
Dysregulated Expression of Transposable Elements in TDP-43M337V Human Motor Neurons That Recapitulate Amyotrophic Lateral Sclerosis In Vitro

 ,
,
Abstract
1. Introduction
2. Results
2.1. Differential Expression of Genes and Transposable Elements in the Mutant iMNs
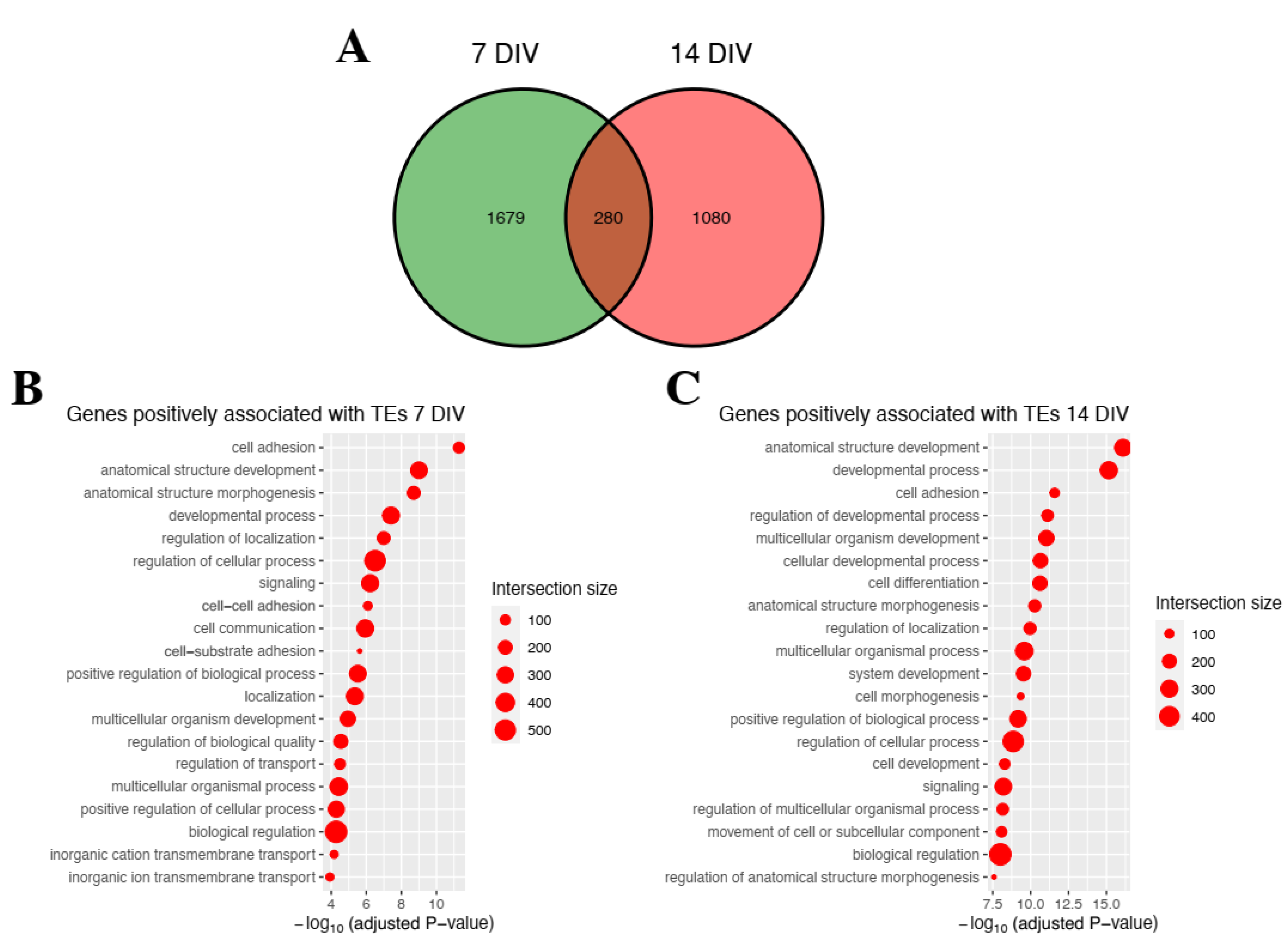
2.2. Genetic Programs Associated with Differentially Expressed Genes
2.3. Impact of Transposable Elements in Genetic Programs
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Suk, T.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Berning, B.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef]
- Khalfallah, Y.; Kuta, R.; Grasmuck, C.; Prat, A.; Durham, H.D.; Velde, C.V. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci. Rep. 2018, 8, 7551. [Google Scholar] [CrossRef]
- Buratti, E. Functional Significance of TDP-43 Mutations in Disease. Adv. Genet. 2015, 91, 1–53. [Google Scholar] [CrossRef]
- Siomi, M.C.; Sato, K.; Pezic, D.; Aravin, A.A. PIWI-interacting small RNAs: The vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 2011, 12, 246–258. [Google Scholar] [CrossRef]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Fagegaltier, D.; Harris, B.T.; Ostrow, L.W.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177.e5. [Google Scholar] [CrossRef]
- Guo, C.; Jeong, H.-H.; Hsieh, Y.-C.; Klein, H.-U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef]
- Sun, W.; Samimi, H.; Gamez, M.; Zare, H.; Frost, B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat. Neurosci. 2018, 21, 1038–1048. [Google Scholar] [CrossRef]
- De Thé, F.B.; Rekaik, H.; Peze-Heidsieck, E.; Massiani-Beaudoin, O.; Joshi, R.L.; Fuchs, J.; Prochiantz, A. Engrailed homeoprotein blocks degeneration in adult dopaminergic neurons through LINE-1 repression. EMBO J. 2018, 37, e97374. [Google Scholar] [CrossRef]
- Calvo, A.C.; Manzano, R.; Mendonça, D.M.F.; Muñoz, M.J.; Zaragoza, P.; Osta, R. Amyotrophic Lateral Sclerosis: A Focus on Disease Progression. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Coufal, N.G.; Garcia-Perez, J.L.; Peng, G.E.; Marchetto, M.C.N.; Muotri, A.R.; Mu, Y.; Carson, C.T.; Macia, A.; Moran, J.V.; Gage, F.H. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20382–20387. [Google Scholar] [CrossRef]
- Muotri, A.R.; Marchetto, M.C.N.; Coufal, N.G.; Oefner, R.; Yeo, G.; Nakashima, K.; Gage, F.H. L1 retrotransposition in neurons is modulated by MeCP2. Nature 2010, 468, 443–446. [Google Scholar] [CrossRef]
