
In Silico Study of the Interactions of Anle138b Isomer, an Inhibitor of Amyloid Aggregation, with Partner Proteins

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Characterization of the AαSyn–Anle138b Isomer Complex
2.2. Characterization of the AαSyn–CypA and Anle138b Isomer Complex
2.3. Molecular Docking: Characterization of the CypA and Anle138b Isomer Complex
2.4. Molecular Dinamics: Characterization of the CypA and Anle138b Isomer Complex
3. Discussion
4. Materials and Methods
4.1. Objects of Study
4.2. Molecular Docking
4.3. Molecular Dynamics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Khoor, A.; Colby, T.V. Amyloidosis of the Lung. Arch. Pathol. Lab. Med. 2017, 141, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Snyder, P.W. Chapter 5—Diseases of Immunity1. In Pathologic Basis of Veterinary Disease, 6th ed.; Zachary, J.F., Ed.; Mosby: Maryland Heights, MO, USA, 2017; pp. 242–285.e5. ISBN 978-0-323-35775-3. [Google Scholar]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2020: Update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2020, 27, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease with the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, E.; Chandrasekhar, G.; Chandrasekar, P.; Anbarasu, K.; Vickram, A.S.; Karunakaran, R.; Rajasekaran, R.; Srikumar, P.S. Alpha-Synuclein Aggregation in Parkinson’s Disease. Front. Med. 2021, 8, 736978. [Google Scholar] [CrossRef]
- Vidović, M.; Rikalovic, M.G. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells 2022, 11, 1732. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef]
- Georgieva, E.R.; Ramlall, T.F.; Borbat, P.P.; Freed, J.H.; Eliezer, D. Membrane-Bound Alpha-Synuclein Forms an Extended Helix: Long-Distance Pulsed ESR Measurements Using Vesicles, Bicelles, and Rod-Like Micelles. J. Am. Chem. Soc. 2008, 130, 12856–12857. [Google Scholar] [CrossRef]
