
Untargeted NMR Metabolomics Reveals Alternative Biomarkers and Pathways in Alkaptonuria

 , , ,
, , ,  and
and 
Abstract
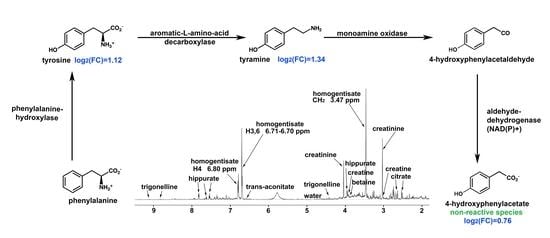
1. Introduction
2. Results and Discussion
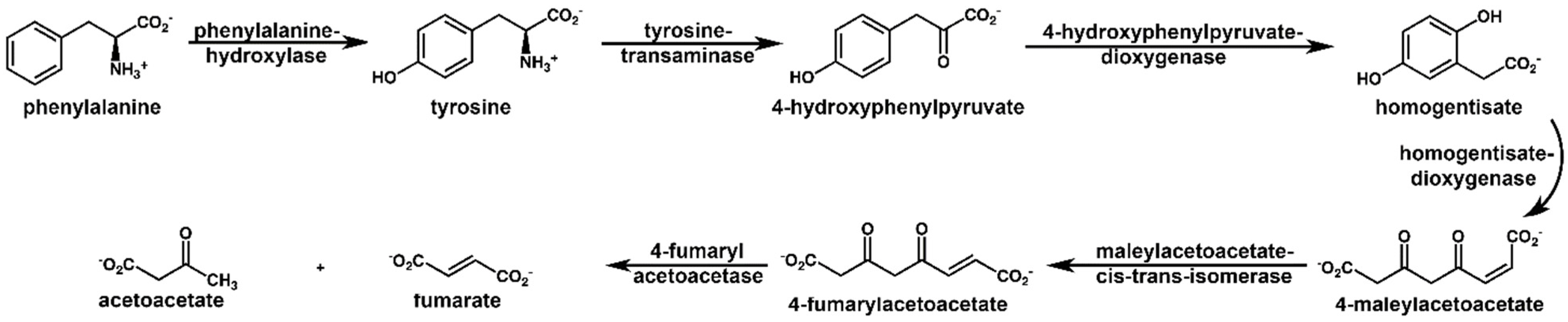
2.1. NMR Spectroscopy
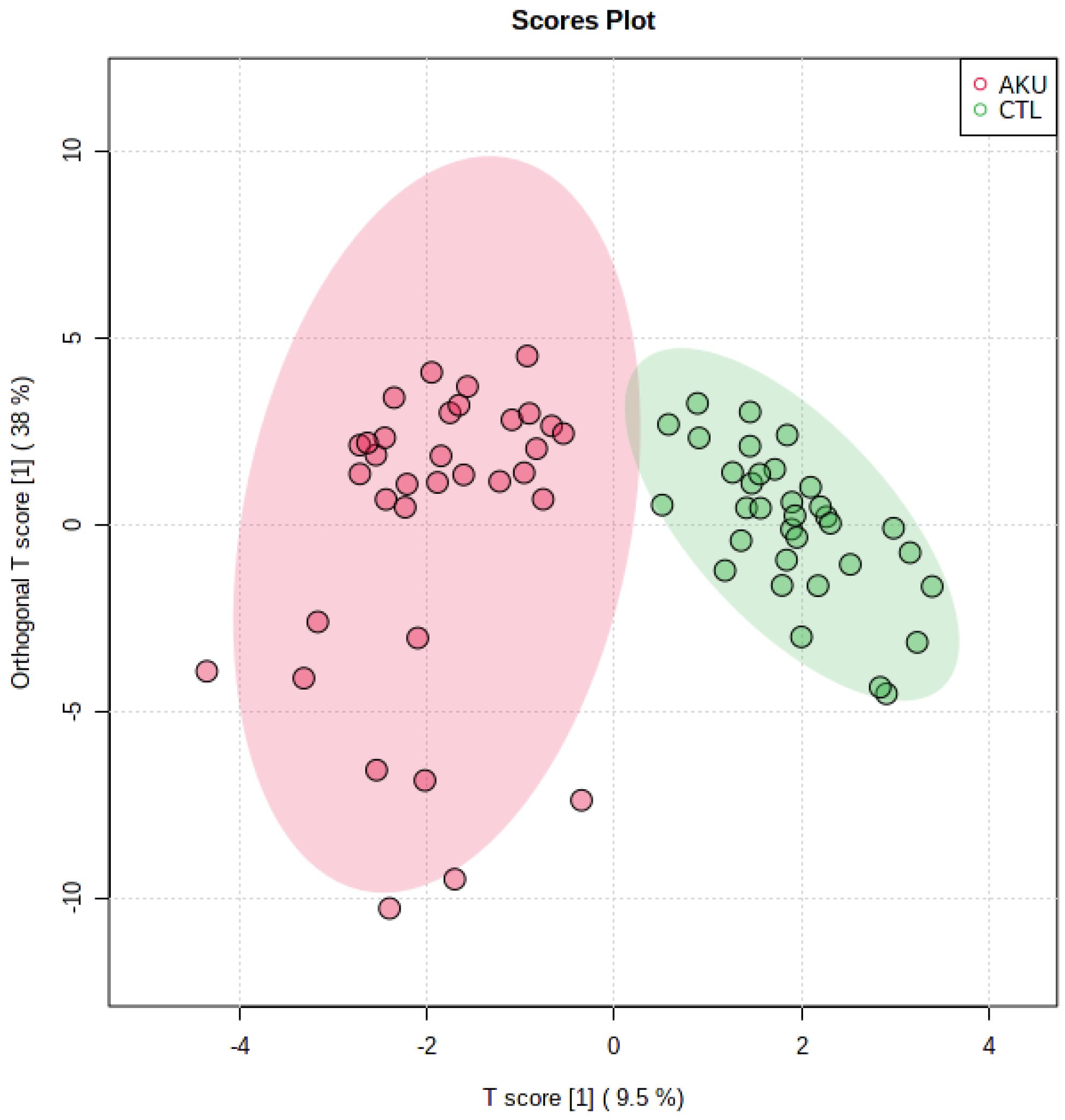
2.2. Multivariate Analysis
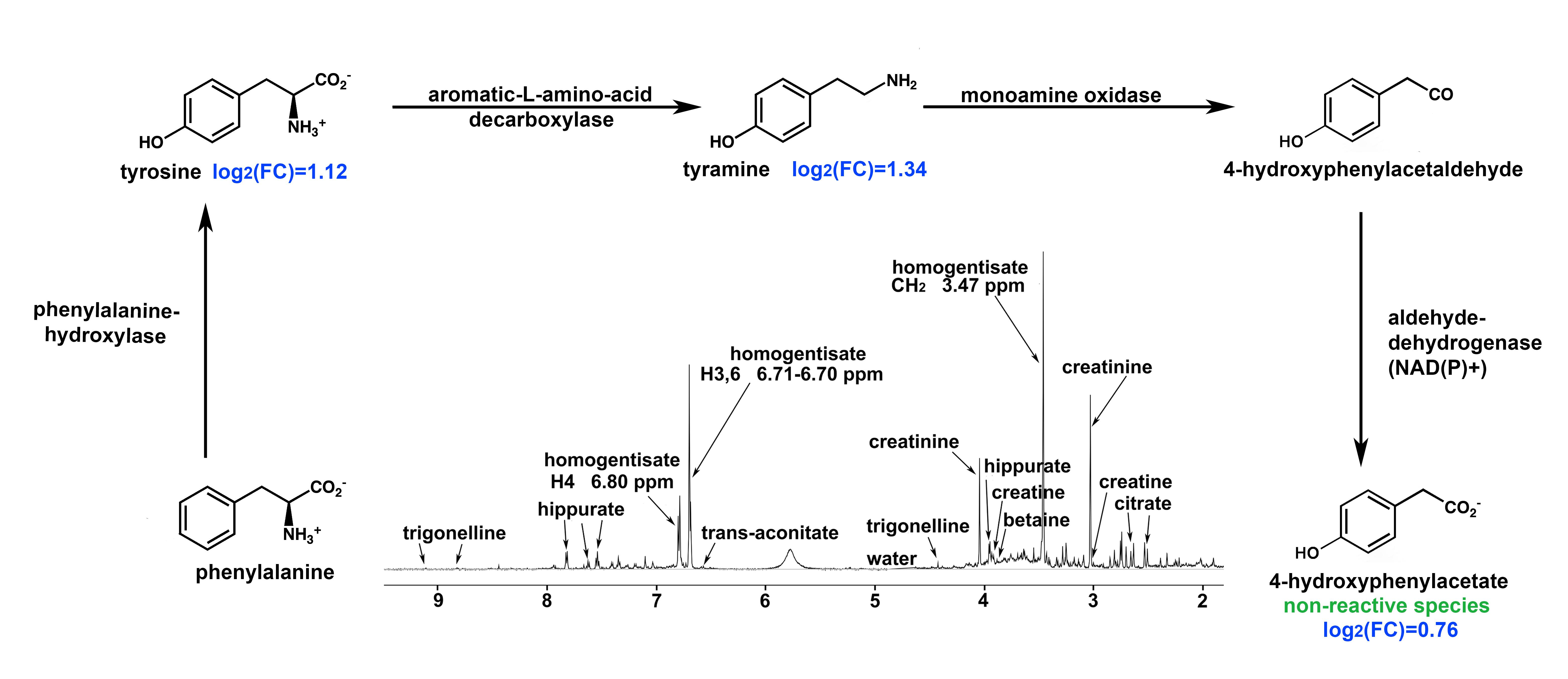
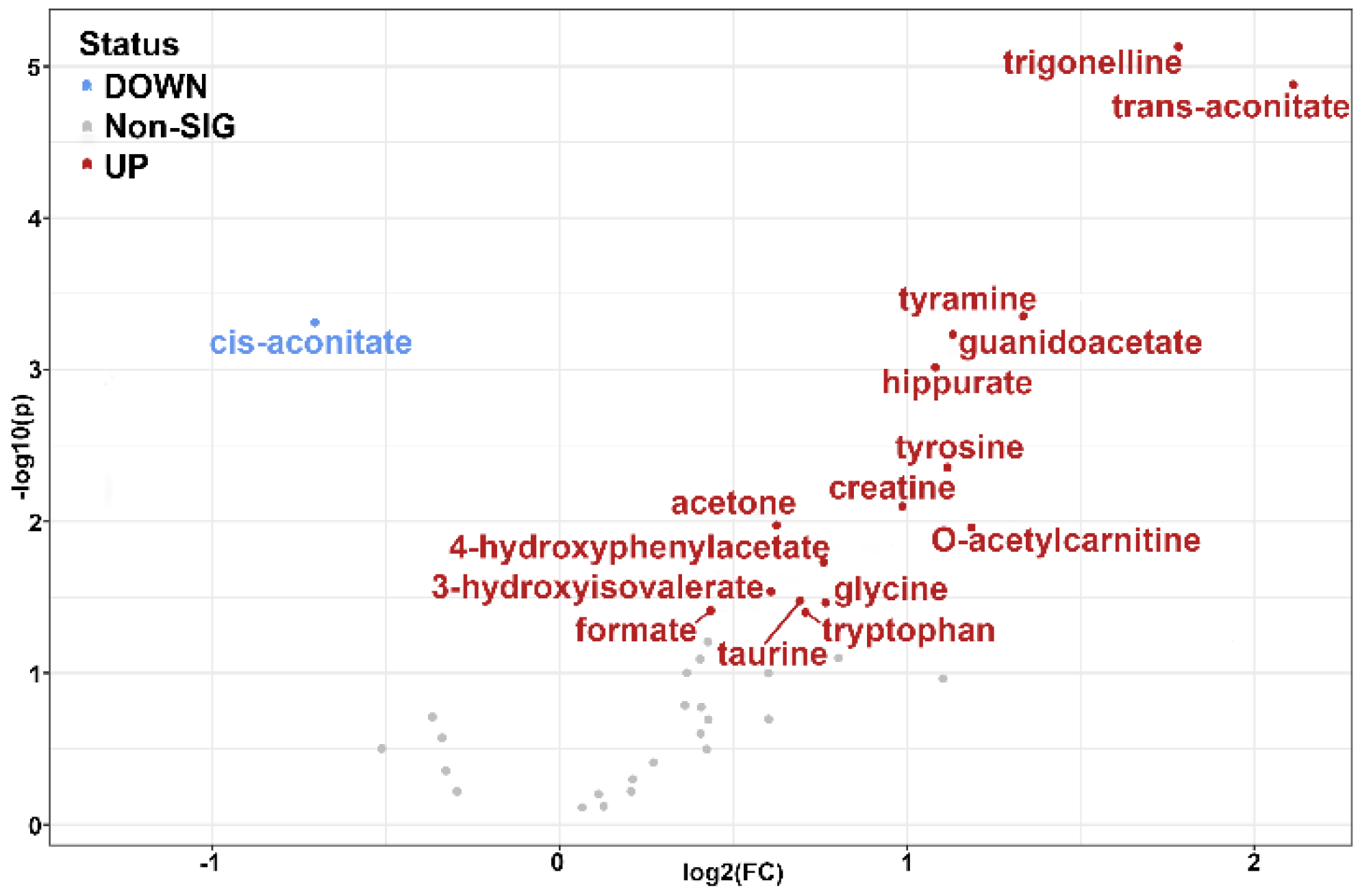
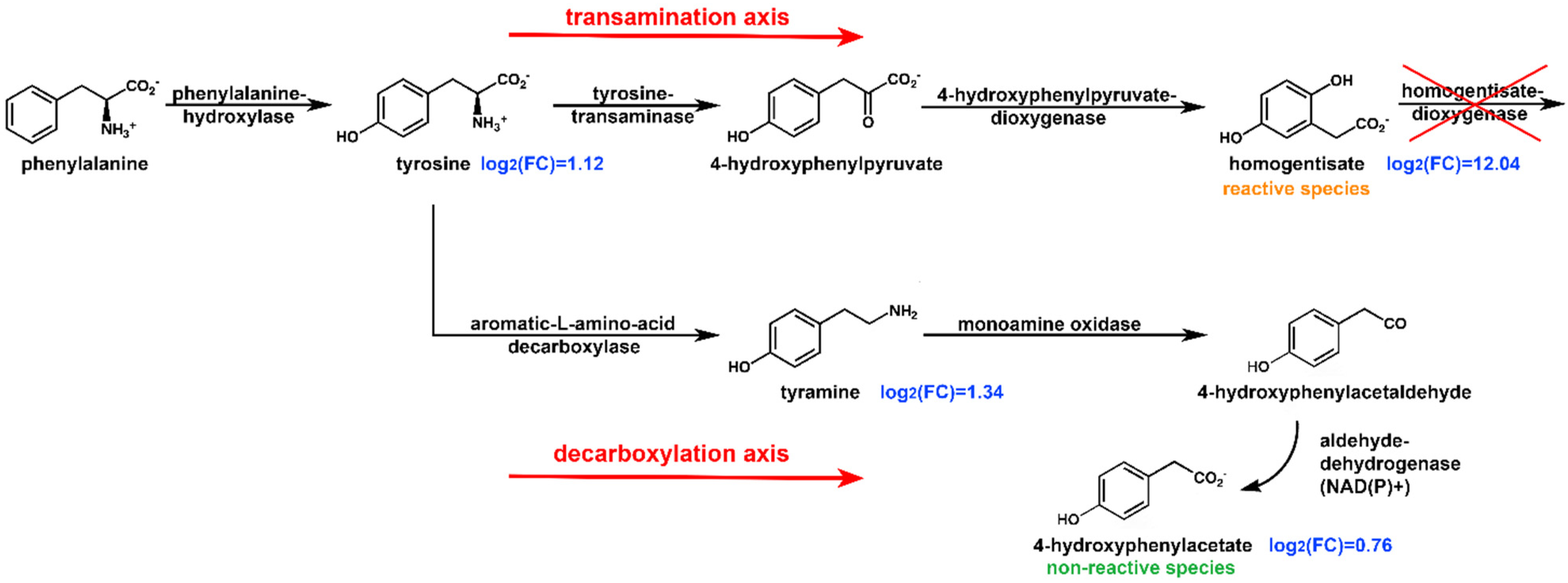
2.3. Pathway Analysis
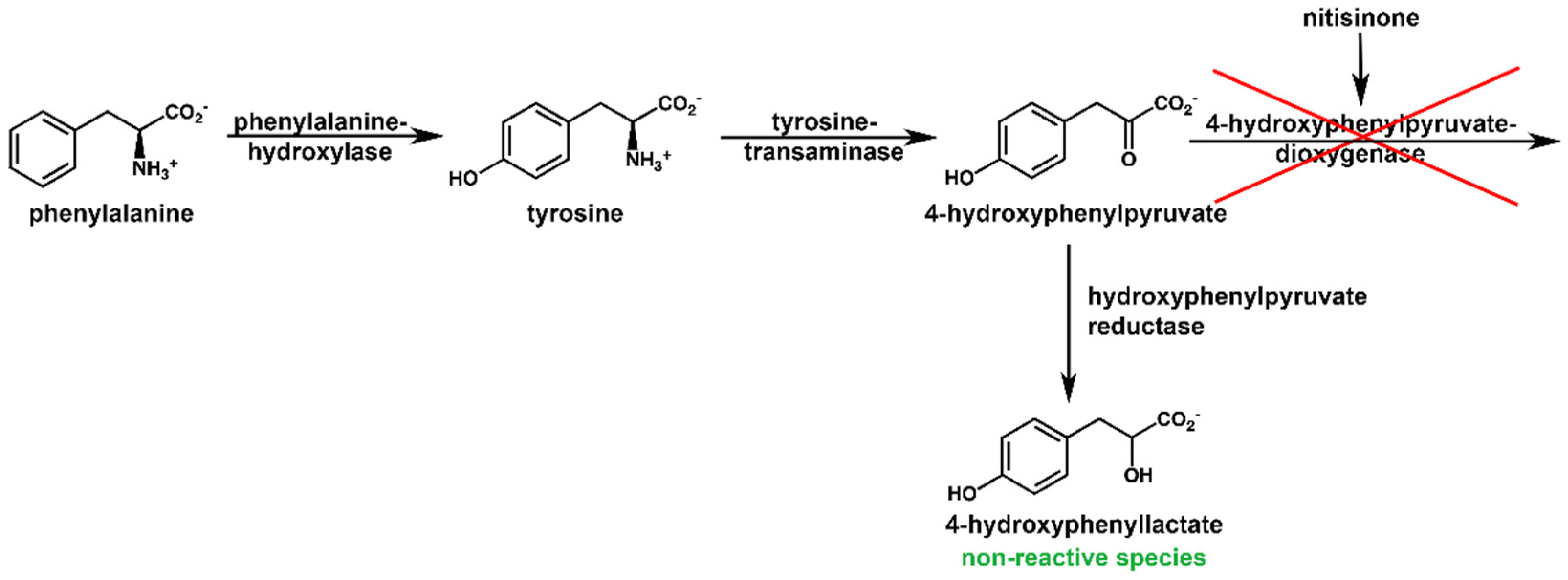
2.4. NMR Metabolic Profile of a Nitisinone-Treated Patient
2.5. Reactivity of HPAA and HPLA
3. Materials and Methods
3.1. Biological Samples
3.2. Sample Preparation
3.3. 1H NMR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zatkova, A.; Ranganath, L.; Kadasi, L. Alkaptonuria: Current Perspectives. Appl. Clin. Genet. 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Canon, J.M.; Penalva, M.A. Molecular Characterization of a Gene Encoding a Homogentisate Dioxygenase from Aspergillus Nidulans and Identification of Its Human and Plant Homologues. J. Biol. Chem 1995, 270, 21199–21205. [Google Scholar] [CrossRef]
