

Normal Pregnancy-Induced Islet Beta Cell Proliferation in Mouse Models That Are Deficient in Serotonin-Signaling

 ,
,
Abstract
1. Introduction
2. Results
2.1. Tph1KO Mice
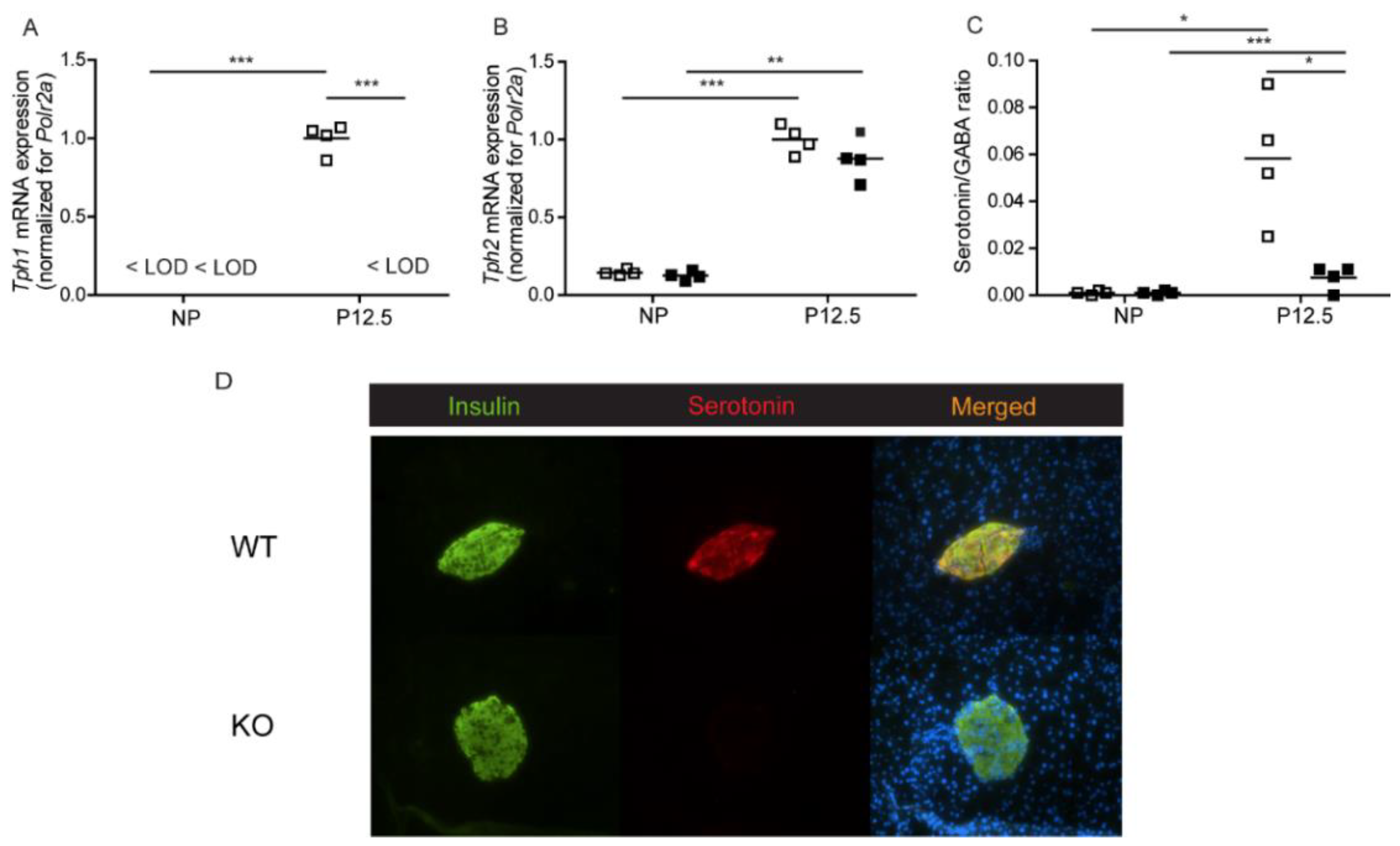
2.1.1. Serotonin Biosynthesis in Pancreatic Islets of Total Tph1KO Mice
2.1.2. Beta Cell Proliferation in Total Tph1KO Mice
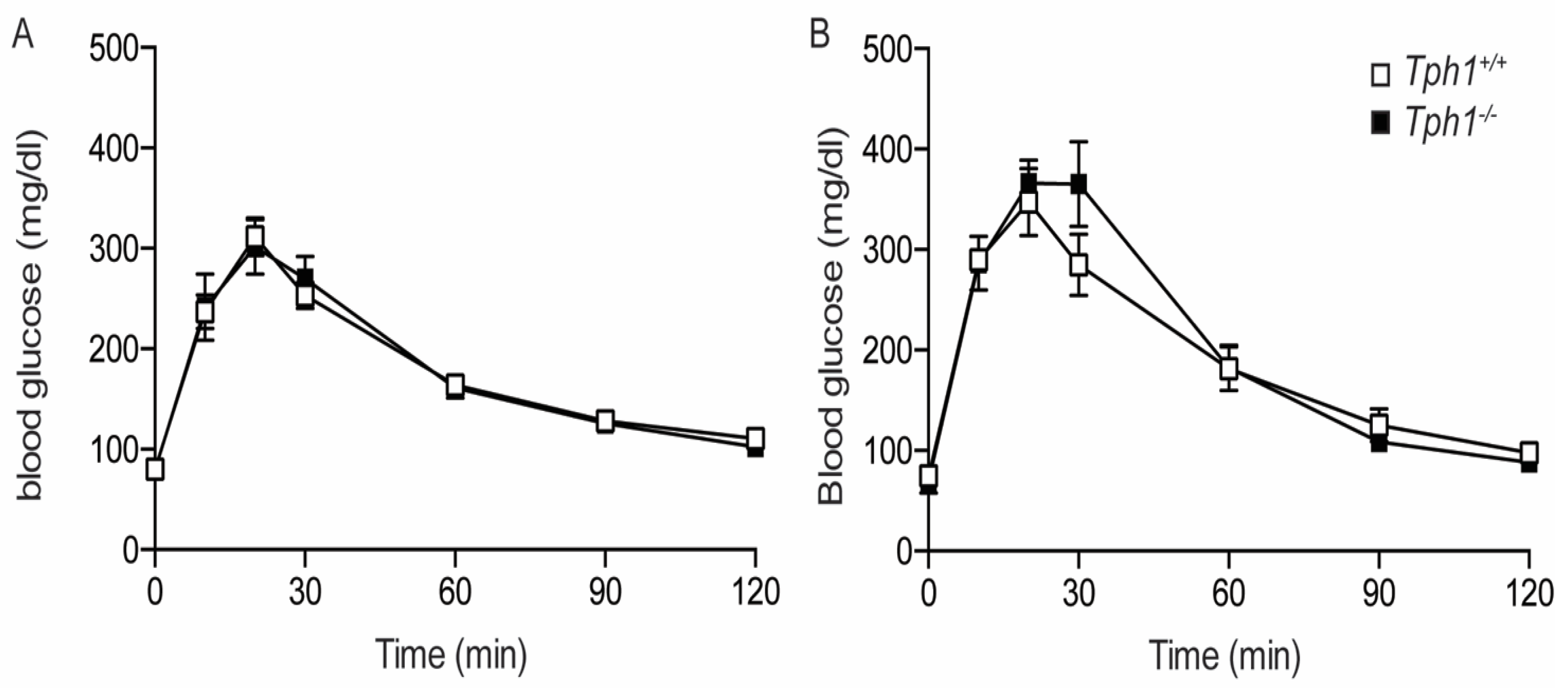
2.1.3. Glucose Tolerance in Total Tph1KO Mice
2.2. 129P2/OlaHsd Mice
2.2.1. Loss of Pregnancy-Induced Regulation Tph1 in 129P2/OlaHsd Mice
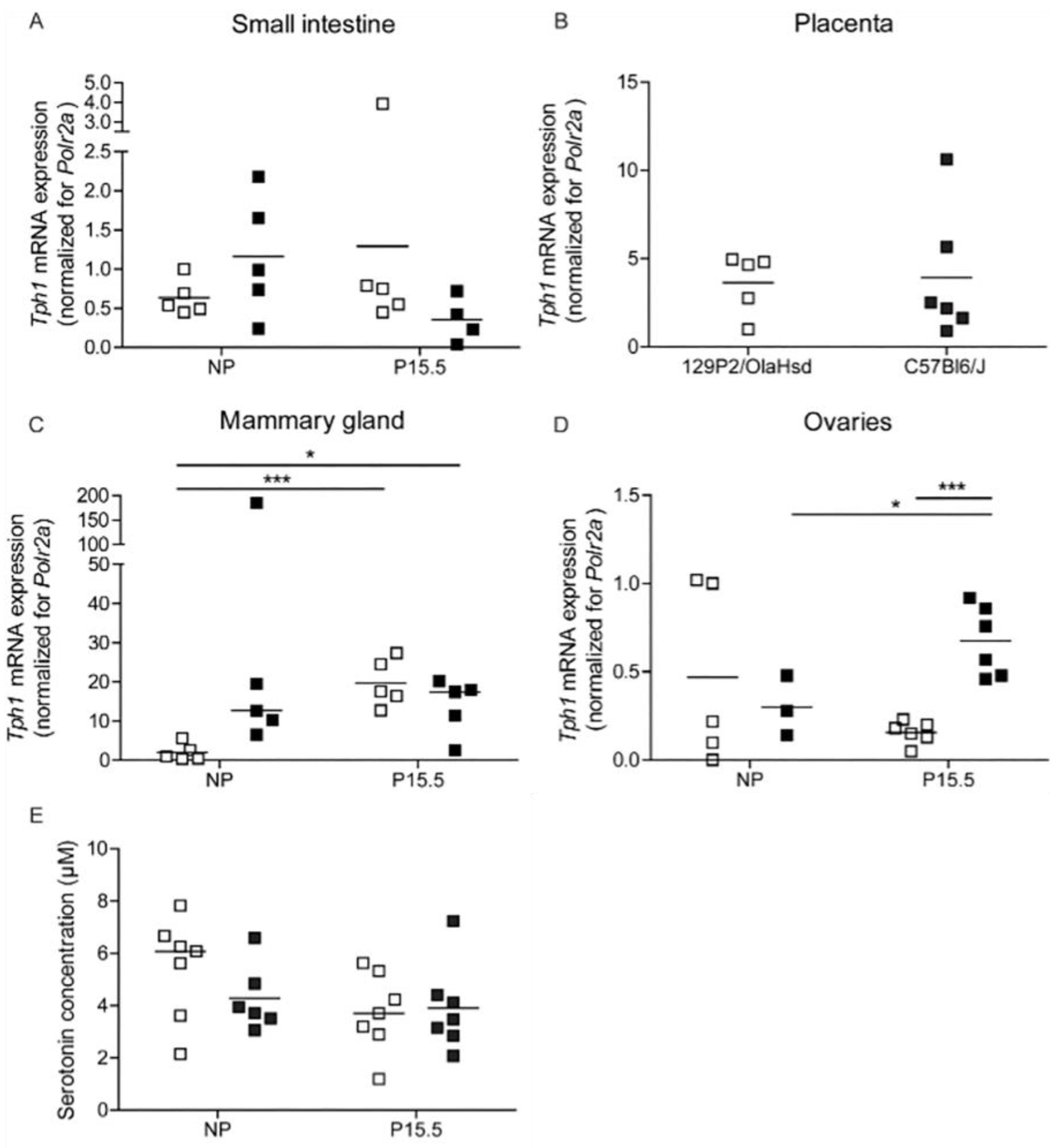
2.2.2. Tph1 Expression in Intestine, Placenta, and Mammary Glands from 129P2/OlaHsd Mice
2.2.3. Beta Cell Proliferation in Pregnant 129P2/OlaHsd Mice
2.3. Htr2b Deficient Mice
2.3.1. Low Expression Levels of Htr2b in Mouse Islets as Compared to Placenta
2.3.2. Expression of Htr2b mRNA, Tph1, and Tph2 mRNA in Htr2b Deficient Mice
2.3.3. Beta Cell Proliferation in Htr2b Deficient Mice
2.3.4. Glucose Tolerance in Htr2b Deficient Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.1.1. Mouse Models
4.1.2. Oral Glucose Tolerance Tests
4.1.3. Isolation of Mouse Pancreatic Islets
4.2. Pancreatic Islet Immunohistochemistry
4.3. Serotonin and GABA Content
4.4. RNA Extraction
4.5. Quantitative RT-PCR
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schraenen, A.; Lemaire, K.; De Faudeur, G.; Hendrickx, N.; Granvik, M.; Van Lommel, L.; Mallet, J.; Vodjdani, G.; Gilon, P.; Binart, N.; et al. Placental lactogens induce serotonin biosynthesis in a subset of mouse beta cells during pregnancy. Diabetologia 2010, 53, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Toyofuku, Y.; Lynn, F.C.; Chak, E.; Uchida, T.; Mizukami, H.; Fujitani, Y.; Kawamori, R.; Miyatsuka, T.; Kosaka, Y.; et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat. Med. 2010, 16, 804–808. [Google Scholar] [CrossRef]