- de Oliveira, G.A.P.; Silva, J.L. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2019, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Chen, S.W.; Dobson, C.M. Structural Characteristics of α-Synuclein Oligomers. Int. Rev. Cell Mol. Biol. 2017, 329, 79–143. [Google Scholar]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Iljina, M.; Garcia, G.A.; Horrocks, M.H.; Tosatto, L.; Choi, M.L.; Ganzinger, K.A.; Abramov, A.Y.; Gandhi, S.; Wood, N.W.; Cremades, N.; et al. Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc. Natl. Acad. Sci. USA 2016, 113, E1206–E1215. [Google Scholar] [CrossRef] [PubMed]
- Pujols, J.; Peña-Díaz, S.; Pallarès, I.; Ventura, S. Chemical Chaperones as Novel Drugs for Parkinson’s Disease. Trends Mol. Med. 2020, 26, 408–421. [Google Scholar] [CrossRef]
- Levin, J.; Sing, N.; Melbourne, S.; Morgan, A.; Mariner, C.; Spillantini, M.G.; Wegrzynowicz, M.; Dalley, J.W.; Langer, S.; Ryazanov, S.; et al. Safety, tolerability and pharmacokinetics of the oligomer modulator anle138b with exposure levels sufficient for therapeutic efficacy in a murine Parkinson model: A randomised, double-blind, placebo-controlled phase 1a trial. eBioMedicine 2022, 80, 104021. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Kuzdas-Wood, D.; Levin, J.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b Partly Ameliorates Motor Deficits Despite Failure of Neuroprotection in a Model of Advanced Multiple System Atrophy. Front. Neurosci. 2016, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Antonschmidt, L.; Matthes, D.; Dervişoğlu, R.; Frieg, B.; Dienemann, C.; Leonov, A.; Nimerovsky, E.; Sant, V.; Ryazanov, S.; Giese, A.; et al. The clinical drug candidate anle138b binds in a cavity of lipidic α-synuclein fibrils. Nat. Commun. 2022, 13, 5385. [Google Scholar] [CrossRef]
- Peña-Díaz, S.; Pujols, J.; Vasili, E.; Pinheiro, F.; Santos, J.; Manglano-Artuñedo, Z.; Outeiro, T.F.; Ventura, S. The small aromatic compound SynuClean-D inhibits the aggregation and seeded polymerization of multiple α-synuclein strains. J. Biol. Chem. 2022, 298, 101902. [Google Scholar] [CrossRef]
- Pujols, J.; Peña-Díaz, S.; Lázaro, D.F.; Peccati, F.; Pinheiro, F.; González, D.; Carija, A.; Navarro, S.; Conde-Giménez, M.; García, J.; et al. Small molecule inhibits α-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef]