- Zatkova, A.; Sedlackova, T.; Radvansky, J.; Polakova, H.; Nemethova, M.; Aquaron, R.; Dursun, I.; Usher, J.L.; Kadasi, L. Identification of 11 Novel Homogentisate 1,2 Dioxygenase Variants in Alkaptonuria Patients and Establishment of a Novel LOVD-Based HGD Mutation Database. JIMD Rep. 2012, 4, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Ascher, D.B.; Spiga, O.; Sekelska, M.; Pires, D.E.V.; Bernini, A.; Tiezzi, M.; Kralovicova, J.; Borovska, I.; Soltysova, A.; Olsson, B.; et al. Homogentisate 1,2-Dioxygenase (HGD) Gene Variants, Their Analysis and Genotype–Phenotype Correlations in the Largest Cohort of Patients with AKU. Eur. J. Hum. Genet. 2019, 27, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Bernini, A.; Galderisi, S.; Spiga, O.; Amarabom, C.O.; Santucci, A. Transient Pockets as Mediators of Gas Molecules Routes inside Proteins: The Case Study of Dioxygen Pathway in Homogentisate 1,2-Dioxygenase and Its Implication in Alkaptonuria Development. Comput. Biol. Chem. 2020, 88, 107356. [Google Scholar] [CrossRef] [PubMed]