- Layden, B.T.; Durai, V.; Newman, M.V.; Marinelarena, A.M.; Ahn, C.W.; Feng, G.; Lin, S.; Zhang, X.; Kaufman, D.B.; Jafari, N.; et al. Regulation of pancreatic islet gene expression in mouse islets by pregnancy. J. Endocrinol. 2010, 207, 265–279. [Google Scholar] [CrossRef]
- Rieck, S.; White, P.; Schug, J.; Fox, A.J.; Smirnova, O.; Gao, N.; Gupta, R.K.; Wang, Z.V.; Scherer, P.E.; Keller, M.P.; et al. The transcriptional response of the islet to pregnancy in mice. Mol. Endocrinol. 2009, 23, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Tyce, G.M. Origin and metabolism of serotonin. J. Cardiovasc. Pharmacol. 1990, 16 (Suppl 3), S1–S7. [Google Scholar] [CrossRef]
- Mercado, C.P.; Kilic, F. Molecular mechanisms of SERT in platelets: Regulation of plasma serotonin levels. Mol. Interv. 2010, 10, 231–241. [Google Scholar] [CrossRef]
- Lesurtel, M.; Graf, R.; Aleil, B.; Walther, D.J.; Tian, Y.; Jochum, W.; Gachet, C.; Bader, M.; Clavien, P.A. Platelet-derived serotonin mediates liver regeneration. Science 2006, 312, 104–107. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef]
- Cote, F.; Fligny, C.; Bayard, E.; Launay, J.M.; Gershon, M.D.; Mallet, J.; Vodjdani, G. Maternal serotonin is crucial for murine embryonic development. Proc. Natl. Acad. Sci. USA 2007, 104, 329–334. [Google Scholar] [CrossRef]