- Wrasidlo, W.; Tsigelny, I.F.; Price, D.L.; Dutta, G.; Rockenstein, E.; Schwarz, T.C.; Ledolter, K.; Bonhaus, D.; Paulino, A.; Eleuteri, S.; et al. A de novo compound targeting α-synuclein improves deficits in models of Parkinson’s disease. Brain 2016, 139, 3217–3236. [Google Scholar] [CrossRef] [PubMed]
- Prots, I.; Grosch, J.; Brazdis, R.-M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O.; et al. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [PubMed]
- Price, D.L.; Koike, M.A.; Khan, A.; Wrasidlo, W.; Rockenstein, E.; Masliah, E.; Bonhaus, D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018, 8, 16165. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.-X.; Kalmar, L.; Tompa, P.; Han, K.-H.; Selenko, P.; Dunker, A.K.; Daughdrill, G.W.; Uversky, V.N. The alphabet of intrinsic disorder: I. Act like a Pro: On the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord. Proteins 2013, 1, e24360. [Google Scholar] [CrossRef]
- Williams, R.M.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: Amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 2001, 89–100. [Google Scholar] [CrossRef]
- Rudnev, V.R.; Kulikova, L.I.; Nikolsky, K.S.; Malsagova, K.A.; Kopylov, A.T.; Kaysheva, A.L. Current Approaches in Supersecondary Structures Investigation. Int. J. Mol. Sci. 2021, 22, 11879. [Google Scholar] [CrossRef]
- Favretto, F.; Baker, J.D.; Strohäker, T.; Andreas, L.B.; Blair, L.J.; Becker, S.; Zweckstetter, M. The Molecular Basis of the Interaction of Cyclophilin A with α-Synuclein. Angew. Chem. Int. Ed Engl. 2020, 59, 5643–5646. [Google Scholar] [CrossRef]
- Daneri-Becerra, C.; Ciucci, S.M.; Mazaira, G.; Galigniana, M.D. Role of Mitochondrial Heat-shock Proteins and Immunophilins in Neuro Degenerative Diseases. Curr. Drug Targets 2021, 22, 1596–1617. [Google Scholar] [CrossRef]
- Zhang, X.-C.; Wang, W.-D.; Wang, J.-S.; Pan, J.-C.; Zou, G.-L. Evidences of monomer, dimer and trimer of recombinant human cyclophilin A. Protein Pept. Lett. 2011, 18, 1188–1193. [Google Scholar] [CrossRef]
