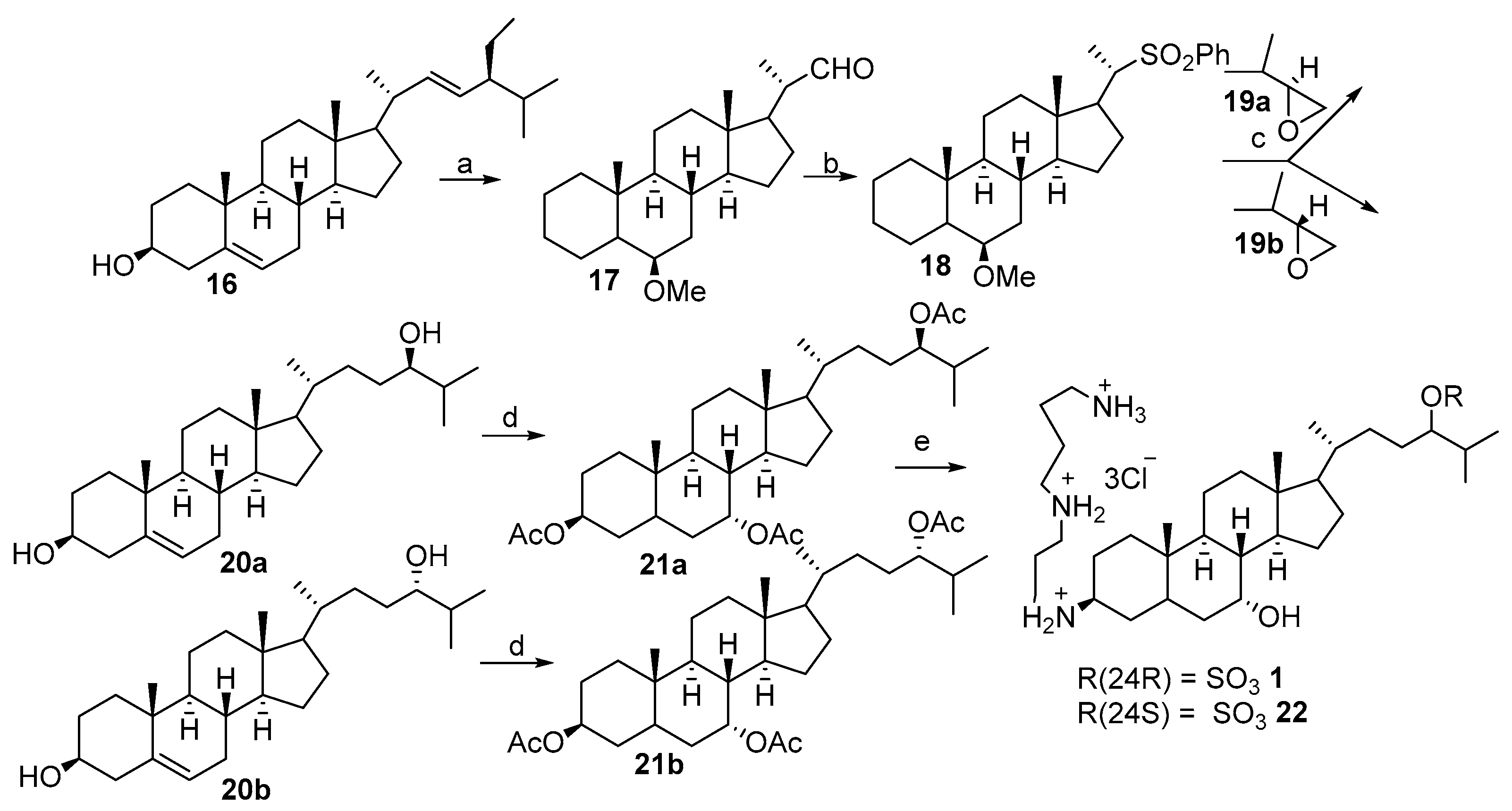{kind=link}
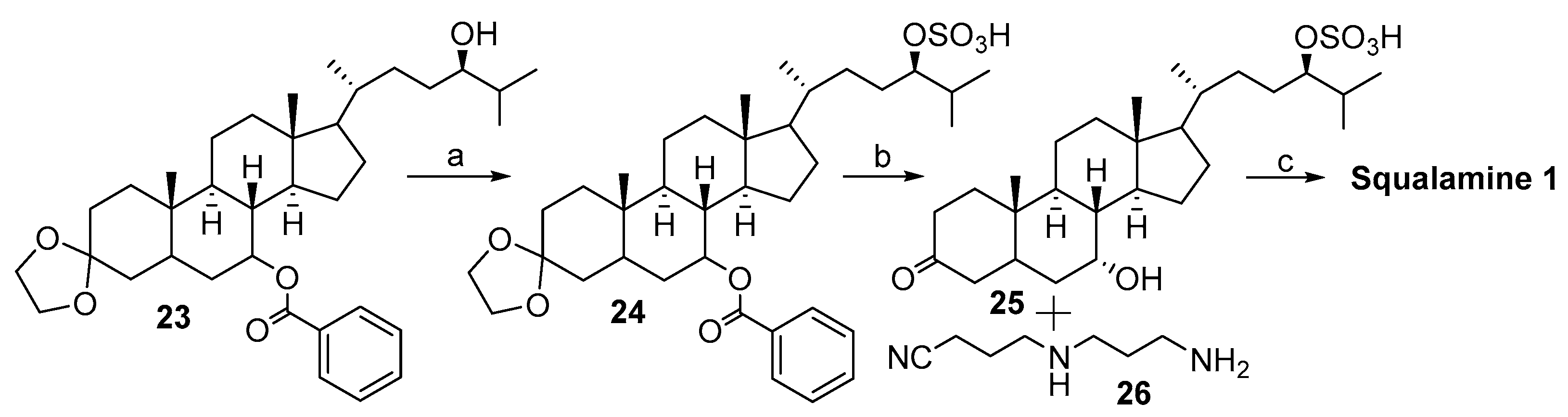{kind=link}
{kind=link}
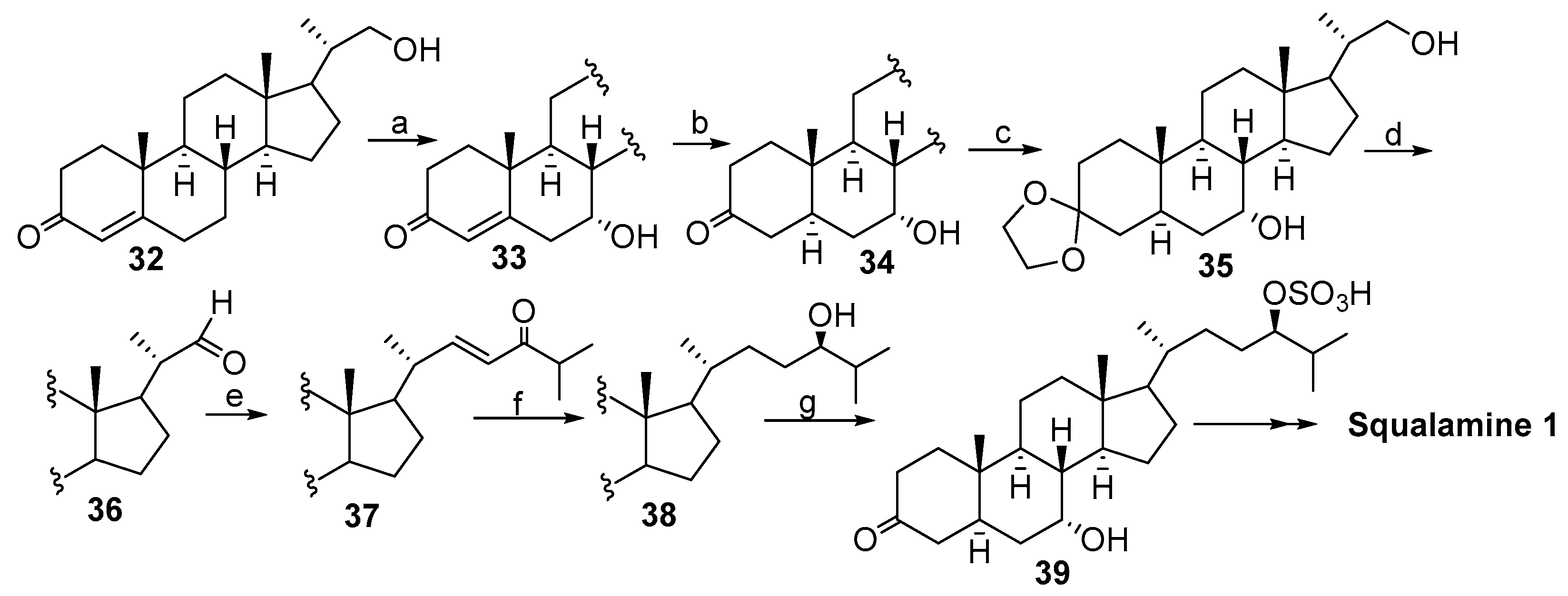{kind=link}
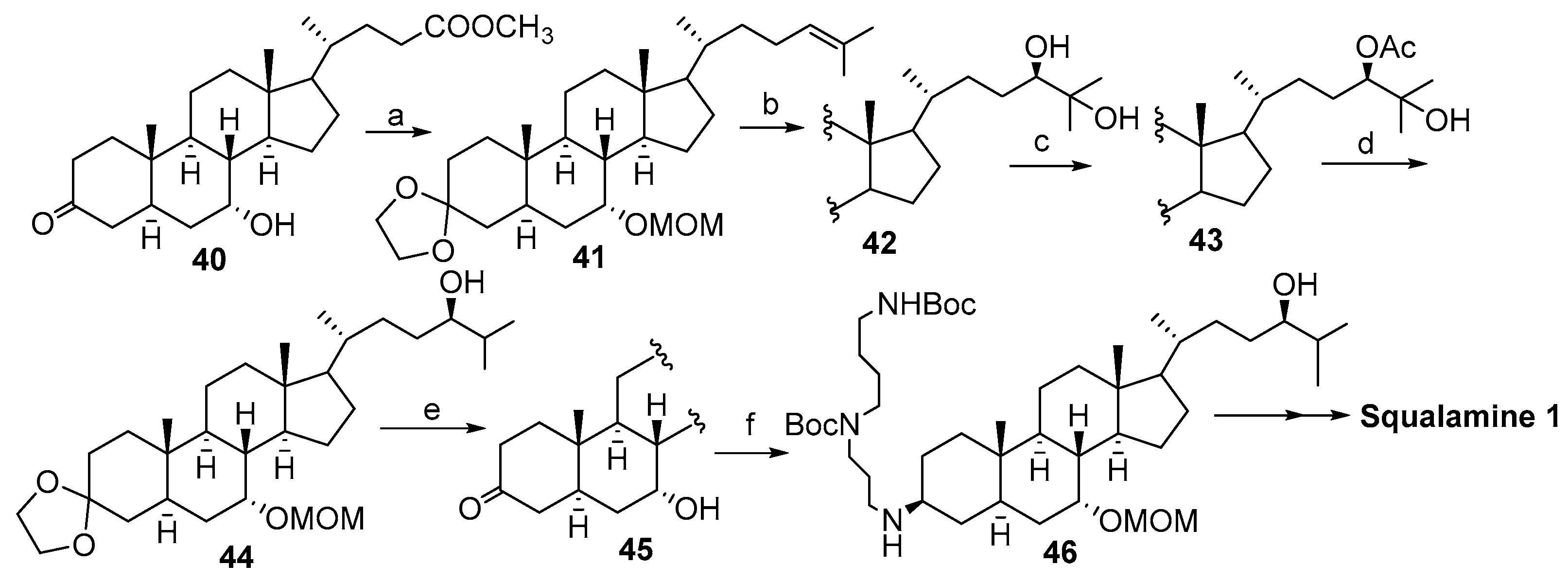{kind=link}
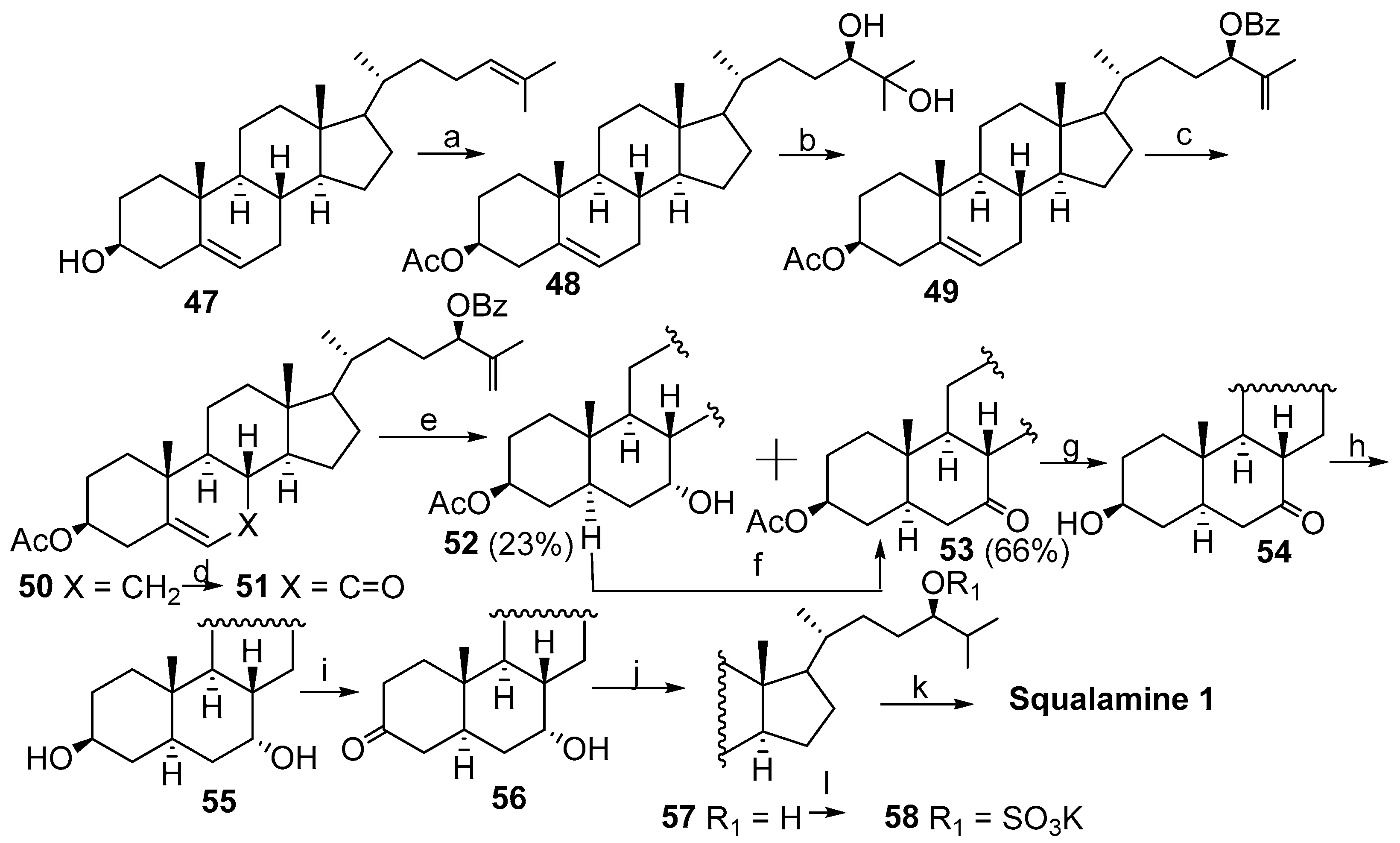{kind=link}
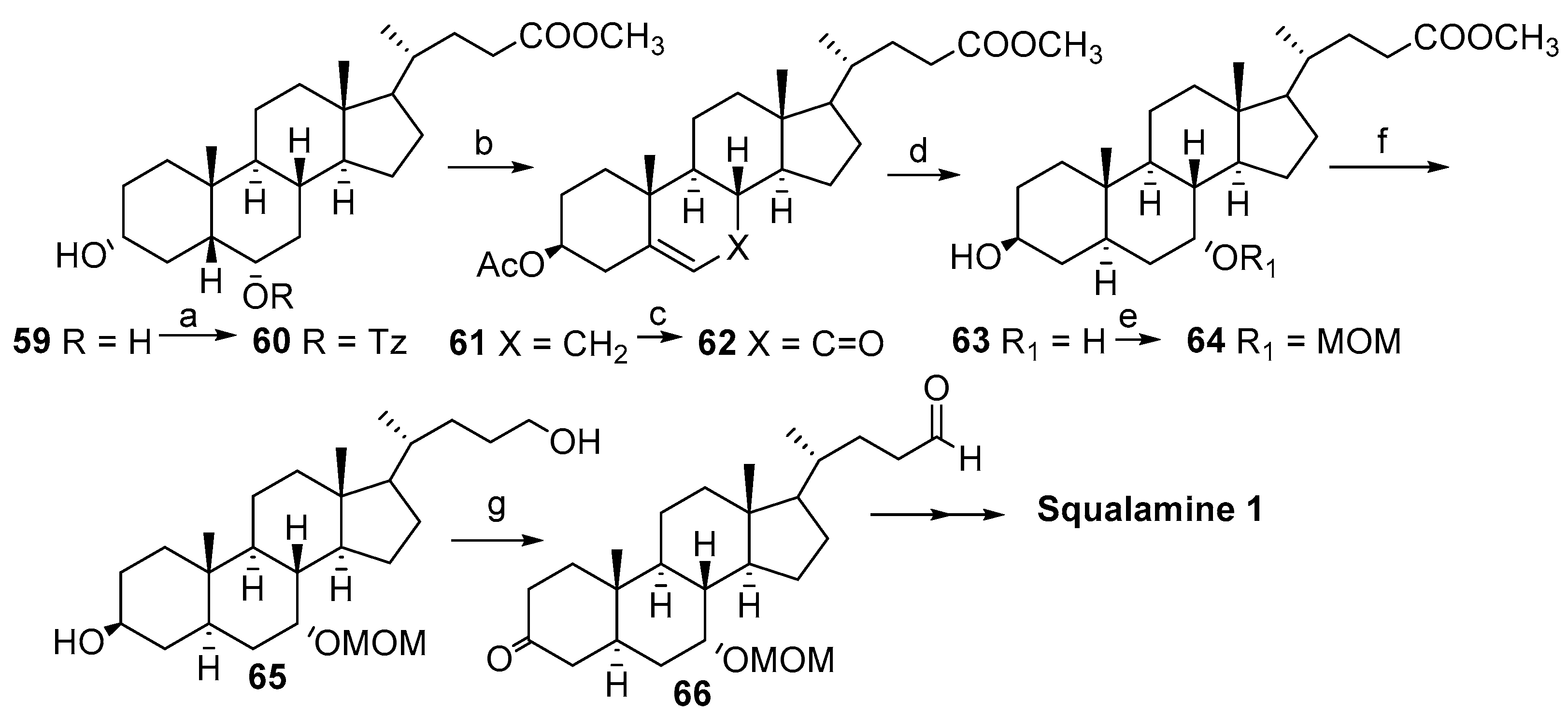{kind=link}
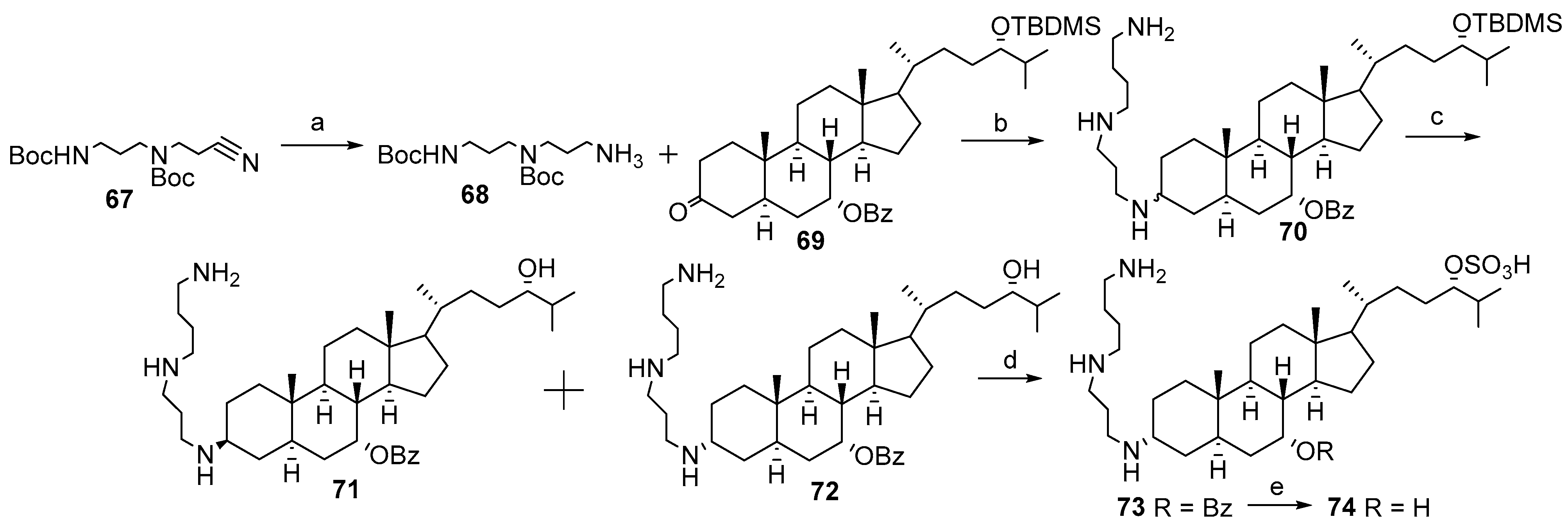{kind=link}
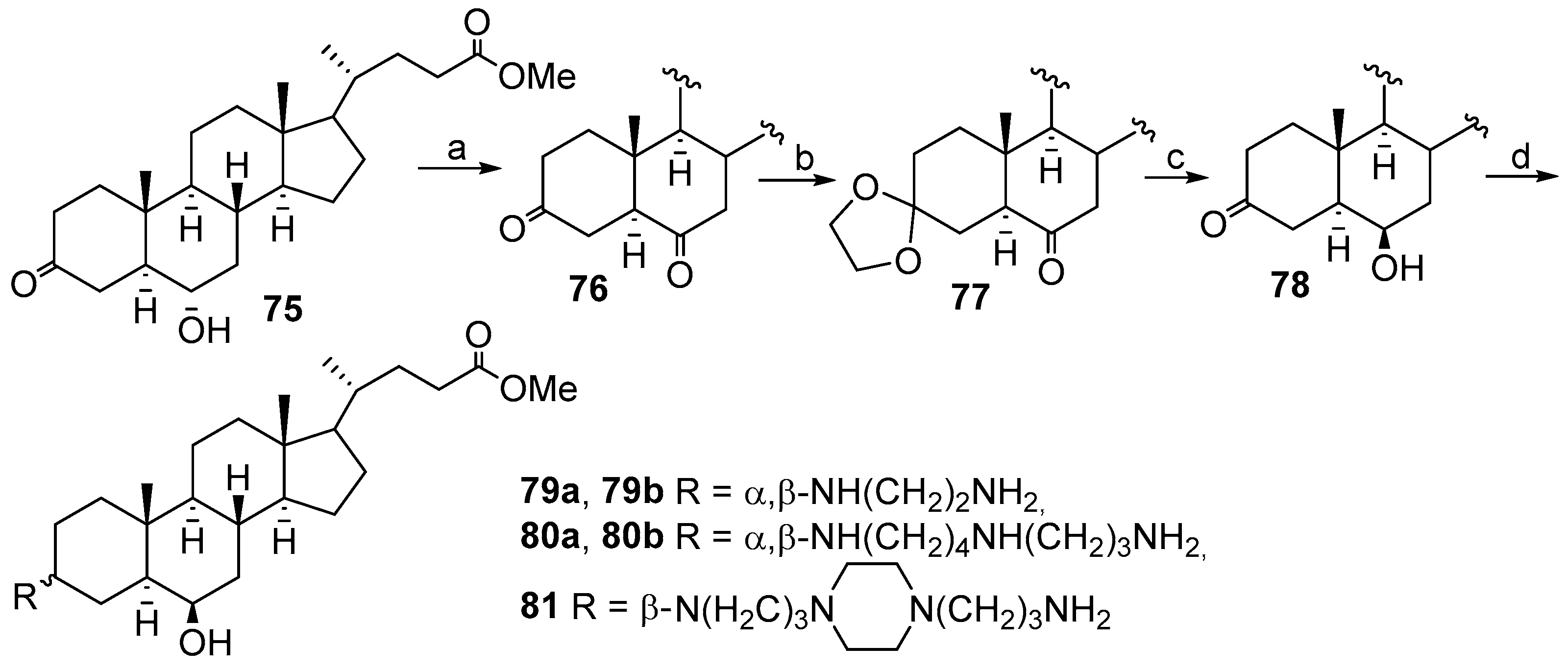{kind=link}
{kind=link}
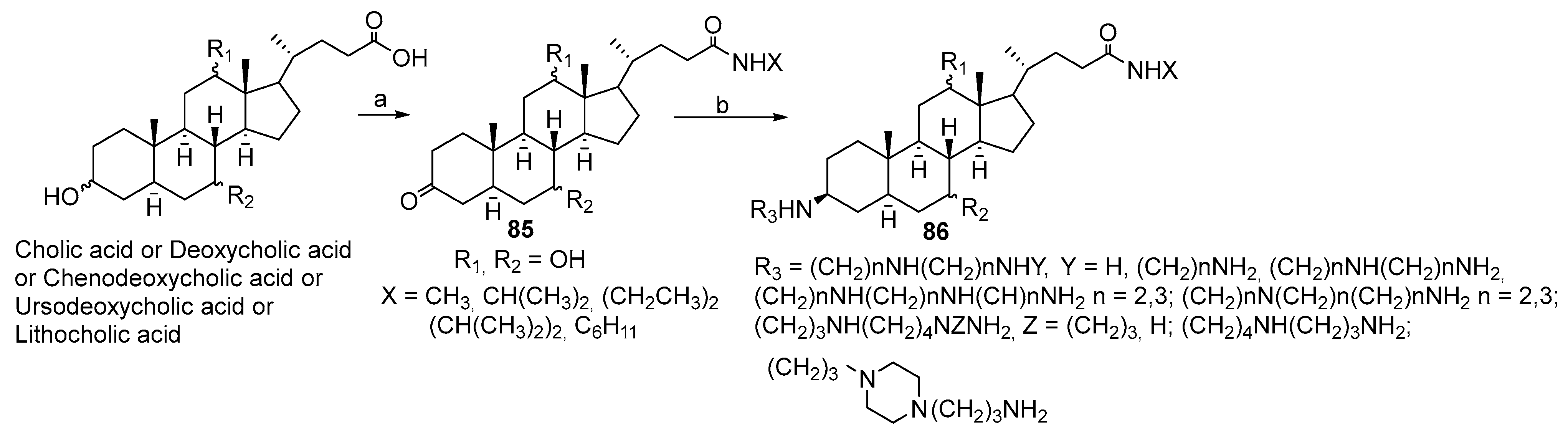{kind=link}
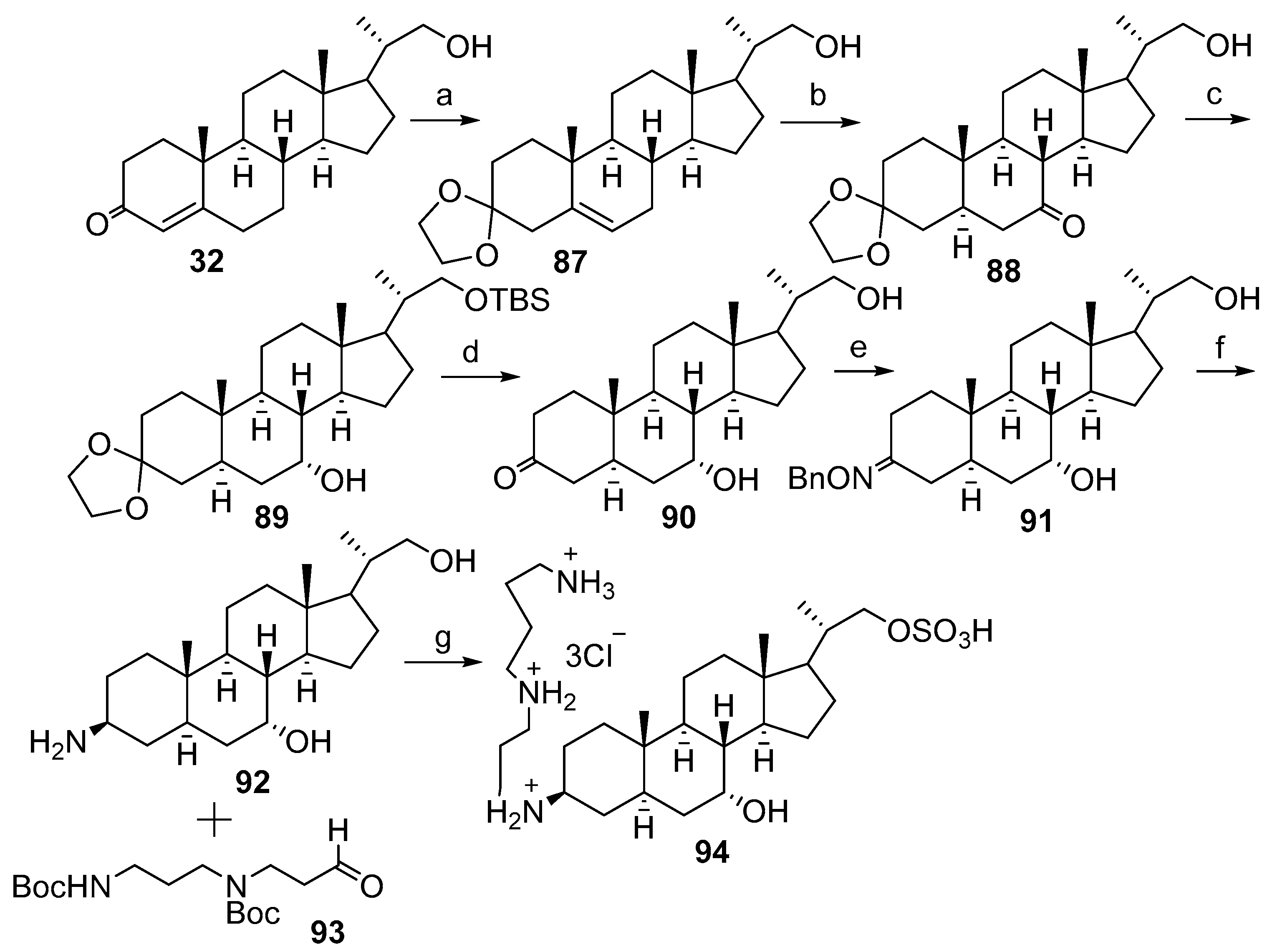{kind=link}
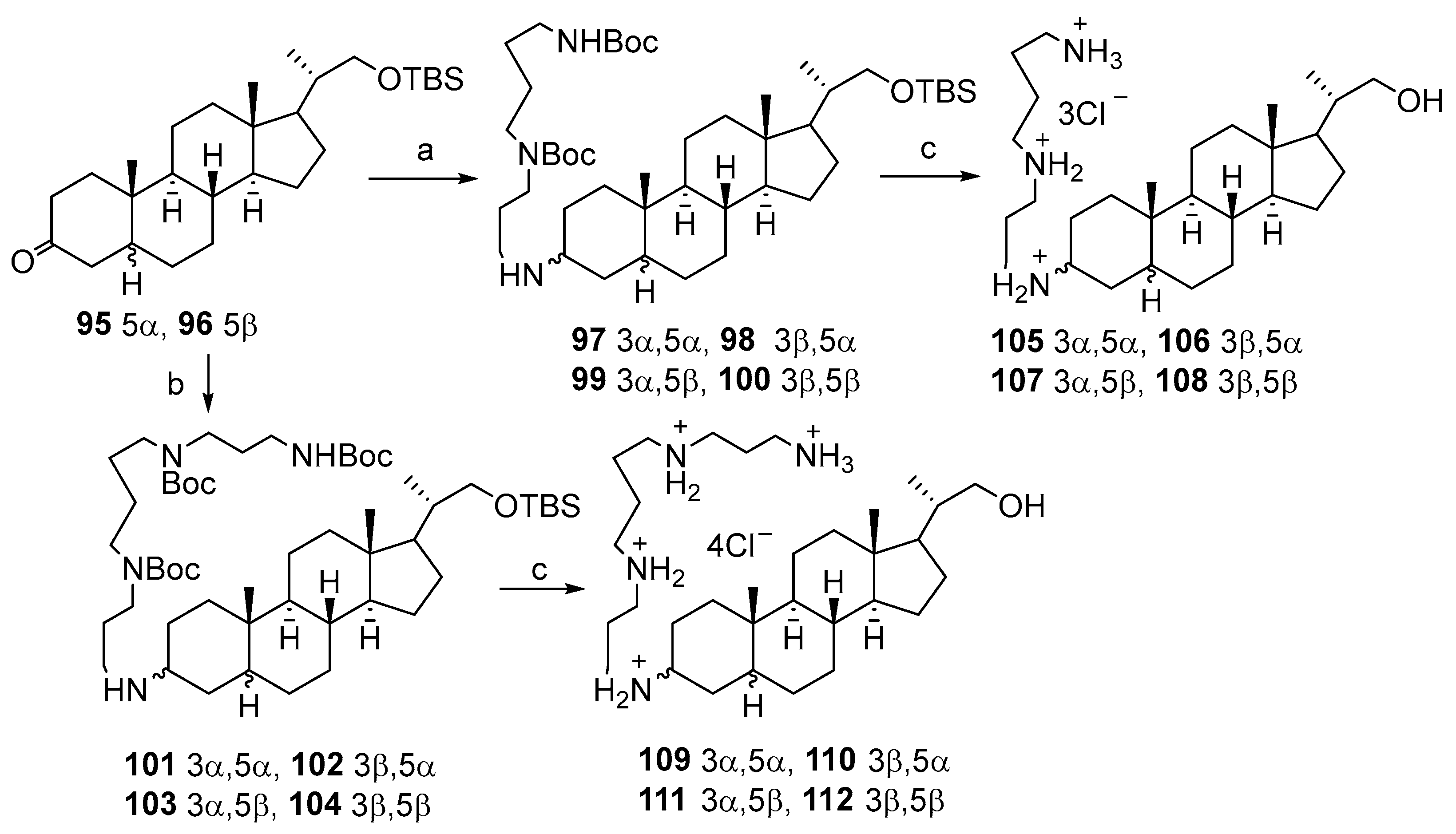{kind=link}
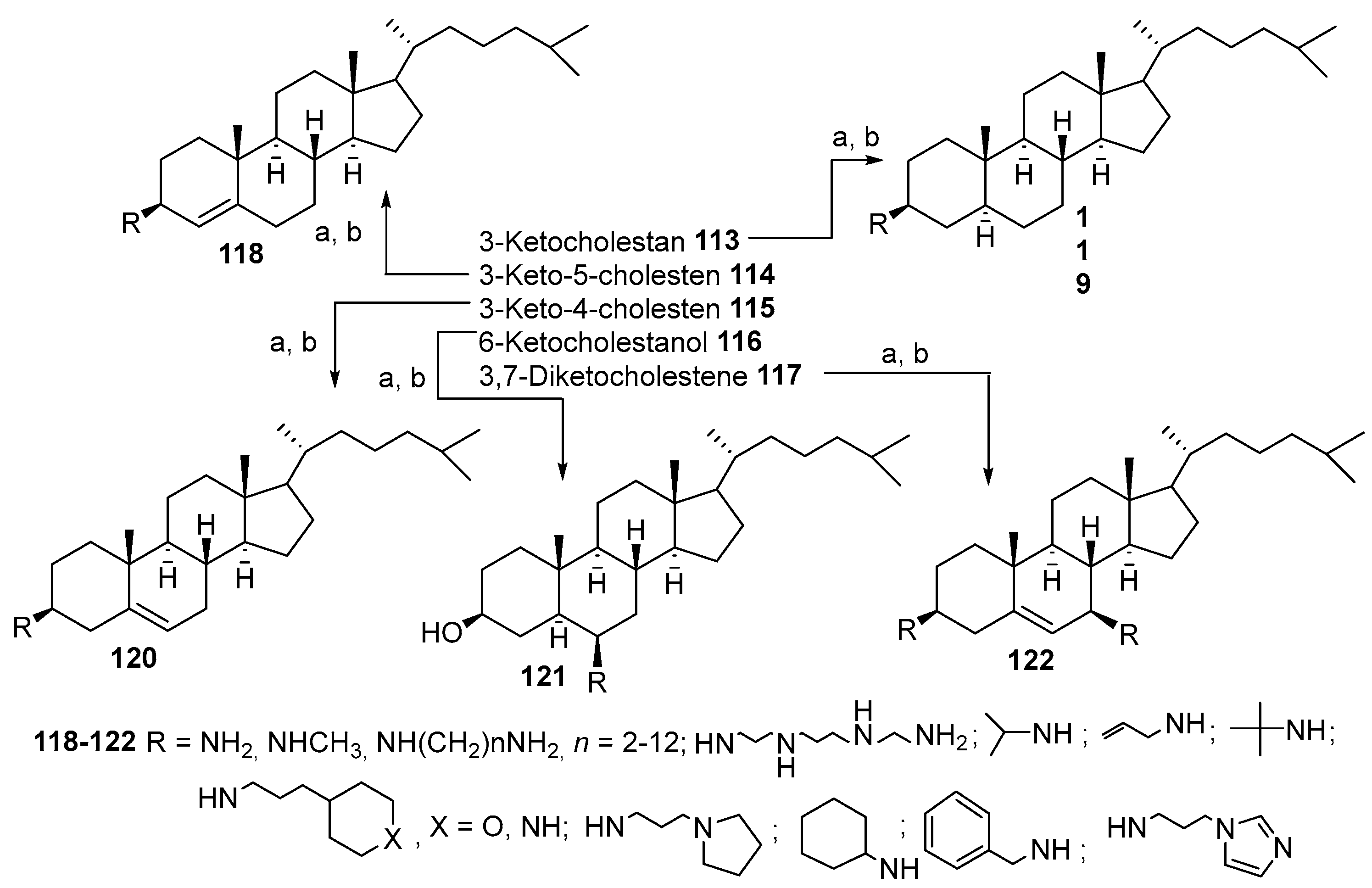{kind=link}
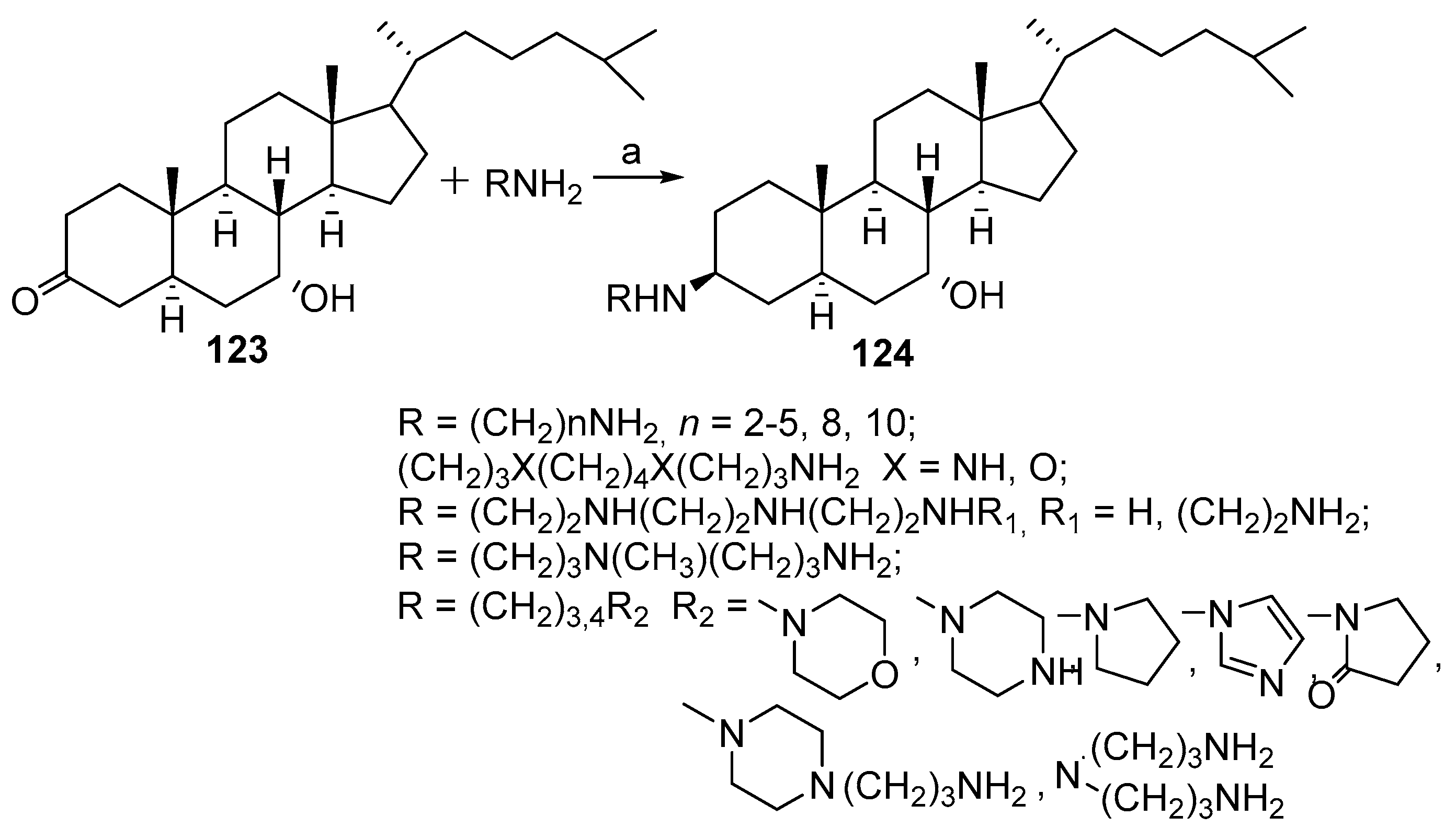{kind=link}
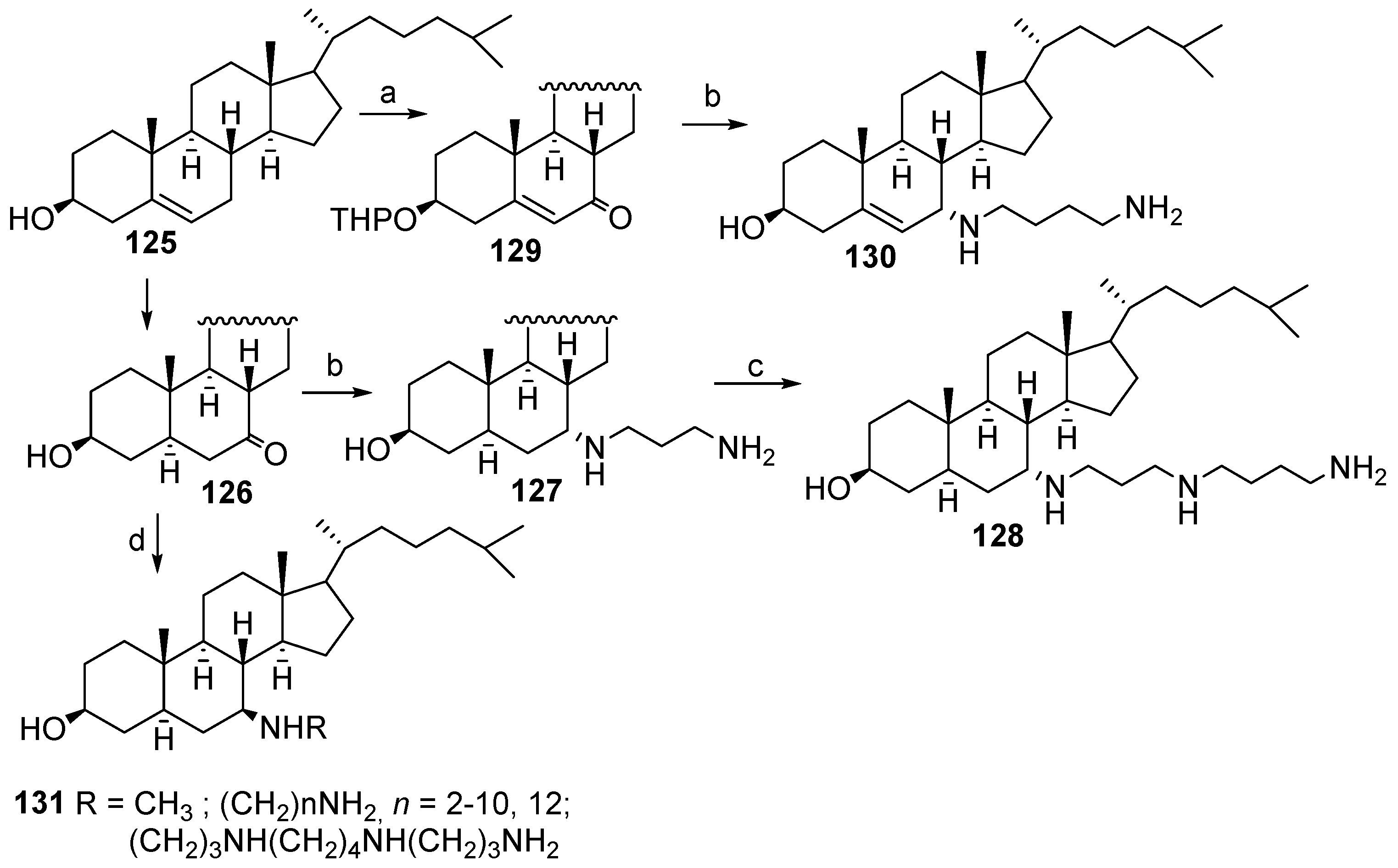{kind=link}
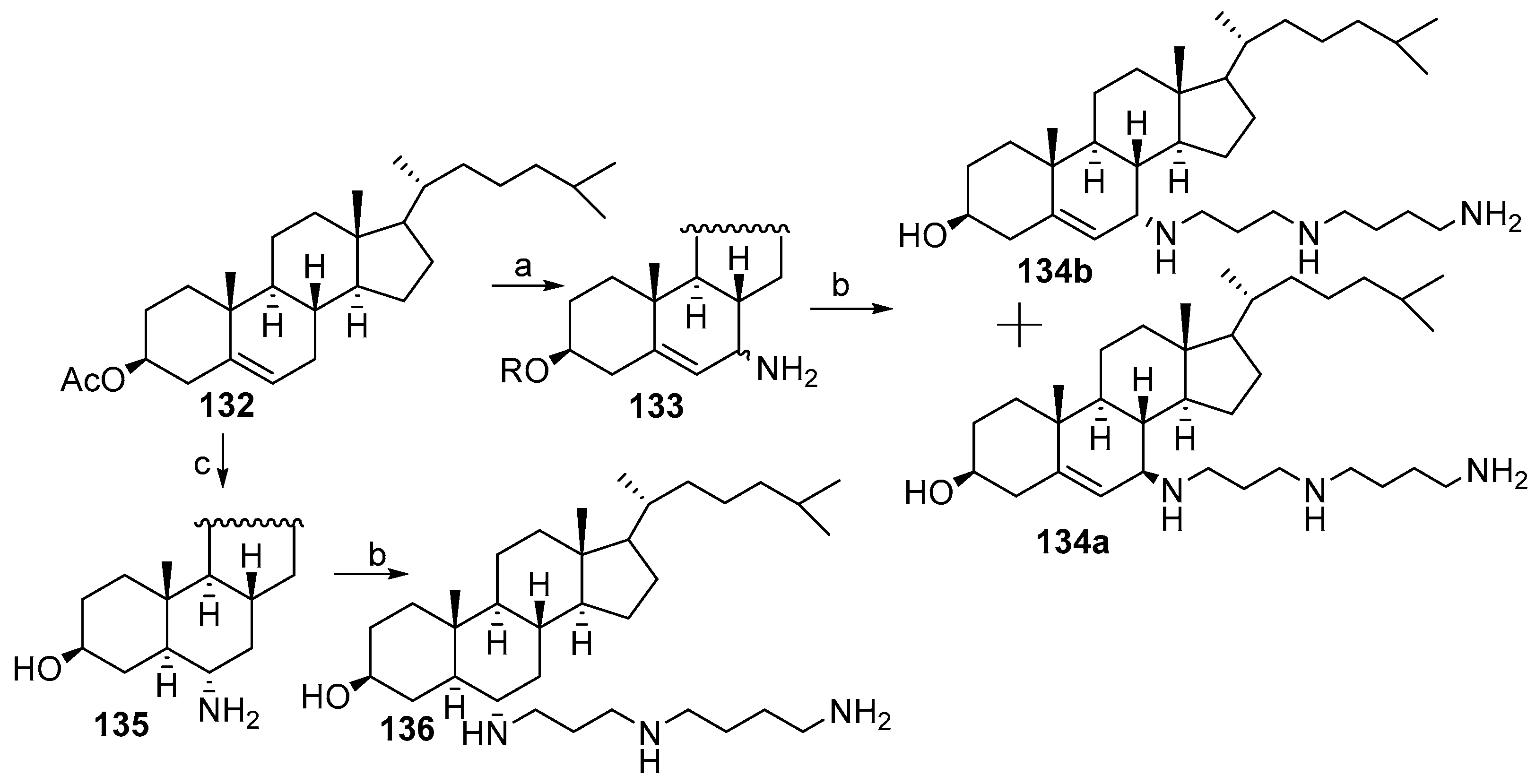{kind=link}