- Matsuda, M.; Imaoka, T.; Vomachka, A.J.; Gudelsky, G.A.; Hou, Z.; Mistry, M.; Bailey, J.P.; Nieport, K.M.; Walther, D.J.; Bader, M.; et al. Serotonin regulates mammary gland development via an autocrine-paracrine loop. Dev. Cell 2004, 6, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.H.; Svensson, C.; Galsgaard, E.D.; Møldrup, A.; Billestrup, N. Beta cell proliferation and growth factors. J. Mol. Med. 1999, 77, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Goyvaerts, L.; Schraenen, A.; Schuit, F. Serotonin competence of mouse beta cells during pregnancy. Diabetologia 2016, 59, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Cote, F.; Thévenot, E.; Fligny, C.; Fromes, Y.; Darmon, M.; Ripoche, M.A.; Bayard, E.; Hanoun, N.; Saurini, F.; Lechat, P.; et al. Disruption of the nonneuronal tph1 gene demonstrates the importance of peripheral serotonin in cardiac function. Proc. Natl. Acad. Sci. USA 2003, 100, 13525–13530. [Google Scholar] [CrossRef] [PubMed]
- Schraenen, A.; de Faudeur, G.; Thorrez, L.; Lemaire, K.; Van Wichelen, G.; Granvik, M.; Van Lommel, L.; in’t Veld, P.; Schuit, F. mRNA expression analysis of cell cycle genes in islets of pregnant mice. Diabetologia 2010, 53, 2579–2588. [Google Scholar] [CrossRef]
- Parsons, J.A.; Brelje, T.C.; Sorenson, R.L. Adaptation of islets of Langerhans to pregnancy: Increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992, 130, 1459–1466. [Google Scholar]
- Sorenson, R.L.; Brelje, T.C. Adaptation of islets of Langerhans to pregnancy: Beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm. Metab. Res. 1997, 29, 301–307. [Google Scholar] [CrossRef]
- Goyvaerts, L.; Lemaire, K.; Arijs, I.; Auffret, J.; Granvik, M.; Van Lommel, L.; Binart, N.; in’t Veld, P.; Schuit, F.; Schraenen, A. Prolactin receptors and placental lactogen drive male mouse pancreatic islets to pregnancy-related mRNA changes. PLoS ONE 2015, 10, e0121868. [Google Scholar] [CrossRef] [PubMed]