- Casale, A.M.; Liguori, F.; Ansaloni, F.; Cappucci, U.; Finaurini, S.; Spirito, G.; Persichetti, F.; Sanges, R.; Gustincich, S.; Piacentini, L. Transposable element activation promotes neurodegeneration in a Drosophila model of Huntington’s disease. iScience 2022, 25, 103702. [Google Scholar] [CrossRef]
- Li, W.; Jin, Y.; Prazak, L.; Hammell, M.; Dubnau, J. Transposable Elements in TDP-43-Mediated Neurodegenerative Disorders. PLoS ONE 2012, 7, e44099. [Google Scholar] [CrossRef]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2010, 69, 141–151. [Google Scholar] [CrossRef]
- Krug, L.; Chatterjee, N.; Borges-Monroy, R.; Hearn, S.; Liao, W.-W.; Morrill, K.; Prazak, L.; Rozhkov, N.; Theodorou, D.; Hammell, M.; et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 2017, 13, e1006635. [Google Scholar] [CrossRef]
- Valdebenito-Maturana, B.; Arancibia, E.; Riadi, G.; Tapia, J.C.; Carrasco, M. Locus-specific analysis of Transposable Elements during the progression of ALS in the SOD1G93A mouse model. PLoS ONE 2021, 16, e0258291. [Google Scholar] [CrossRef]
- Prudencio, M.; Gonzales, P.K.; Cook, C.N.; Gendron, T.F.; Daughrity, L.M.; Song, Y.; Ebbert, M.T.; van Blitterswijk, M.; Zhang, Y.-J.; Jansen-West, K.; et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. 2017, 26, 3421–3431. [Google Scholar] [CrossRef]
- Ye, L.; Swingen, C.; Zhang, J. Induced Pluripotent Stem Cells and Their Potential for Basic and Clinical Sciences. Curr. Cardiol. Rev. 2013, 9, 63–72. [Google Scholar] [CrossRef]
- Richard, J.-P.; Maragakis, N.J. Induced pluripotent stem cells from ALS patients for disease modeling. Brain Res. 2015, 1607, 15–25. [Google Scholar] [CrossRef]
- E Cáceres, D.; Torres, F.C.; A Castillo, C.; Maureira, A.; A Franco-Campos, F.; Osorio, M.; Segovia, R.; Tapia, J.C.; A Carrasco, M. A Platform to Understand Amyotrophic Lateral Sclerosis (ALS) and Extend Human Motor Neurons Longevity. Int. J. Morphol. 2019, 37, 1203–1209. [Google Scholar] [CrossRef]
- Ishii, T.; Kawakami, E.; Endo, K.; Misawa, H.; Watabe, K. Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS ONE 2017, 12, e0179375. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef]
- Nahm, M.; Lim, S.M.; Kim, Y.-E.; Park, J.; Noh, M.-Y.; Lee, S.; Roh, J.E.; Hwang, S.-M.; Park, C.-K.; Kim, Y.H.; et al. ANXA11 mutations in ALS cause dysregulation of calcium homeostasis and stress granule dynamics. Sci. Transl. Med. 2020, 12, aax3993. [Google Scholar] [CrossRef]
- Lin, J.; Huang, P.; Chen, W.; Ye, C.; Su, H.; Yao, X. Key Molecules and Pathways Underlying Sporadic Amyotrophic Lateral Sclerosis: Integrated Analysis on Gene Expression Profiles of Motor Neurons. Front. Genet. 2020, 11, 578143. [Google Scholar] [CrossRef]