- Bernini, A.; Galderisi, S.; Spiga, O.; Bernardini, G.; Niccolai, N.; Manetti, F.; Santucci, A. Toward a Generalized Computational Workflow for Exploiting Transient Pockets as New Targets for Small Molecule Stabilizers: Application to the Homogentisate 1,2-Dioxygenase Mutants at the Base of Rare Disease Alkaptonuria. Comput. Biol. Chem. 2017, 70, 133–141. [Google Scholar] [CrossRef]
- Braconi, D.; Millucci, L.; Bernini, A.; Spiga, O.; Lupetti, P.; Marzocchi, B.; Niccolai, N.; Bernardini, G.; Santucci, A. Homogentisic Acid Induces Aggregation and Fibrillation of Amyloidogenic Proteins. Biochim. Biophys. Acta BBA Gen. Subj. 2017, 1861, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Spiga, O.; Cicaloni, V.; Bernini, A.; Zatkova, A.; Santucci, A. ApreciseKUre: An Approach of Precision Medicine in a Rare Disease. BMC Med. Inform. Decis. Mak. 2017, 17, 42. [Google Scholar] [CrossRef]
- Braconi, D.; Bernardini, G.; Paffetti, A.; Millucci, L.; Geminiani, M.; Laschi, M.; Frediani, B.; Marzocchi, B.; Santucci, A. Comparative Proteomics in Alkaptonuria Provides Insights into Inflammation and Oxidative Stress. Int. J. Biochem. Cell Biol. 2016, 81, 271–280. [Google Scholar] [CrossRef]