- Goffin, V.; Binart, N.; Touraine, P.; Kelly, P.A. Prolactin: The new biology of an old hormone. Annu. Rev. Physiol. 2002, 64, 47–67. [Google Scholar] [CrossRef] [PubMed]
- Nebigil, C.G.; Choi, D.S.; Dierich, A.; Hickel, P.; Le Meur, M.; Messaddeq, N.; Launay, J.M.; Maroteaux, L. Serotonin 2B receptor is required for heart development. Proc. Natl. Acad. Sci. USA 2000, 97, 9508–9513. [Google Scholar] [CrossRef]
- Iida, H.; Ogihara, T.; Min, M.; Hara, A.; Kim, Y.G.; Fujimaki, K.; Tamaki, M.; Fujitani, Y.; Kim, H.; Watada, H. Expression mechanism of tryptophan hydroxylase 1 in mouse islets during pregnancy. J. Mol. Endocrinol. 2015, 55, 41–53. [Google Scholar] [CrossRef]
- Kahle, M.; Horsch, M.; Fridrich, B.; Seelig, A.; Schultheiß, J.; Leonhardt, J.; Irmler, M.; Beckers, J.; Rathkolb, B.; Wolf, E.; et al. Phenotypic comparison of common mouse strains developing high-fat diet-induced hepatosteatosis. Mol. Metab. 2013, 2, 435–446. [Google Scholar] [CrossRef]
- Roseboom, P.H.; Namboodiri, M.A.; Zimonjic, D.B.; Popescu, N.C.; Rodriguez, I.R.; Gastel, J.A.; Klein, D.C. Natural melatonin ‘knockdown’ in C57BL/6J mice: Rare mechanism truncates serotonin N-acetyltransferase. Brain Res. Mol. Brain Res. 1998, 63, 189–197. [Google Scholar] [CrossRef]
- Loric, S.; Launay, J.M.; Colas, J.F.; Maroteaux, L. New mouse 5-HT2-like receptor. Expression in brain, heart and intestine. FEBS Lett. 1992, 312, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Toye, A.A.; Lippiat, J.D.; Proks, P.; Shimomura, K.; Bentley, L.; Hugill, A.; Mijat, V.; Goldsworthy, M.; Moir, L.; Haynes, A.; et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 2005, 48, 675–686. [Google Scholar] [CrossRef]