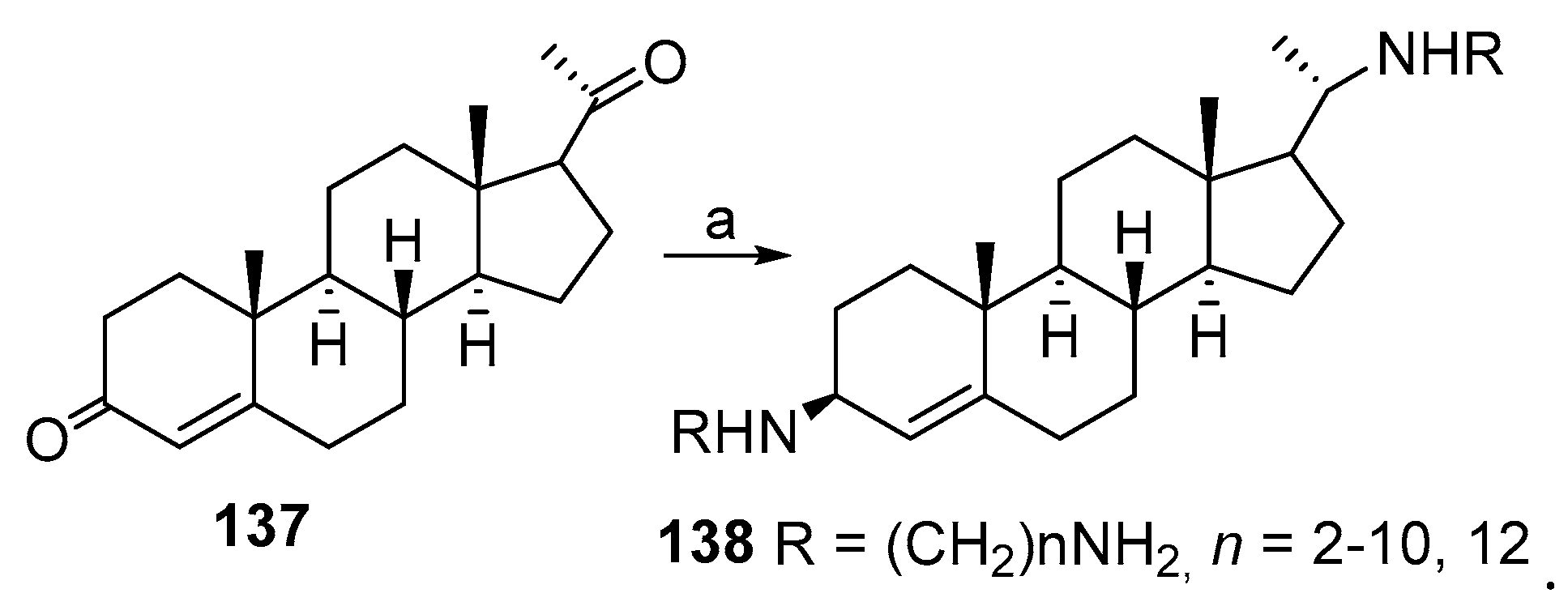{kind=link}
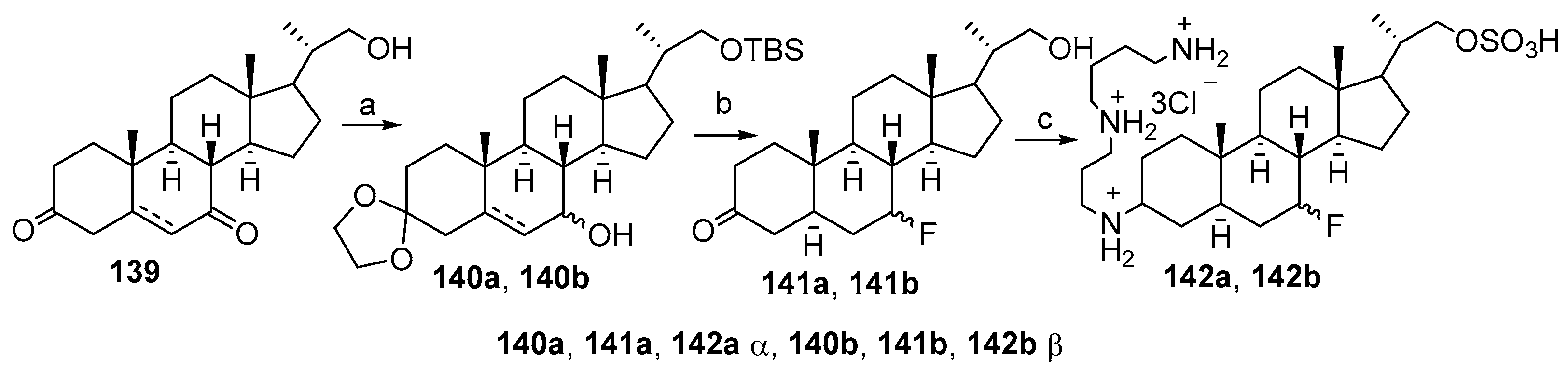{kind=link}
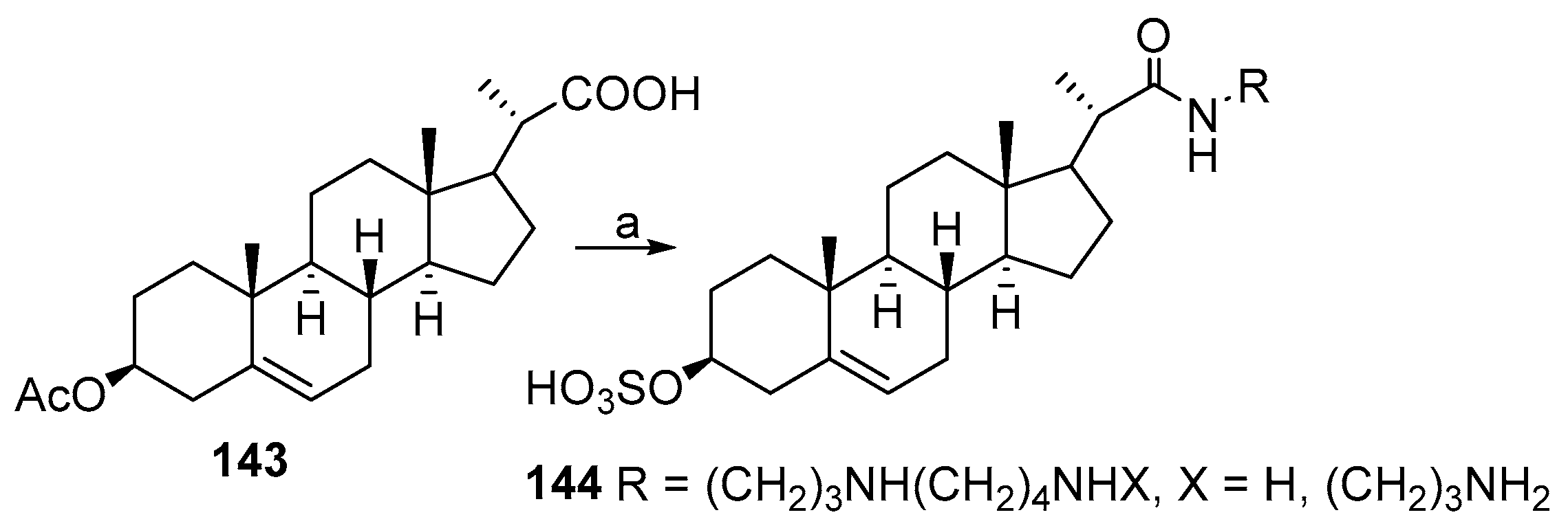{kind=link}
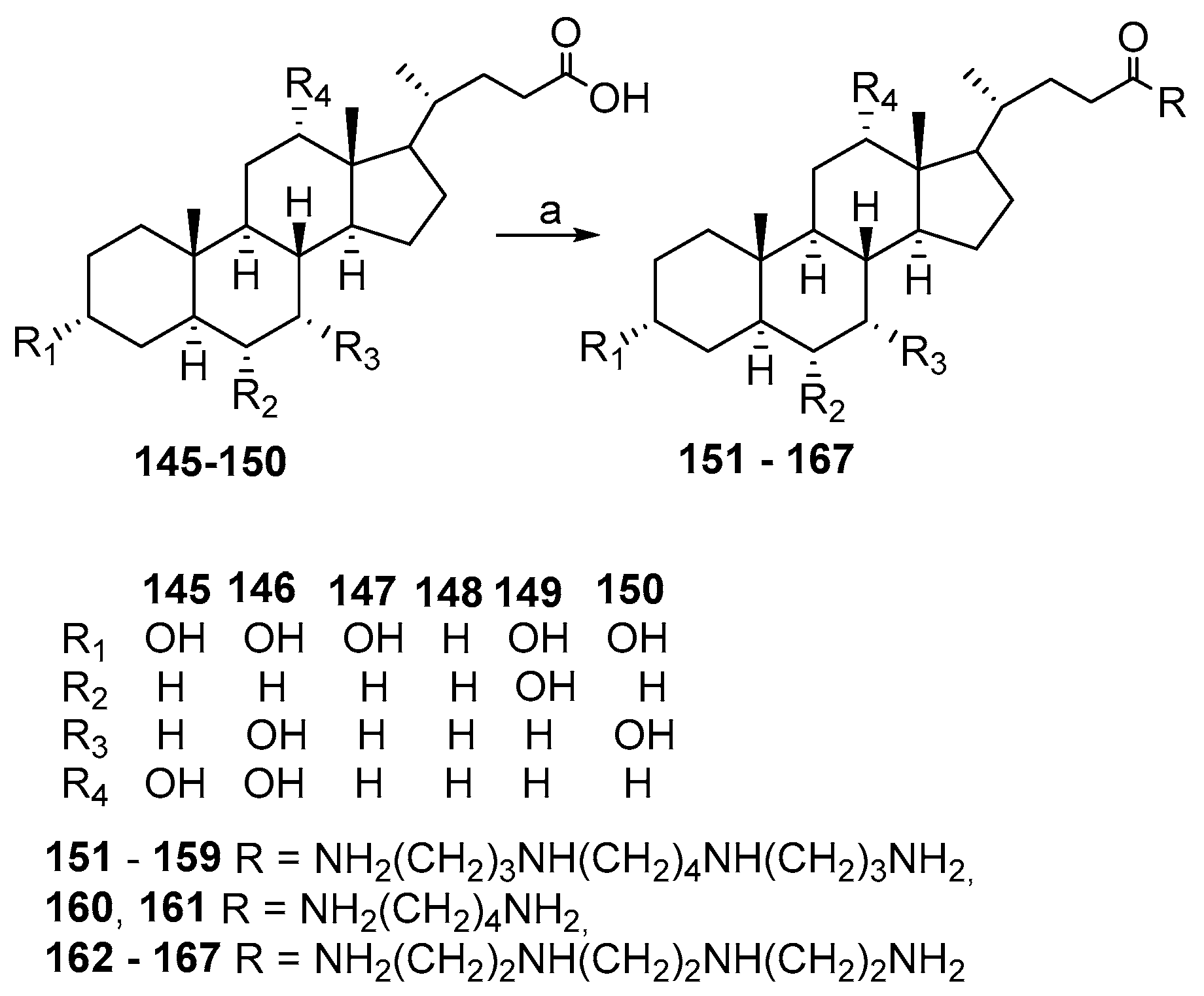{kind=link}
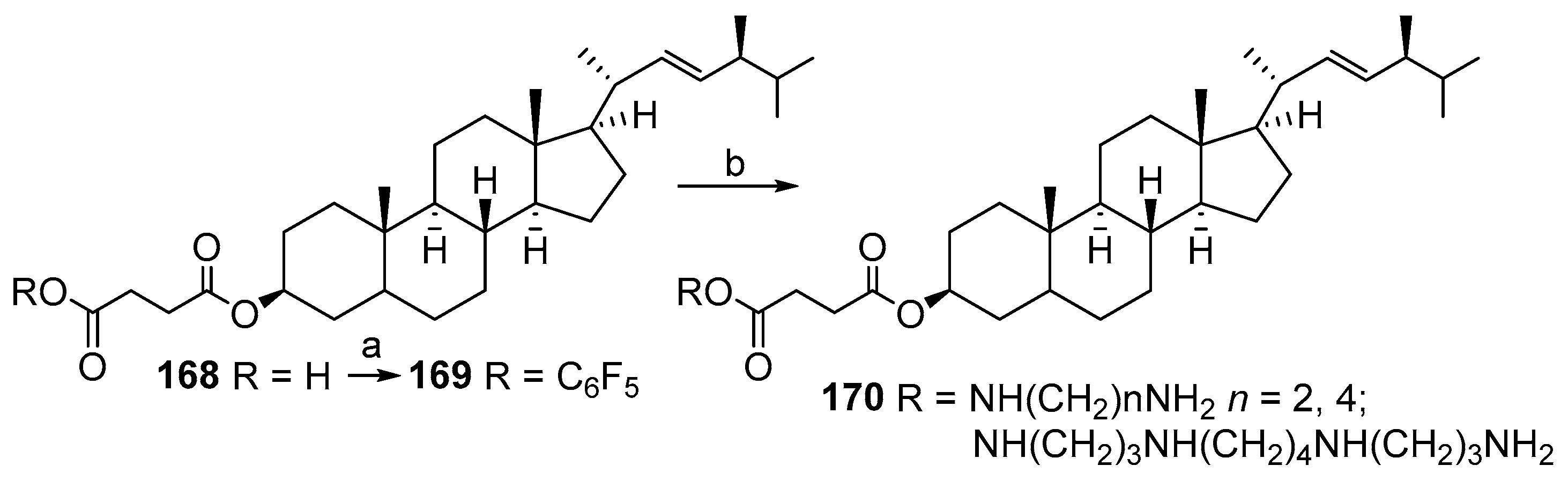{kind=link}
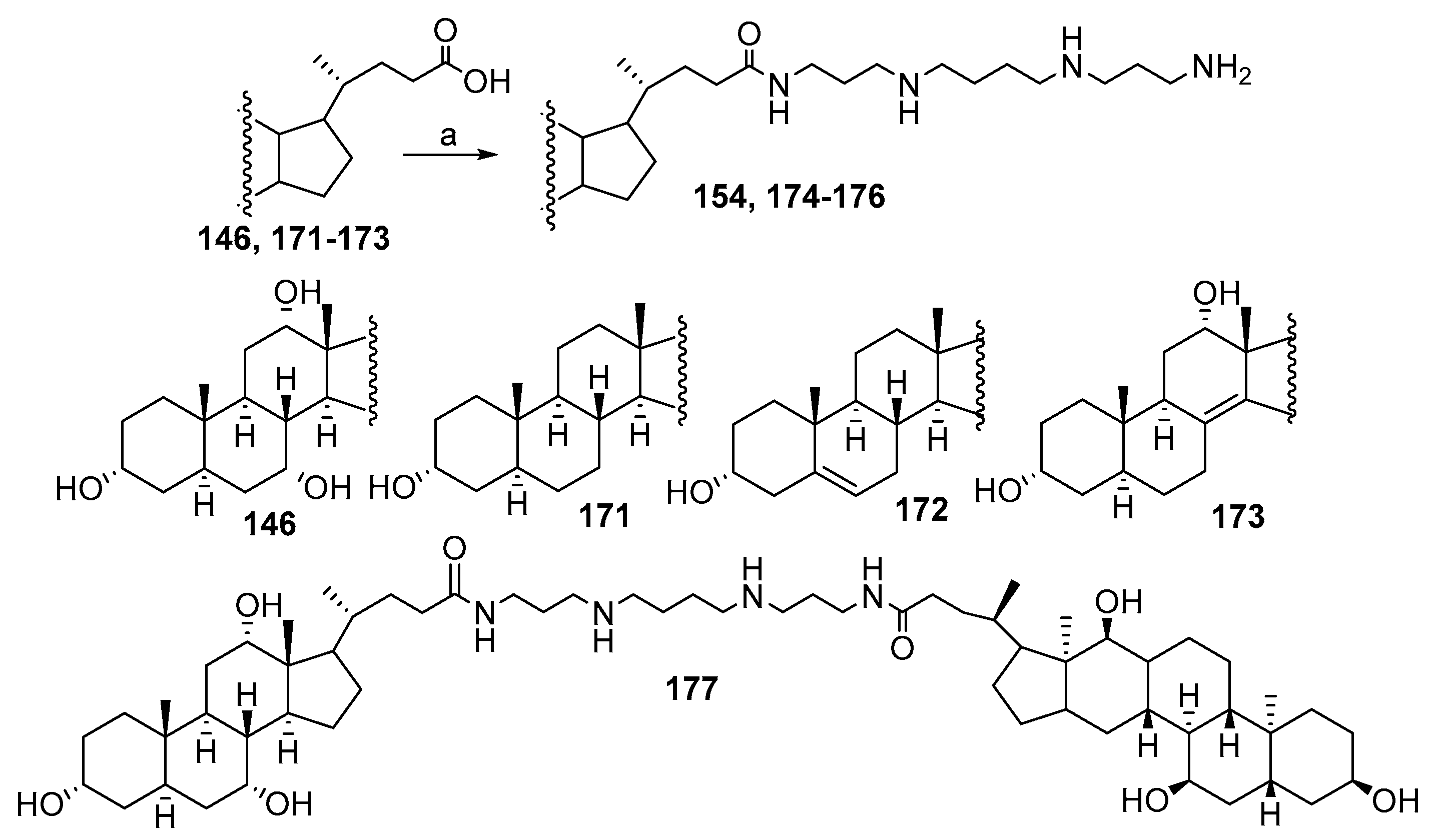{kind=link}
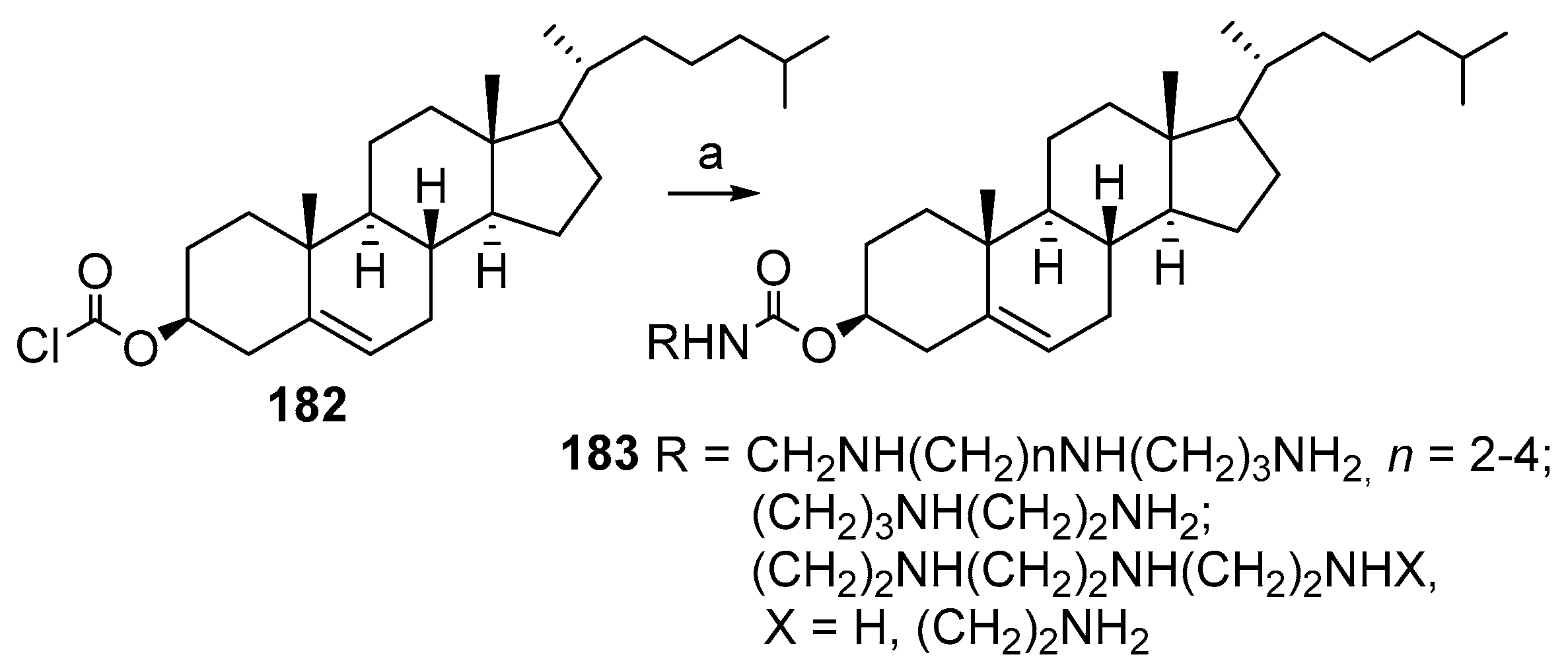{kind=link}
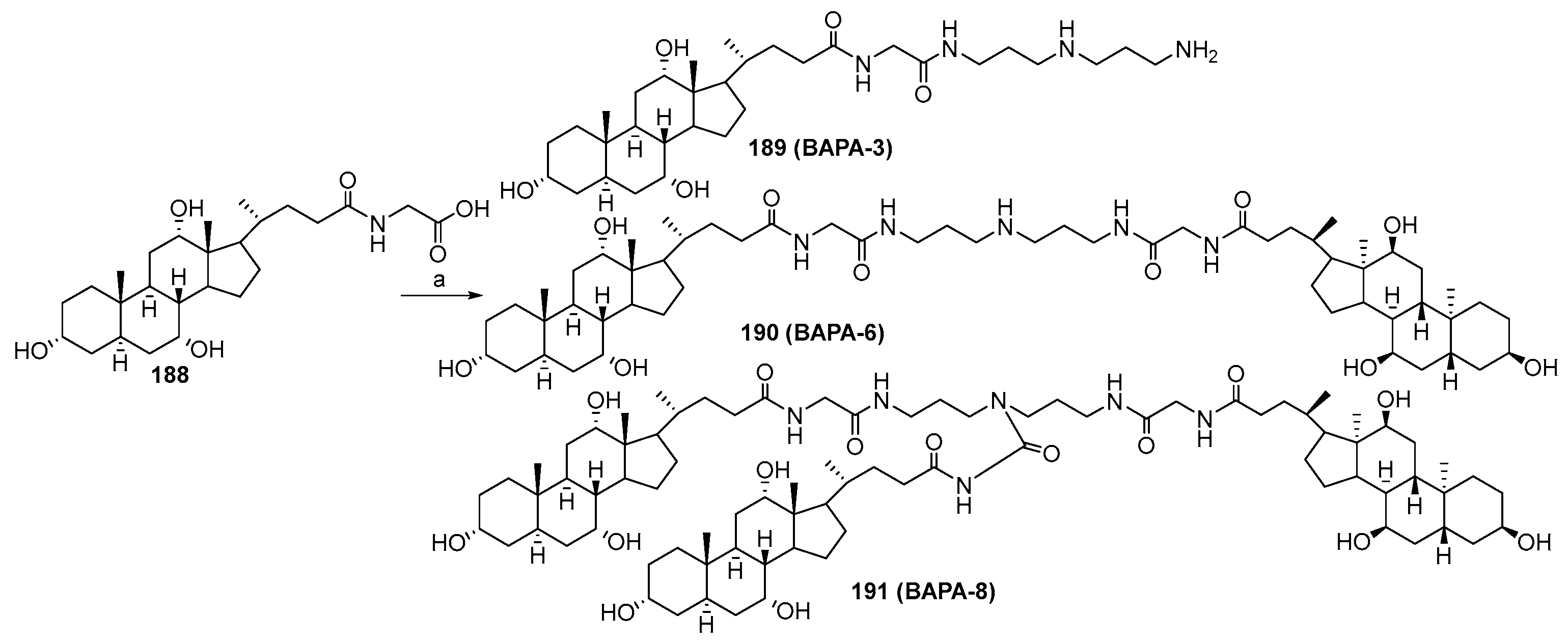{kind=link}
{kind=link}
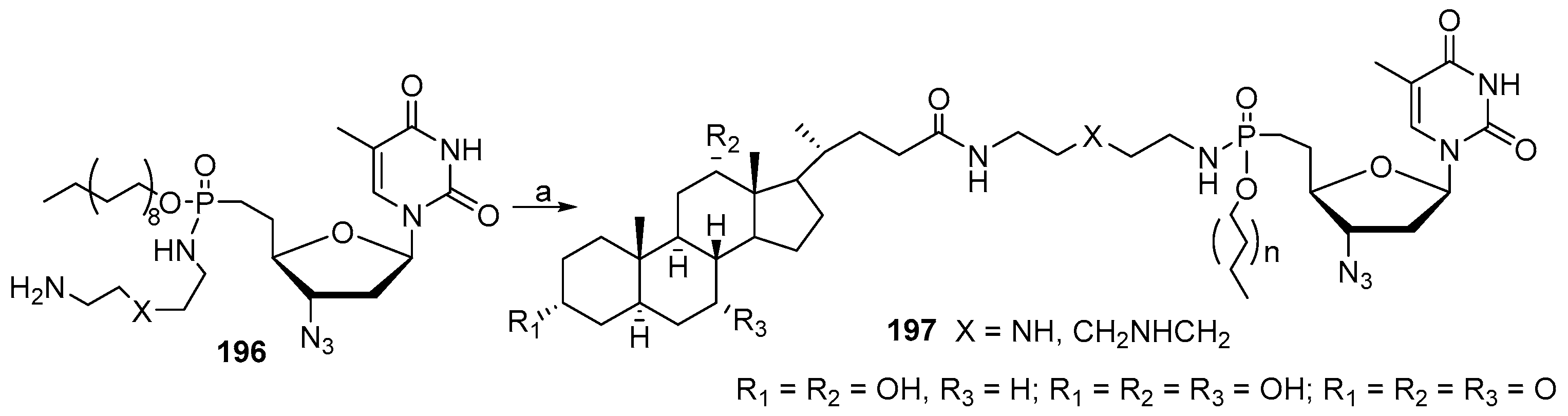{kind=link}
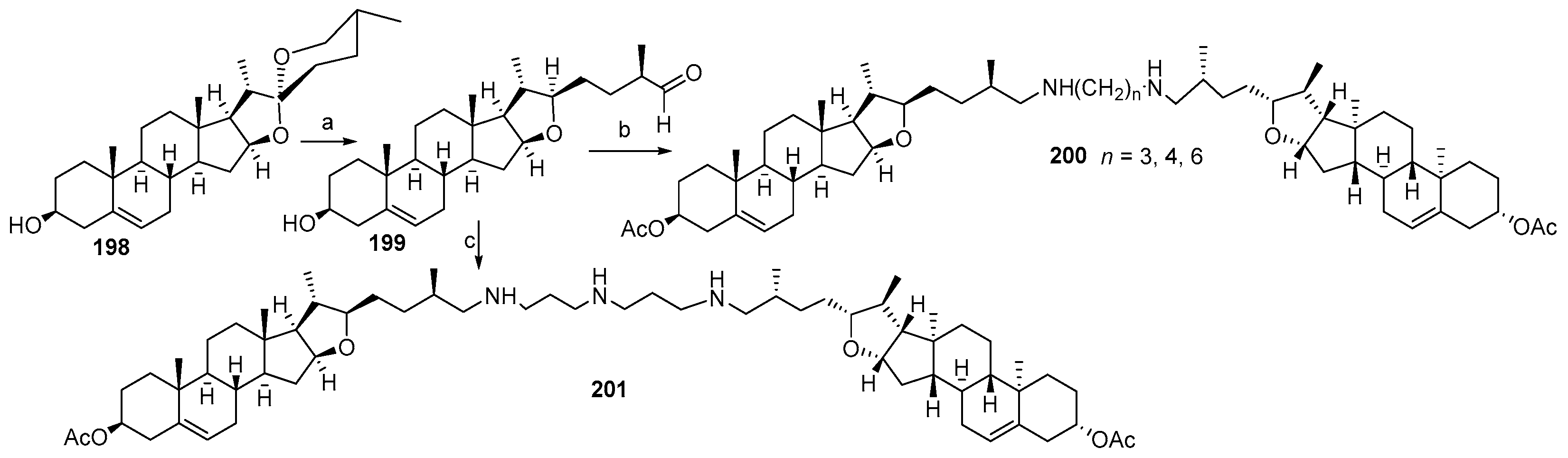{kind=link}
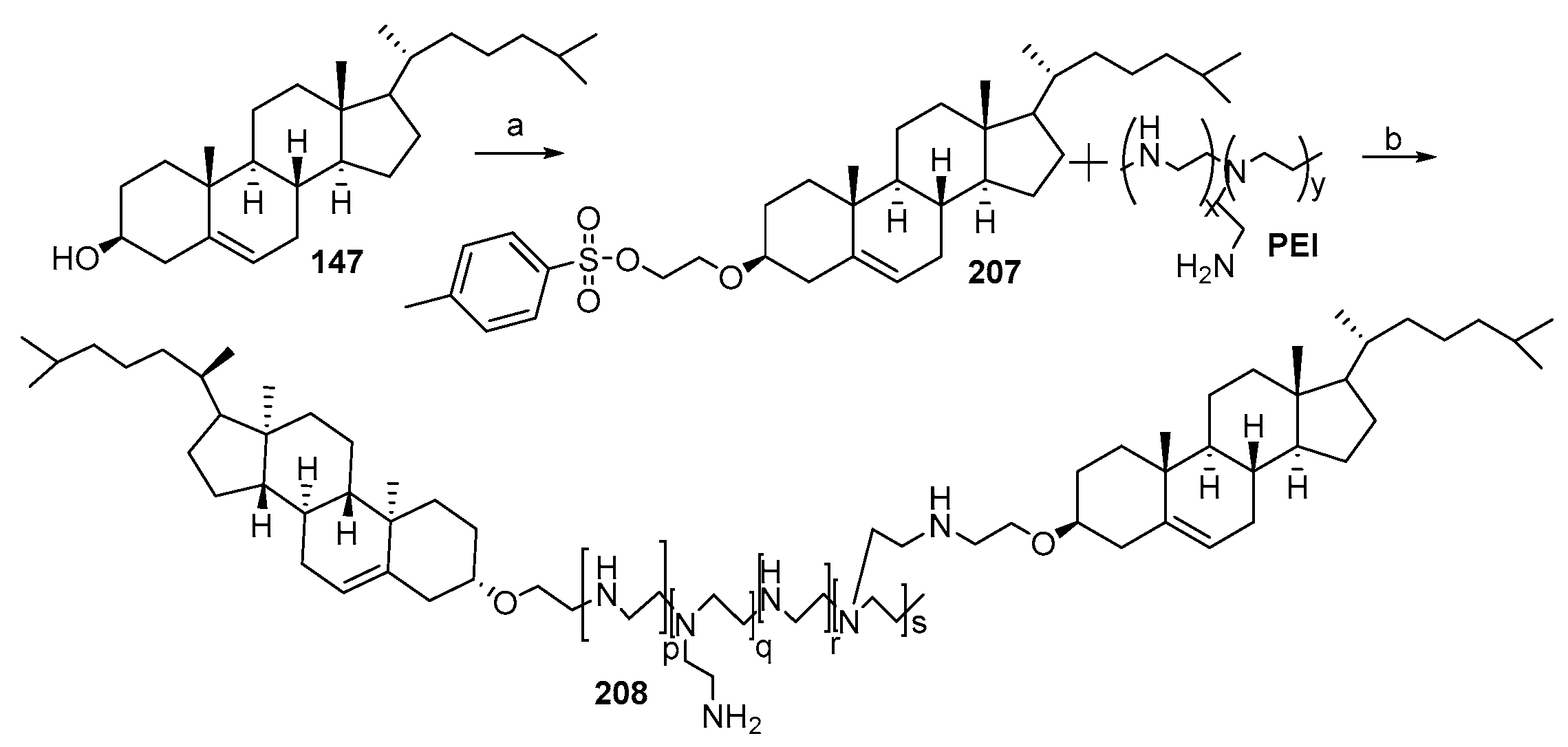{kind=link}
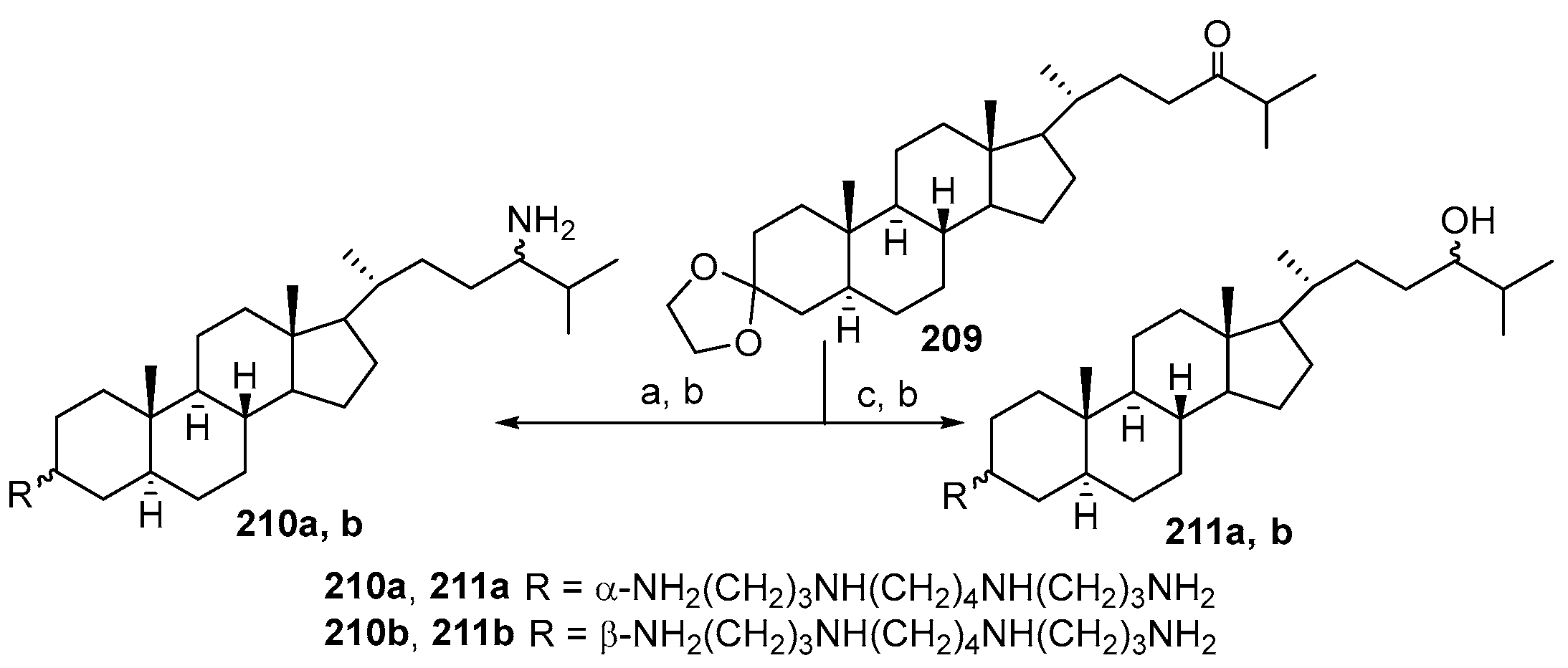{kind=link}
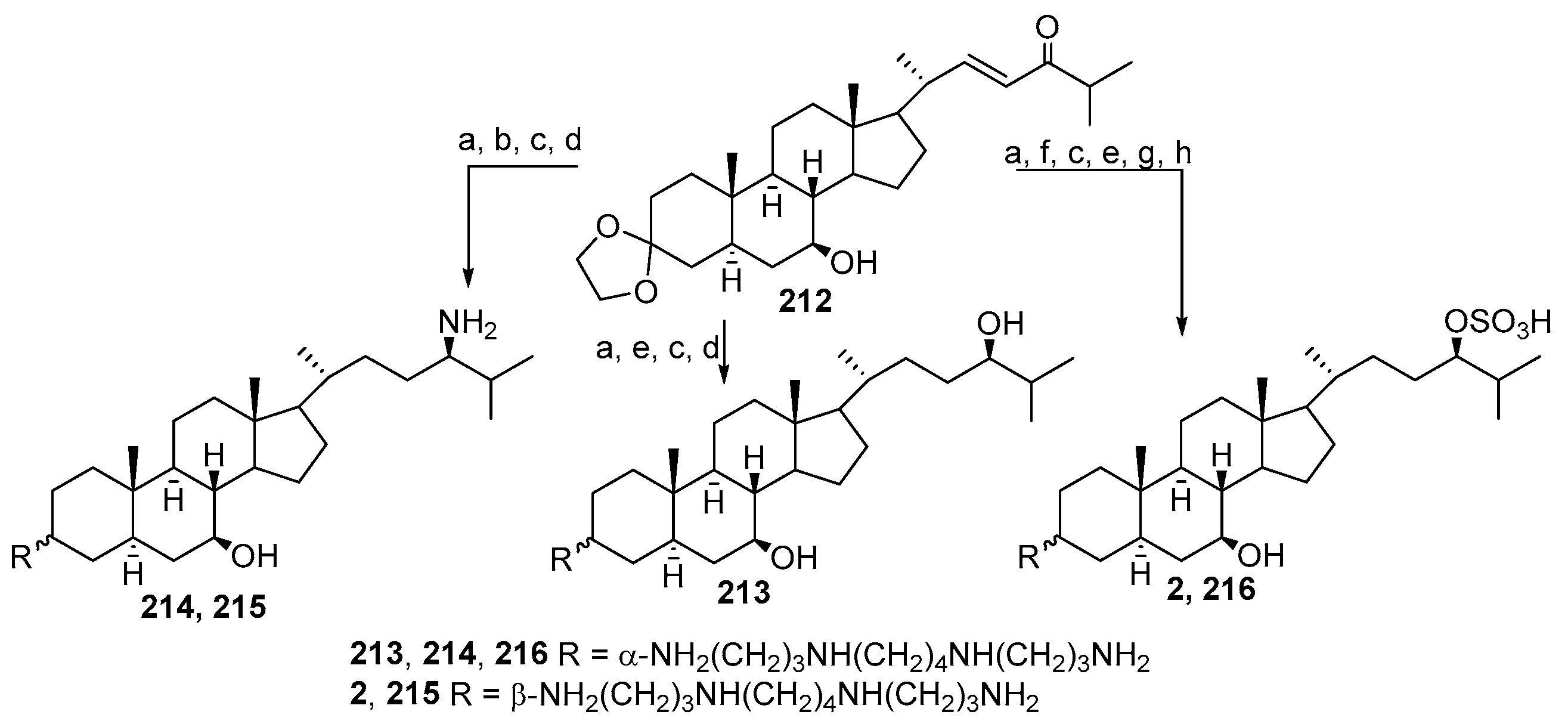{kind=link}
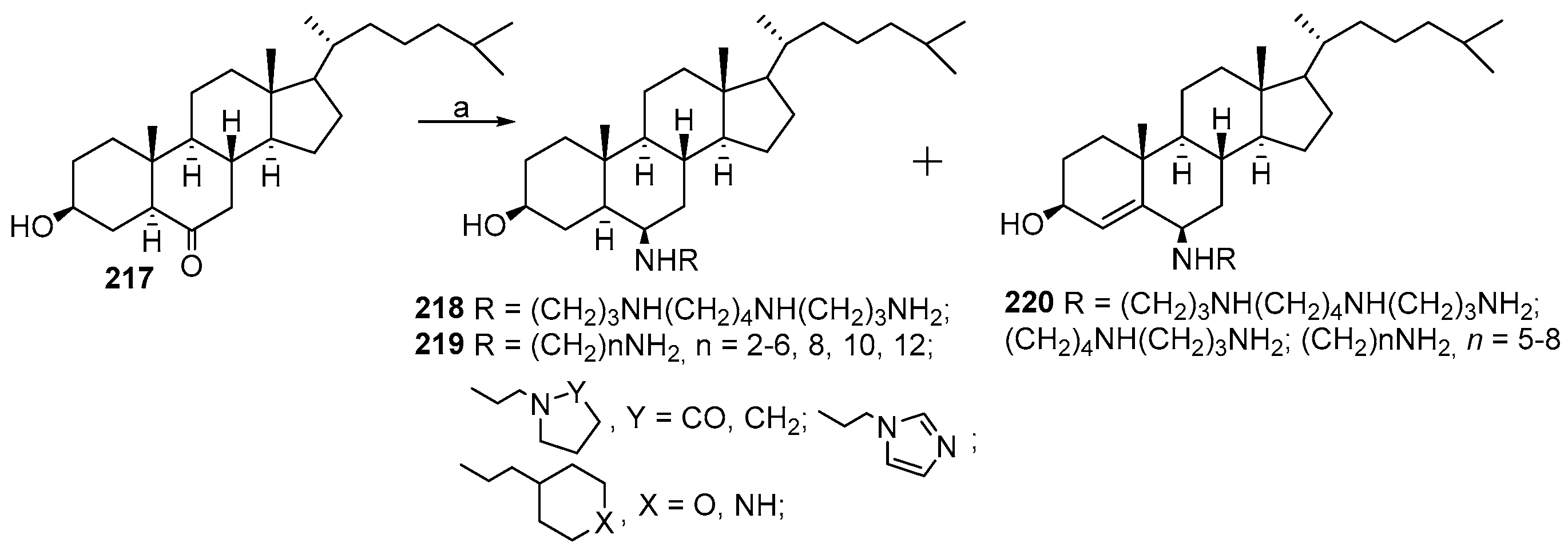{kind=link}
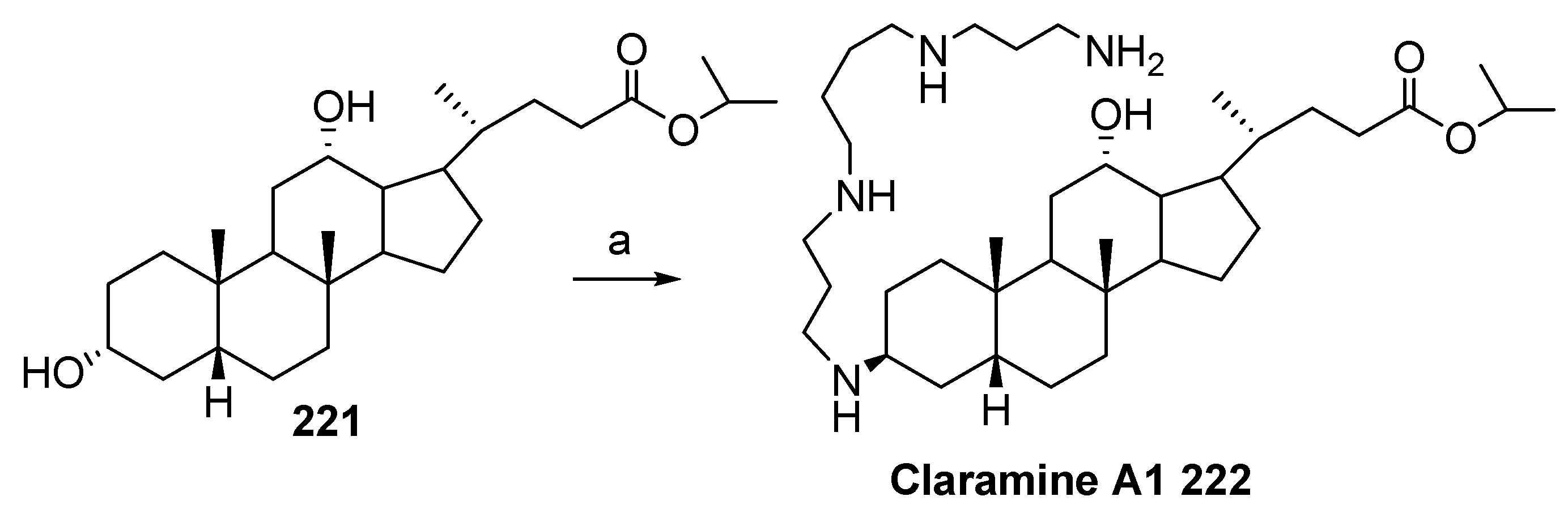{kind=link}