- Lutomski, C.A.; El-Baba, T.J.; Bolla, J.R.; Robinson, C.V. Multiple Roles of SARS-CoV-2 N Protein Facilitated by Proteoform-Specific Interactions with RNA, Host Proteins, and Convalescent Antibodies. JACS Au 2021, 1, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.M.; Sboner, A.; Xia, Y.; Gerstein, M. The role of disorder in interaction networks: A structural analysis. Mol. Syst. Biol. 2008, 4, 179. [Google Scholar] [CrossRef] [PubMed]
- Gandass, N.; Kajal; Salvi, P. Intrinsically disordered protein, DNA binding with one finger transcription factor (OsDOF27) implicates thermotolerance in yeast and rice. Front. Plant Sci. 2022, 13, 956299. [Google Scholar] [CrossRef] [PubMed]
- Wallin, S. Intrinsically disordered proteins: Structural and functional dynamics. Res. Rep. Biol. 2017, 8, 7–16. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins in Chronic Diseases. Biomolecules 2019, 9, 147. [Google Scholar] [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Hirano, M.; Stoessl, A.J.; Imamichi, Y.; Ikeda, A.; Li, Y.; Funayama, M.; Yamada, I.; Nakamura, Y.; Sossi, V.; et al. Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol. Aging 2017, 57, 248.e7–248.e12. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Koros, C.; Strohäker, T.; Schulte, C.; Bozi, M.; Varvaresos, S.; Ibáñez de Opakua, A.; Simitsi, A.M.; Bougea, A.; Voumvourakis, K.; et al. A Novel SNCA A30G Mutation Causes Familial Parkinson’s Disease. Mov. Disord. 2021, 36, 1624–1633. [Google Scholar] [CrossRef]
- Oliveira, L.M.A.; Gasser, T.; Edwards, R.; Zweckstetter, M.; Melki, R.; Stefanis, L.; Lashuel, H.A.; Sulzer, D.; Vekrellis, K.; Halliday, G.M.; et al. Alpha-synuclein research: Defining strategic moves in the battle against Parkinson’s disease. Npj Park. Dis. 2021, 7, 1–23. [Google Scholar] [CrossRef]
- Lin, W.; Bonin, M.; Boden, A.; Wieduwild, R.; Murawala, P.; Wermke, M.; Andrade, H.; Bornhäuser, M.; Zhang, Y. Peptidyl prolyl cis/trans isomerase activity on the cell surface correlates with extracellular matrix development. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Dominguez-Meijide, A.; Vasili, E.; König, A.; Cima-Omori, M.-S.; Ibáñez de Opakua, A.; Leonov, A.; Ryazanov, S.; Zweckstetter, M.; Griesinger, C.; Outeiro, T.F. Effects of pharmacological modulators of α-synuclein and tau aggregation and internalization. Sci. Rep. 2020, 10, 12827. [Google Scholar] [CrossRef]