- Spreafico, A.; Millucci, L.; Ghezzi, L.; Geminiani, M.; Braconi, D.; Amato, L.; Chellini, F.; Frediani, B.; Moretti, E.; Collodel, G.; et al. Antioxidants Inhibit SAA Formation and Pro-Inflammatory Cytokine Release in a Human Cell Model of Alkaptonuria. Rheumatology 2013, 52, 1667–1673. [Google Scholar] [CrossRef]
- Millucci, L.; Bernardini, G.; Marzocchi, B.; Braconi, D.; Geminiani, M.; Gambassi, S.; Laschi, M.; Frediani, B.; Galvagni, F.; Orlandini, M.; et al. Angiogenesis in Alkaptonuria. J Inherit Metab Dis 2016, 39, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Bernardini, G.; Spreafico, A.; Orlandini, M.; Braconi, D.; Laschi, M.; Geminiani, M.; Lupetti, P.; Giorgetti, G.; Viti, C.; et al. Histological and Ultrastructural Characterization of Alkaptonuric Tissues. Calcif. Tissue Int. 2017, 101, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Ghezzi, L.; Paccagnini, E.; Giorgetti, G.; Viti, C.; Braconi, D.; Laschi, M.; Geminiani, M.; Soldani, P.; Lupetti, P.; et