- Salomon, D.; Meda, P. Heterogeneity and contact-dependent regulation of hormone secretion by individual B cells. Exp. Cell Res. 1986, 162, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Schuit, F.C.; in’t Veld, P.A.; Pipeleers, D.G. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3865–3869. [Google Scholar] [CrossRef]
- Heimberg, H.; De Vos, A.; Vandercammen, A.; Van Schaftingen, E.; Pipeleers, D.; Schuit, F. Heterogeneity in glucose sensitivity among pancreatic beta-cells is correlated to differences in glucose phosphorylation rather than glucose transport. EMBO J. 1993, 12, 2873–2879. [Google Scholar] [CrossRef] [PubMed]
- Van Schoors, J.; Viaene, J.; Van Wanseele, Y.; Smolders, I.; Dejaegher, B.; Vander Heyden, Y.; Van Eeckhaut, A. An improved microbore UHPLC method with electrochemical detection for the simultaneous determination of low monoamine levels in in vivo brain microdialysis samples. J. Pharm. Biomed. Anal. 2016, 127, 136–146. [Google Scholar] [CrossRef]
- Smismans, A.; Schuit, F.; Pipeleers, D. Nutrient regulation of gamma-aminobutyric acid release from islet beta cells. Diabetologia 1997, 40, 1411–1415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smolders, I.; Sarre, S.; Michotte, Y.; Ebinger, G. The analysis of excitatory, inhibitory and other amino acids in rat brain microdialysates using microbore liquid chromatography. J. Neurosci. Methods 1995, 57, 47–53. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer/Probe | Sequence |
|---|---|---|
| Tph1 | Forward | 5′-ATGCACAGCACCACATTTA-3′ |
| Reverse | 5′-CAGTACGGTAAACTCACATGA-3′ | |
| Probe | 5′-(6FAM)TCAACTGTTCTCGGCTGATGTCGCAGTCA(TAMRA)-3′ | |
| Tph2 | Taqman assay | Mm00557717_m1 (Applied Biosystems) |
| Htr2b | Forward | 5′-TGCATTCATCAAGATTACAGTGGTA-3′ |