- Braun, A.R.; Liao, E.E.; Horvath, M.; Kalra, P.; Acosta, K.; Young, M.C.; Kochen, N.N.; Lo, C.H.; Brown, R.; Evans, M.D.; et al. Potent inhibitors of toxic alpha-synuclein identified via cellular time-resolved FRET biosensors. Npj Park. Dis. 2021, 7, 1–17. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinf. Oxf. Engl. 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- In Silico Study of 3-Hydroxypyrimidine-2,4-diones as Inhibitors of HIV RT-Associated RNase H Using Molecular Docking, Molecular Dynamics, 3D-QSAR, and Pharmacophore Models—New Journal of Chemistry (RSC Publishing). Available online: https://pubs.rsc.org/en/content/articlelanding/2019/nj/c9nj03353j (accessed on 1 December 2022).
- Protein-Ligand Complex. Available online: http://www.mdtutorials.com/gmx/complex/index.html (accessed on 1 December 2022).
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Sajid Jamal, Q.M.; Alharbi, A.H.; Ahmad, V. Identification of doxorubicin as a potential therapeutic against SARS-CoV-2 (COVID-19) protease: A molecular docking and dynamics simulation studies. J. Biomol. Struct. Dyn. 2022, 40, 7960–7974. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
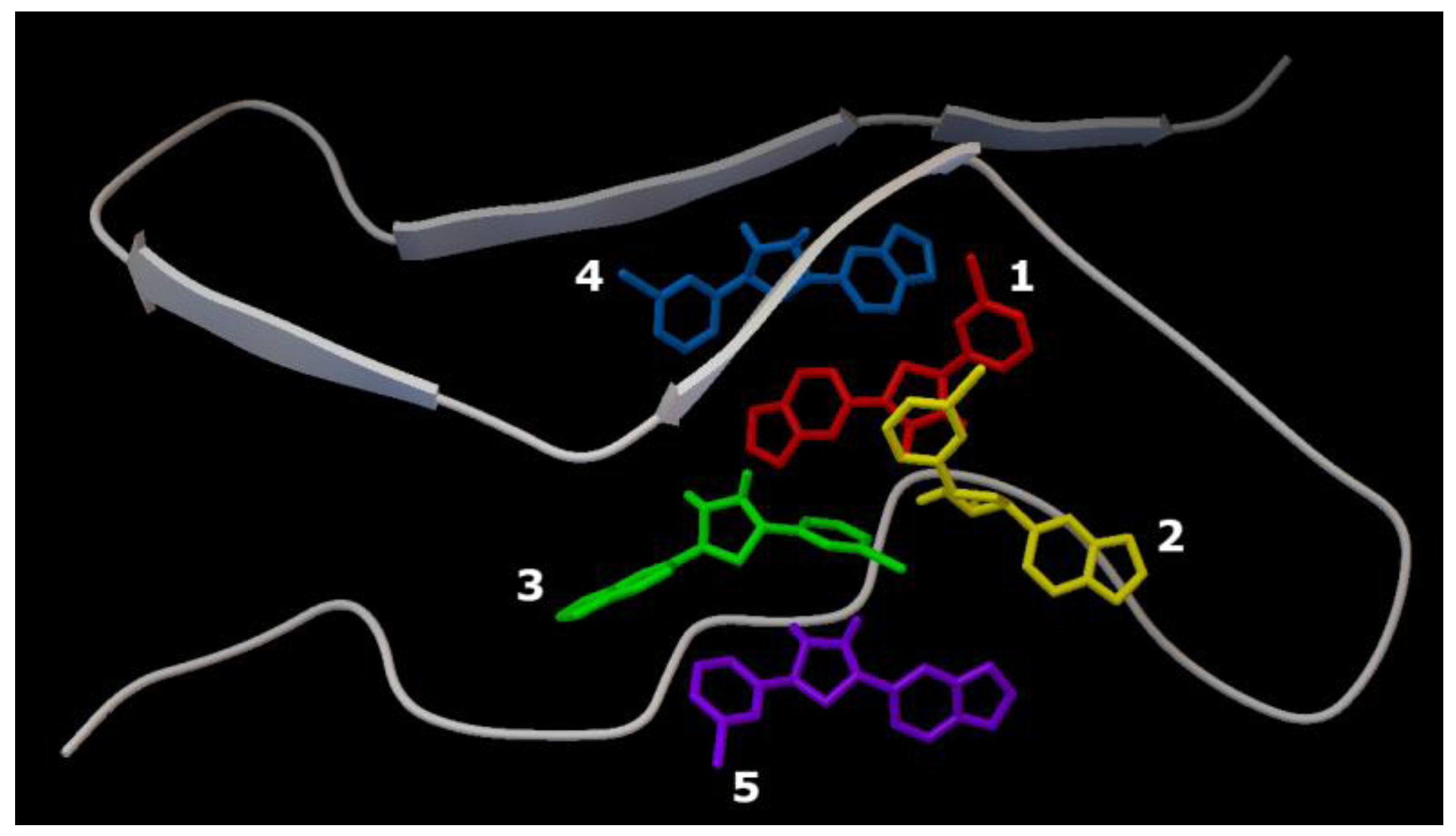{kind=link}
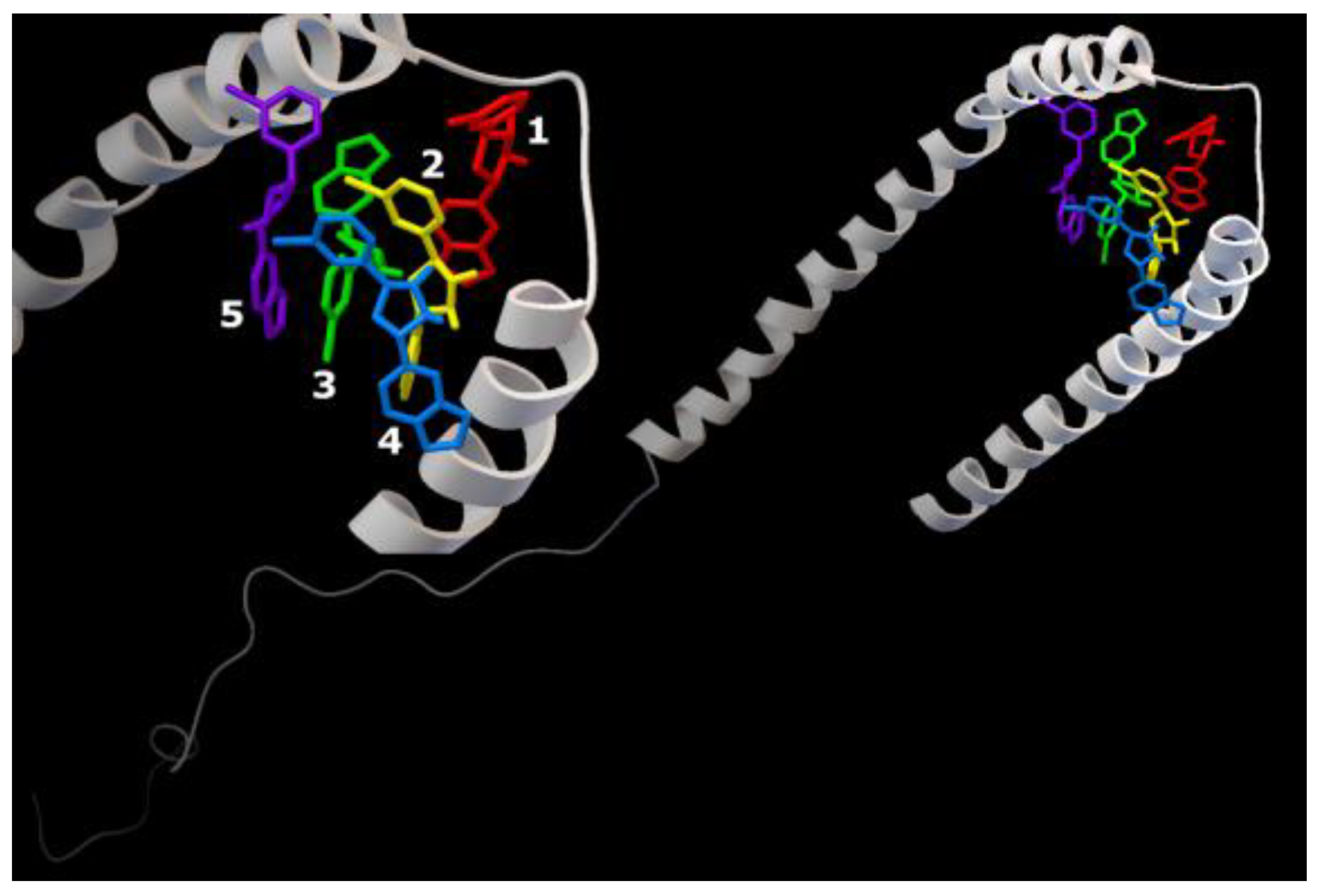{kind=link}
{kind=link}
{kind=link}
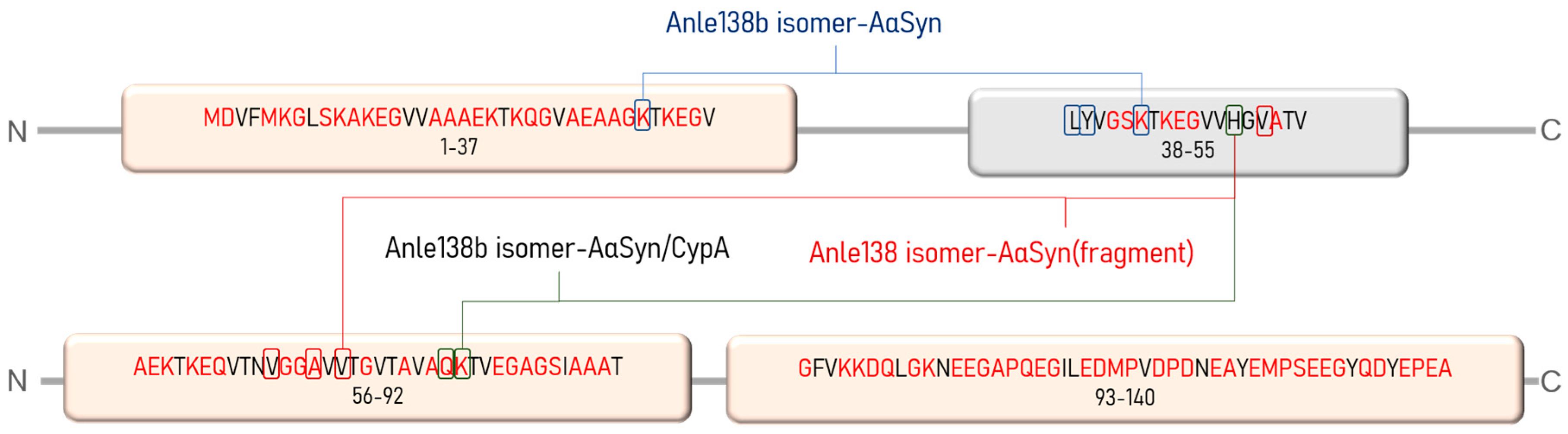{kind=link}
{kind=link}
| Interactions | Residue | AA | Distance, Å | Angle | Ligand Atom | Affinity (kcal/mol) |
|---|---|---|---|---|---|---|
| First ligand | ||||||
| Hydrophobic | 66A | Val | 3.86 | – | 95 | −5.8 |
| 66A | Val | 3.60 | – | 95 | ||
| 69 A | Ala | 3.77 | – | 95 | ||
| 71 A | Val | 3.61 | – | 115 | ||
| 71 A | Val | 3.87 | – | 108 | ||
| Hydrogen bonds | 50 A | His | 3.35 | 121.47 | – | |
| 52 A | Val | 2.18 | 156.21 | – | ||
| 52 A | Val | 1.84 | 157.17 | – | ||
| Second ligand | ||||||
| Hydrophobic | 53 A | Ala | 3.58 | – | 84 | −6.9 |
| 55 A | Val | 3.94 | – | 91 | ||
| 71 A | Val | 3.63 | – | 75 | ||
| 71 A | Val | 3.77 | – | 76 | ||
| Hydrogen bonds | 50 A | His | 2.50 | 113.40 | – | |
| 52 A | Val | 2.30 | 130.98 | – | ||
| 54 A | Thr | 3.00 | 136.63 | – | ||