al. Amyloidosis, Inflammation, and Oxidative Stress in the Heart of an Alkaptonuric Patient. Mediat. Inflamm 2014, 2014, 258471. [Google Scholar] [CrossRef] [PubMed]
- Millucci, L.; Ghezzi, L.; Braconi, D.; Laschi, M.; Geminiani, M.; Amato, L.; Orlandini, M.; Benvenuti, C.; Bernardini, G.; Santucci, A. Secondary Amyloidosis in an Alkaptonuric Aortic Valve. Int. J. Cardiol. 2014, 172, e121–e123. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.T.; Milan, A.M.; Christensen, P.; Ross, G.; Davison, A.S.; Gallagher, J.A.; Dutton, J.J.; Ranganath, L.R. Urine Homogentisic Acid and Tyrosine: Simultaneous Analysis by Liquid Chromatography Tandem Mass Spectrometry. Journal of Chromatography B 2014, 963, 106–112. [Google Scholar] [CrossRef]
- Boedeker, C.W. Über Das Alcapton; Ein Neuer Beitrag Zur Frage: Welche Stoffe Des Harns Können Kupferreduktion Bewirken? Z. Ration. Med. 1859, 7, 130–145. [Google Scholar]
- Benedek, T.G. Rudolph Virchow on Ochronosis. Arthritis Rheum 1966, 9, 66–71. [Google Scholar] [CrossRef]
- Wolkow, M.; Baumann, E. Über Das Wesen Der Alkaptonurie. Z. Physiol. Chem. 1891, 15, 228–285. [Google Scholar]