| Reverse | 5′-TCAGCTCACAGGTGACATTG-3′ | |
| Probe | 5′-(6-FAM)CAATAGGCATCGCCATCCCAGTCCCTAT(TAMRA)-3′ | |
| Polr2a | Forward | 5′-GCACCACGTCCAATGATATTGTG-3′ |
| Reverse | 5′-GGAGATGACATGGTACAGTTCTCG-3′ | |
| Probe | 5′-(6-FAM)CTTCCGCACAGCCTCAATGCCCAGT(TAMRA)-3′ | |
| mKi67 | Forward | 5′-AGTTCTACCAATCCAACTC-3′ |
| Reverse | 5′-TTGGCTTGCTTCCATCCT-3′ | |
| Probe | 5′-(6-FAM)AGACATAATAACCATCATTGACCGCTCCTT(TAMRA)-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goyvaerts, L.; Schraenen, A.; Lemaire, K.; Veld, P.i.; Smolders, I.; Maroteaux, L.; Schuit, F. Normal Pregnancy-Induced Islet Beta Cell Proliferation in Mouse Models That Are Deficient in Serotonin-Signaling. Int. J. Mol. Sci. 2022, 23, 15816. https://doi.org/10.3390/ijms232415816
Goyvaerts L, Schraenen A, Lemaire K, Veld Pi, Smolders I, Maroteaux L, Schuit F. Normal Pregnancy-Induced Islet Beta Cell Proliferation in Mouse Models That Are Deficient in Serotonin-Signaling. International Journal of Molecular Sciences. 2022; 23(24):15816. https://doi.org/10.3390/ijms232415816
Chicago/Turabian StyleGoyvaerts, Lotte, Anica Schraenen, Katleen Lemaire, Peter in’t Veld, Ilse Smolders, Luc Maroteaux, and Frans Schuit. 2022. "Normal Pregnancy-Induced Islet Beta Cell Proliferation in Mouse Models That Are Deficient in Serotonin-Signaling" International Journal of Molecular Sciences 23, no. 24: 15816. https://doi.org/10.3390/ijms232415816
APA StyleGoyvaerts, L., Schraenen, A., Lemaire, K., Veld, P. i., Smolders, I., Maroteaux, L., & Schuit, F. (2022). Normal Pregnancy-Induced Islet Beta Cell Proliferation in Mouse Models That Are Deficient in Serotonin-Signaling. International Journal of Molecular Sciences, 23(24), 15816. https://doi.org/10.3390/ijms232415816