| Third ligand | ||||||
| Hydrophobic | 47 A | Gly | 3.40 | 122.20 | – | −6.3 |
| 72 A | Thr | 2.41 | 114.07 | – | ||
| 72 A | Thr | 2.91 | 105.00 | – | ||
| Fourth ligand | ||||||
| Hydrophobic | 52 A | Val | 3.48 | – | 45 | −6.3 |
| 66 A | Val | 3.60 | – | 45 | ||
| 72 A | Thr | 3.92 | – | 30 | ||
| Hydrogen bonds | 70 A | Val | 2.13 | 149.27 | – | |
| Fifth ligand | ||||||
| Hydrophobic | 46 A | Glu | 3.53 | – | 3 | −5.7 |
| 49 A | Val | 3.64 | – | 23 | ||
| Hydrogen bonds | 48 A | Val | 2.11 | 163.86 | – | |
| π-cation | 50 A | His | 4.16 | – | 17–21 | |
| Interactions | Residue | AA | Distance, Å | Angle | Ligand Atom | Affinity (kcal/mol) |
|---|---|---|---|---|---|---|
| First ligand | ||||||
| Hydrophobic | 40 A | Val | 3.94 | – | 108 | −6.7 |
| 43 A | Lys | 3.84 | – | 114 | ||
| Hydrogen bonds | 32 A | Lys | 3.40 | 111.29 | – | |
| 40 A | Val | 3.36 | 149.31 | – | ||
| 40 A | Val | 2.61 | 108.87 | – | ||
| 43 A | Lys | 2.56 | 112.70 | – | ||
| π-stacking | 39 A | Tyr | 4.71 | 78.53 | 94–99 | |
| π-cation | 43 A | Lys | 5.48 | – | 107–109, 111, 114, 115 | |
| 43 A | Lys | 5.43 | – | 109–113 | ||
| Second ligand | ||||||
| Hydrophobic | 33 A | Thr | 3.79 | – | 91 | −5.8 |
| Hydrogen bonds | 32 A | Lys | 2.47 | 138.24 | – | |
| 36 A | Gly | 2.51 | 128.51 | – | ||
| Third ligand | ||||||
| Hydrophobic | 43 A | Lys | 3.96 | – | 62 | −6.2 |
| 48 A | Val | 3.79 | – | 68 | ||
| 48 A | Val | 3.96 | – | 62 | ||
| 52 A | Val | 3.90 | – | 68 | ||
| Hydrogen bonds | 32 A | Lys | 2.37 | 129.57 | – | |
| 52 A | Val | 3.29 | 112.57 | – | ||
| Fourth ligand | ||||||
| Hydrophobic | 45 A | Lys | 3.76 | – | 6 | −5.5 |
| 48 A | Val | 3.55 | – | 6 | ||
| 49 A | Val | 3.99 | – | 7 | ||
| Interactions | Residue | AA | Distance, Å | Angle | Ligand Atom | Affinity (kcal/mol) |
|---|---|---|---|---|---|---|
| First ligand | ||||||
| Hydrophobic | 81 A | Glu | 3.58 | – | 107 | −7.1 |
| 82 A | Lys | 3.63 | – | 108 | ||
| 107 A | Thr | 3.68 | – | 96 | ||
| Hydrogen bonds | 81 A | Glu | 2.07 | 171.41 | – | |
| 82 A | Lys | 3.13 | 146.9 | – | ||
| 82 A | Lys | 2.43 | 119.68 | – | ||
| π-stacking | 50 B | His | 4.00 | 15.64 | 107–111, 114, 115 | |