- Kotni, M.K.; Zhao, M.; Wei, D.-Q. Gene expression profiles and protein-protein interaction networks in amyotrophic lateral sclerosis patients with C9orf72 mutation. Orphanet J. Rare Dis. 2016, 11, 148. [Google Scholar] [CrossRef]
- Valdebenito-Maturana, B.; Guatimosim, C.; Carrasco, M.A.; Tapia, J.C. Spatially Resolved Expression of Transposable Elements in Disease and Somatic Tissue with SpatialTE. Int. J. Mol. Sci. 2021, 22, 13623. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Bendall, M.L.; de Mulder, M.; Iñiguez, L.P.; Lecanda-Sánchez, A.; Pérez-Losada, M.; Ostrowski, M.A.; Jones, R.B.; Mulder, L.C.F.; Reyes-Terán, G.; Crandall, K.A.; et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol. 2019, 15, e1006453. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Karakülah, G.; Arslan, N.; Yandim, C.; Suner, A. TEffectR: An R package for studying the potential effects of transposable elements on gene expression with linear regression model. PeerJ 2019, 7, e8192. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Timepoint | Up-Regulated Genes | Down-Regulated Genes | Up-Regulated TEs | Down-Regulated TEs |
|---|---|---|---|---|
| 7 DIV | 2625 | 881 | 16,668 | 16,956 |
| 14 DIV | 2617 | 717 | 13,599 | 11,123 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdebenito-Maturana, B.; Rojas-Tapia, M.I.; Carrasco, M.; Tapia, J.C. Dysregulated Expression of Transposable Elements in TDP-43M337V Human Motor Neurons That Recapitulate Amyotrophic Lateral Sclerosis In Vitro. Int. J. Mol. Sci. 2022, 23, 16222. https://doi.org/10.3390/ijms232416222
Valdebenito-Maturana B, Rojas-Tapia MI, Carrasco M, Tapia JC. Dysregulated Expression of Transposable Elements in TDP-43M337V Human Motor Neurons That Recapitulate Amyotrophic Lateral Sclerosis In Vitro. International Journal of Molecular Sciences. 2022; 23(24):16222. https://doi.org/10.3390/ijms232416222
Chicago/Turabian StyleValdebenito-Maturana, Braulio, Matias Ignacio Rojas-Tapia, Mónica Carrasco, and Juan Carlos Tapia. 2022. "Dysregulated Expression of Transposable Elements in TDP-43M337V Human Motor Neurons That Recapitulate Amyotrophic Lateral Sclerosis In Vitro" International Journal of Molecular Sciences 23, no. 24: 16222. https://doi.org/10.3390/ijms232416222
APA StyleValdebenito-Maturana, B., Rojas-Tapia, M. I., Carrasco, M., & Tapia, J. C. (2022). Dysregulated Expression of Transposable Elements in TDP-43M337V Human Motor Neurons That Recapitulate Amyotrophic Lateral Sclerosis In Vitro. International Journal of Molecular Sciences, 23(24), 16222. https://doi.org/10.3390/ijms232416222