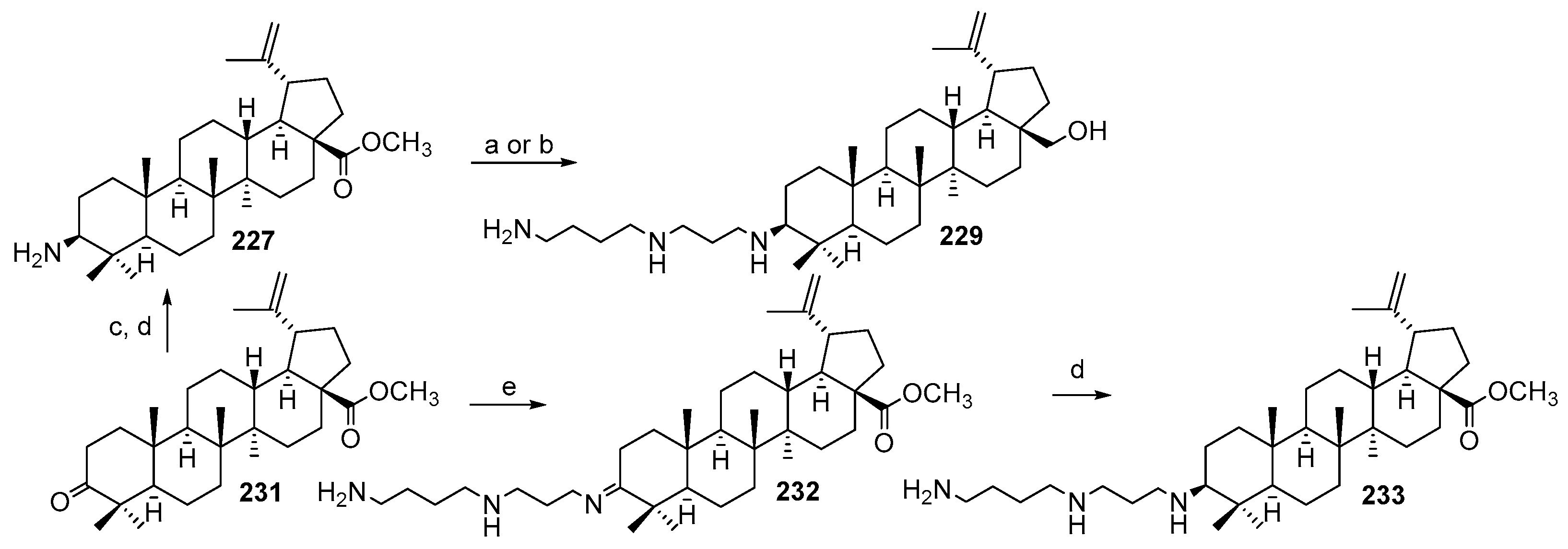{kind=link}
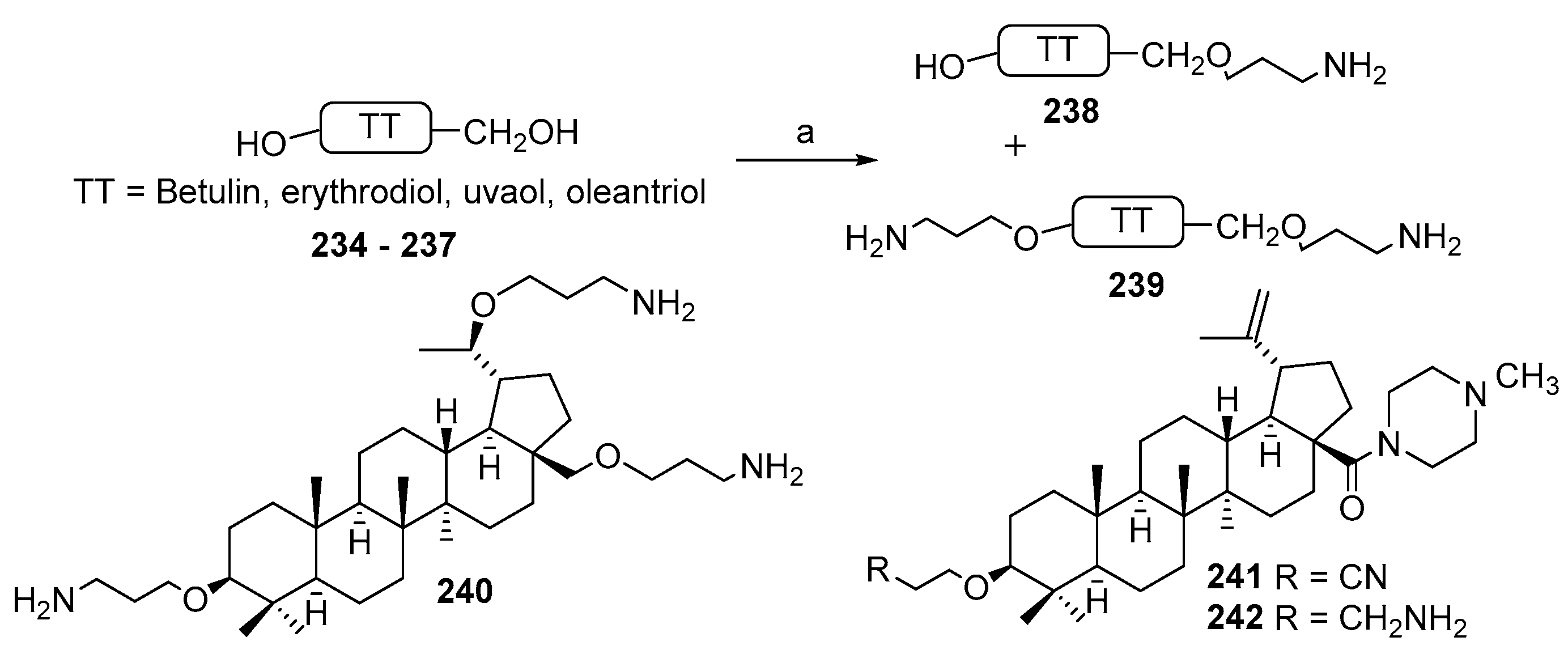{kind=link}
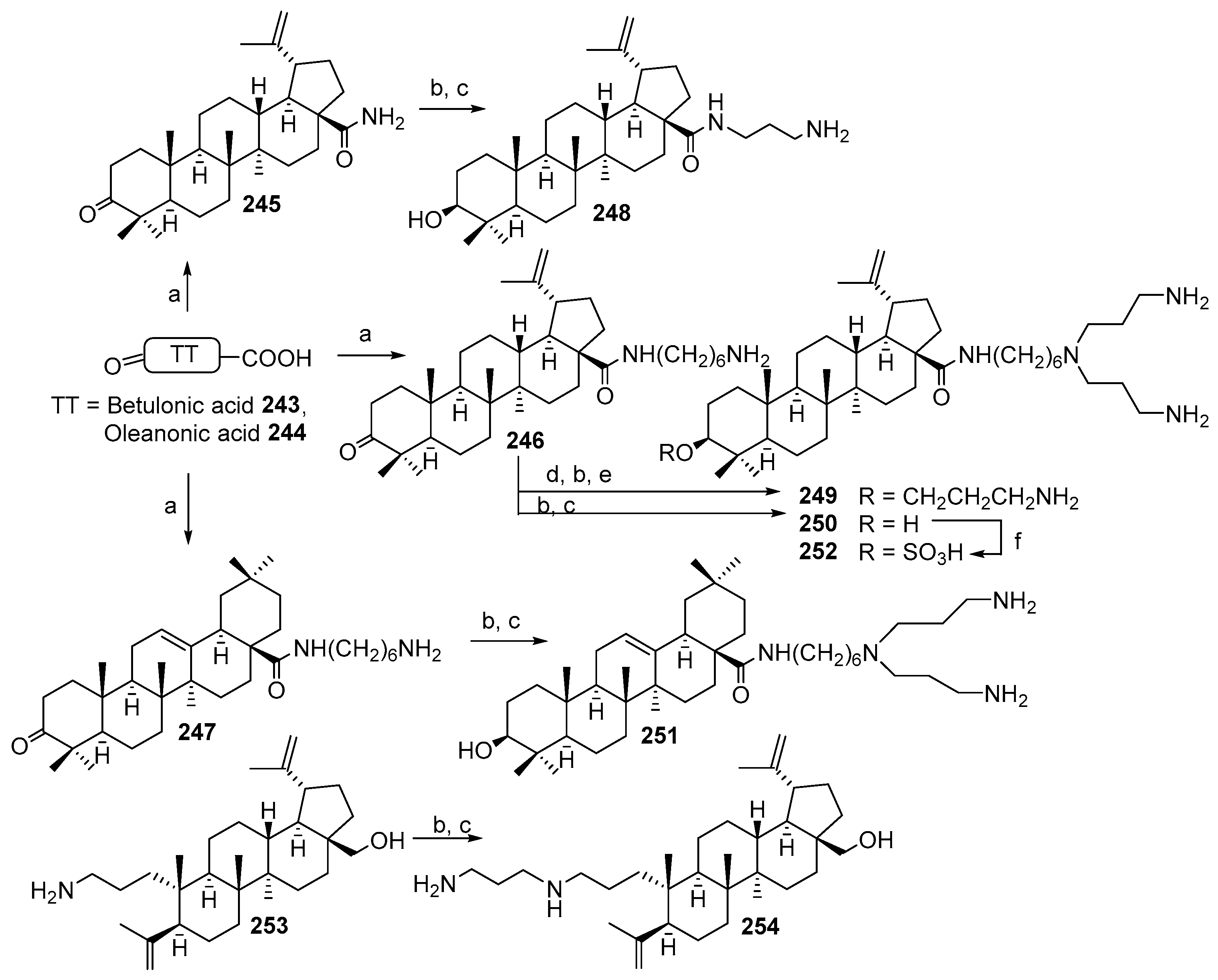{kind=link}
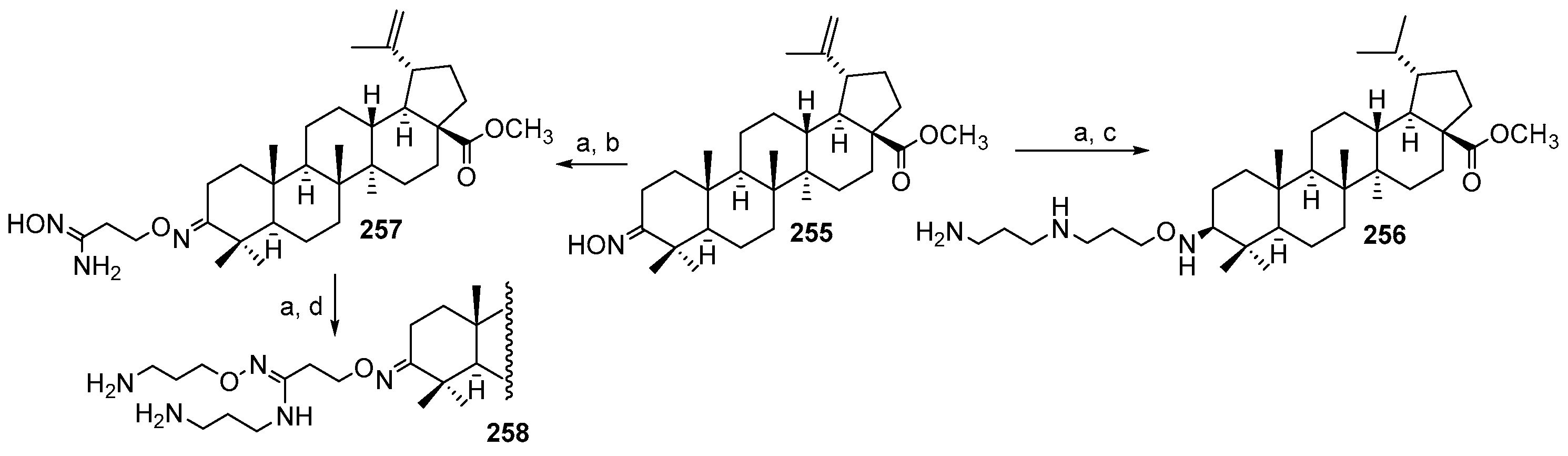{kind=link}
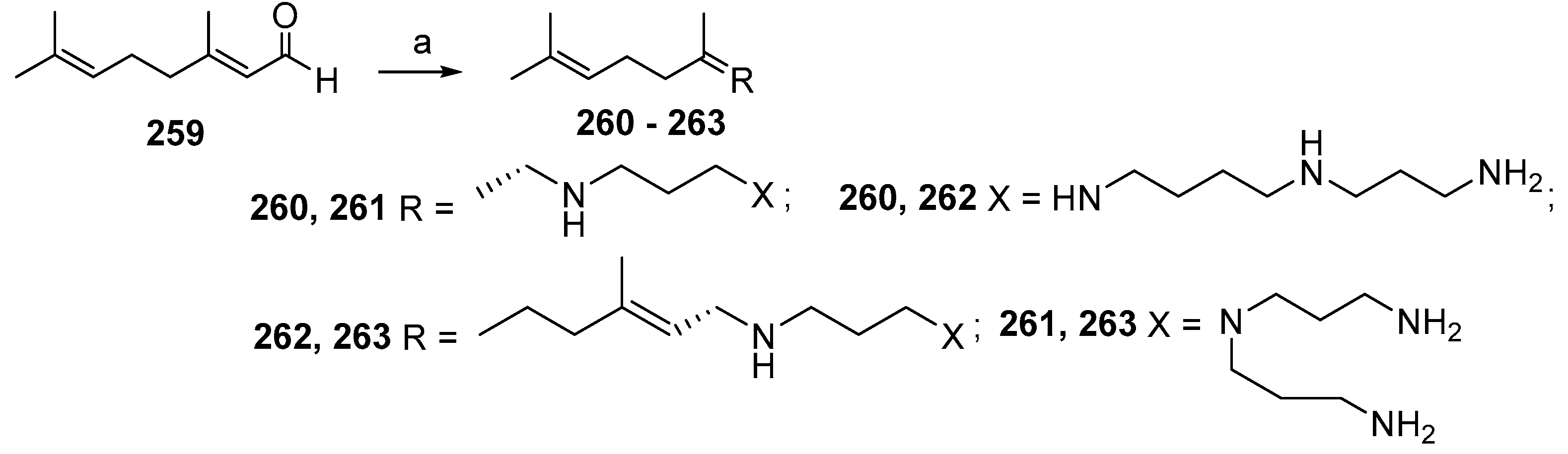{kind=link}
{kind=link}
| Primary Target | Pharmacological Action | Evidence Level | |||
|---|---|---|---|---|---|
| Squalamine | Trodusquemine | Claramine | Ceragenins | ||
| Phospholipid membranes | Antibacterial | In vitro MIC 1–8 μg/mL: E. coli, P. aeruginosa, S. aureus, S. faecalis, P. vulgaris, K. pneumoniae [38], A. baumannii [132]; MIC 0.5–1 μg/mL Methanobrevibacter spp. [130,131]; in animal studies as monotherapy or as sensitizer for conventional antibiotics against resistant strains of S. aureus, E. coli, P. aeruginosa, E. aerogenes, K. pneumoniae [103,128,142] | In vitro MIC 1–4 μg/mL: S. aureus, P. aeruginosa, C. albicans [7] | In vitro MIC 2–16 μg/mL: E. coli, E. aerogenes, E. cloacae, P. aeruginosa, K. pneumoniae, A. baumannii [120], and as an adjuvant for doxycycline against P. aeruginosa PAO1 and E. aerogenes EA28 [121] | In vitro MIC 2–4 μg/mL: E. coli, S. aureus, S. pneumoniae, S. pyogenes, H. influenza, P. aeruginosa, N. meningitides, K. pneumoniae, L. pneumophila, B. subtilis [226,227,229,230,231,239,242,248,249,250,251,252,253]; in animal studies as sensitizer for conventional antibiotics [99,228,229,235,243] |
| Antifungal | In vitro MIC 12.6–25 μM: C. albicans, C. krusei, C. glabrata, A. fumigatus, A. niger, Fusarium spp.) and protozoa (P. caudatum) [127] | In vitro MIC in 0.5–4 µg/mL: Candida spp., C. neoformans, A. fumigatus [245,254,255,256] | |||
| Antiviral | In vitro: Dengue virus, hepatitis B, yellow fever, herpesviruses [146,147]; in animal models: yellow fever, eastern equine encephalitis, cytomegalovirus [30] | In vitro: HIV [167] | In vitro: vaccinia virus [224] | ||
| Misfolded proteins | Neuroprotective | In vitro: alpha-synuclein, amyloid-beta [148,149]; in animal models of Parkinson’s disease [15,153]; phase 2 clinical trilas of squalamine phosphate for Parkinson’s disease [22,158] | In vitro: alpha-synuclein, amyloid-beta, HypF-N [119,152,153,154,155,157] In animal models of Parkinson’s and Alzheimer’s diseases (presumably PTP1B is also involved) [157,159,160,161] | ||
| NHE3 and mitogen signalling | Antiangiogenic | As an adjuvant to cytostatic drugs in animal tumor models [14,152,164,165,166]; as