| Second ligand | ||||||
| Hydrophobic | 76 A | Lys | 3.92 | – | 73 | −7.0 |
| 76 A | Lys | 3.74 | – | 72 | ||
| Hydrogen bonds | 68 A | Thr | 2.33 | 128.37 | – | |
| 75 A | Gly | 3.49 | 108.72 | – | ||
| 75 A | Gly | 2.54 | 109.73 | – | ||
| Halogen bonds | 80 A | Gly | 3.72 | 150.45 | – | |
| Third ligand | ||||||
| Hydrophobic | 46 A | Phe | 3.79 | – | 52 | −7.0 |
| 49 A | Lys | 3.24 | – | 62 | ||
| 67 A | Phe | 3.47 | – | 52 | ||
| Hydrogen bonds | 47 A | Gly | 2.94 | 115.96 | – | |
| 47 A | Gly | 2.73 | 111.32 | – | ||
| 49 A | Lys | 2.52 | 129.52 | – | ||
| 49 A | Lys | 2.94 | 108.80 | – | ||
| π-cation | 49 A | Lys | 4.74 | – | 63–97 | |
| Fourth ligand | ||||||
| Hydrophobic | 26 A | Ala | 3.74 | – | 45 | −6.7 |
| 31 A | Lys | 3.44 | – | 625 | ||
| 79 A | Tyr | 3.57 | – | 29 | ||
| π-cation | 37 A | Arg | 5.36 | – | 40–44 | |
| Fifth ligand | ||||||
| Hydrophobic | 88 A | Phe | 3.72 | – | 4 | −6.6 |
| 105 A | Pro | 3.48 | – | 2 | ||
| 125 A | Lys | 3.94 | – | 15 | ||
| 125 A | Lys | 3.62 | – | 23 | ||
| 125 A | Lys | 3.59 | – | 14 | ||
| Hydrogen bonds | 85 A | Asp | 3.18 | 121.95 | – | |
| 102 A | Asn | 3.26 | 127.71 | – | ||
| 104 A | Gly | 3.06 | 154.03 | – | ||
| Ligand Anle138b and Target | Van der Waal Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | SASA Energy (kJ/mol) | Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| AαSyn–CypA complex (PDB ID 6I42) | −67.96 ± 34.5 | −26.05 ± 29.6 | 53.2 ± 34.47 | −7.7 ± 3.85 | −48.44 ± 26.11 |
| CypA dimer | −151.56 ± 18.9 | −34.54 ± 13.6 | 102.59 ± 18.1 | −15 ± 1.33 | −98.53 ± 22.74 |
| Dimer of the AαSyn–CypA complex | −91.36 ± 33.8 | −16.58 ± 2.3 | 71.63 ± 10.2 | −9.87 ± 3.36 | −46.11 ± 24.69 |
| AαSyn (filament, PDB ID 7V4C) | −140.93 ± 47.68 | −14.66 ± 5 | 78.87 ± 28.95 | −14.37 ± 3.32 | −91 ± 27.29 |
| Ligand | IUPAC (Formula) | Class | Mw, Da | Donor HB * | Acceptor HB | SASA **, Å2 | Dipole Moment, Debay | Sizes | Clinical Phase | IC50, nM *** | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Anle138b isomer | (3-(1,3-benzodioxol-5-yl)-5-(3-bromophenyl)-1H-pyrazolidine | pyrazolidine | 347.21 | 2 | 4 | 313.74 | 1.61 | 13.514 Å 6.402 Å 4.381 Å | Preclinical | 900 | [51] |
| Target | PDB ID (WebLink) | Uniprot ID | Shape of Protein | Resolution, Å | Ligand (Locus, a.a.) | Method | Length, a.a. |
|---|---|---|---|---|---|---|---|
| AαSyn (micelle-bounded) | 1XQ8 (https://www.rcsb.org/structure/1XQ8, accessed on 25 November 2022) | P37840 | IDP * | – | – | NMR ** | 140 |
| AαSyn (filament) | 7V4C (https://www.rcsb.org/structure/7V4C, accessed on 25 November 2022) | P37840 | 3.30 | – | EM *** | 140 **** | |
| CypA | 6I42 (https://www.rcsb.org/structure/6I42, accessed on 25 November 2022) | P62937 | Globular | 1.38 | AαSyn (1–13) | X-ray | 164 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondratyev, M.S.; Rudnev, V.R.; Nikolsky, K.S.; Petrovsky, D.V.; Kulikova, L.I.; Malsagova, K.A.; Stepanov, A.A.; Kopylov, A.T.; Kaysheva, A.L. In Silico Study of the Interactions of Anle138b Isomer, an Inhibitor of Amyloid Aggregation, with Partner Proteins. Int. J. Mol. Sci. 2022, 23, 16096. https://doi.org/10.3390/ijms232416096
Kondratyev MS, Rudnev VR, Nikolsky KS, Petrovsky DV, Kulikova LI, Malsagova KA, Stepanov AA, Kopylov AT, Kaysheva AL. In Silico Study of the Interactions of Anle138b Isomer, an Inhibitor of Amyloid Aggregation, with Partner Proteins. International Journal of Molecular Sciences. 2022; 23(24):16096. https://doi.org/10.3390/ijms232416096
Chicago/Turabian StyleKondratyev, Maxim S., Vladimir R. Rudnev, Kirill S. Nikolsky, Denis V. Petrovsky, Liudmila I. Kulikova, Kristina A. Malsagova, Alexander A. Stepanov, Arthur T. Kopylov, and Anna L. Kaysheva. 2022. "In Silico Study of the Interactions of Anle138b Isomer, an Inhibitor of Amyloid Aggregation, with Partner Proteins" International Journal of Molecular Sciences 23, no. 24: 16096. https://doi.org/10.3390/ijms232416096
APA StyleKondratyev, M. S., Rudnev, V. R., Nikolsky, K. S., Petrovsky, D. V., Kulikova, L. I., Malsagova, K. A., Stepanov, A. A., Kopylov, A. T., & Kaysheva, A. L. (2022). In Silico Study of the Interactions of Anle138b Isomer, an Inhibitor of Amyloid Aggregation, with Partner Proteins. International Journal of Molecular Sciences, 23(24), 16096. https://doi.org/10.3390/ijms232416096