a monotherapy in animal retinopathy models [167,168,169]; squalamine lactate as monotherapy or in combination with mAb to VEGF in phase 2 clinical trilas for age-related macular edema [210,211,212,213,214]; combinatorial therapy in phase 2 clinical trilas for non-small cell lung cancer [210,211,212] | |||
| PTP1B | Antidiabetic via insulin and leptin receptors signalling | As monotherapy in diabetic ob/ob and db/db mice [173,174,175] and DIO mice [181,220]; as monotherapy in phase 1 clinical study for type 2 diabetes mellitus (NCT00606112, NCT00806338) | As monotherapy in diabetic CaMK2aCre/LMO4flox mice [218] | ||
| Anticancer via growth factor receptors signalling | As monotherapy in animal models of solid tumors [180,186,188,189,190,191,192,193,194,195]; as monotherapy in phase 1 clinical study for HER-2 positive metastatic breast cancer (NCT02524951) | As monotherapy in animal models of glioblastoma and colorectal carcinoma [219] | |||
| Atherosclerosis | In animal models of LDLR−/− atherosclerosis [199]; phase 1 study has been announced | ||||
| Anxiolytic | In animal models of anxiety and schizophrenia [205,206] | ||||
| Regenerative | In animal models of trauma and myocardial infarction [201,202] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazakova, O.; Giniyatullina, G.; Babkov, D.; Wimmer, Z. From Marine Metabolites to the Drugs of the Future: Squalamine, Trodusquemine, Their Steroid and Triterpene Analogues. Int. J. Mol. Sci. 2022, 23, 1075. https://doi.org/10.3390/ijms23031075
Kazakova O, Giniyatullina G, Babkov D, Wimmer Z. From Marine Metabolites to the Drugs of the Future: Squalamine, Trodusquemine, Their Steroid and Triterpene Analogues. International Journal of Molecular Sciences. 2022; 23(3):1075. https://doi.org/10.3390/ijms23031075
Chicago/Turabian StyleKazakova, Oxana, Gulnara Giniyatullina, Denis Babkov, and Zdenek Wimmer. 2022. "From Marine Metabolites to the Drugs of the Future: Squalamine, Trodusquemine, Their Steroid and Triterpene Analogues" International Journal of Molecular Sciences 23, no. 3: 1075. https://doi.org/10.3390/ijms23031075
APA StyleKazakova, O., Giniyatullina, G., Babkov, D., & Wimmer, Z. (2022). From Marine Metabolites to the Drugs of the Future: Squalamine, Trodusquemine, Their Steroid and Triterpene Analogues. International Journal of Molecular Sciences, 23(3), 1075. https://doi.org/10.3390/ijms23031075