- Bernini, A.; Petricci, E.; Atrei, A.; Baratto, M.C.; Manetti, F.; Santucci, A. A Molecular Spectroscopy Approach for the Investigation of Early Phase Ochronotic Pigment Development in Alkaptonuria. Sci. Rep. 2021, 11, 22562. [Google Scholar] [CrossRef]
- Mistry, J.B.; Bukhari, M.; Taylor, A.M. Alkaptonuria. Rare Dis. 2013, 1, e27475. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Psarelli, E.E.; Arnoux, J.B.; Braconi, D.; Briggs, M.; Bröijersén, A.; Loftus, N.; Bygott, H.; Cox, T.F.; Davison, A.S.; et al. Efficacy and Safety of Once-Daily Nitisinone for Patients with Alkaptonuria (SONIA 2): An International, Multicentre, Open-Label, Randomised Controlled Trial. Lancet Diabetes Endocrinol. 2020, 8, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Norman, B.P.; Davison, A.S.; Hughes, J.H.; Sutherland, H.; Wilson, P.J.; Berry, N.G.; Hughes, A.T.; Milan, A.M.; Jarvis, J.C.; Roberts, N.B.; et al. Metabolomic Studies in the Inborn Error of Metabolism Alkaptonuria Reveal New Biotransformations in Tyrosine Metabolism. Genes Dis. 2021, 9, 1129–1142. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Cox, T.F.; Psarelli, E.E.; Szamosi, J.; Hughes, A.T.; Milan, A.M.; Hall, A.K.; Rovensky, J.; Ranganath, L.R. Relationship Between Serum Concentrations of Nitisinone and Its Effect on Homogentisic Acid and Tyrosine in Patients with Alkaptonuria. JIMD Rep. 2015, 24, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, L.R.; Milan, A.M.; Hughes, A.T.; Dutton, J.J.; Fitzgerald, R.; Briggs, M.C.; Bygott, H.; Psarelli, E.E.; Cox, T.F.; Gallagher, J.A.; et al. Suitability of Nitisinone in Alkaptonuria 1 (SONIA 1): An International, Multicentre, Randomised, Open-Label, No-Treatment Controlled, Parallel-Group, Dose-Response Study to Investigate the Effect of Once Daily Nitisinone on 24-h Urinary Homogentisic Acid Excretion in Patients with Alkaptonuria after 4 Weeks of Treatment. Ann. Rheum. Dis. 2016, 75, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Laschi, M.; Bernardini, G.; Dreassi, E.; Millucci, L.; Geminiani, M.; Braconi, D.; Marzocchi, B.; Botta, M.; Manetti, F.; Santucci, A. Inhibition of Para-Hydroxyphenylpyruvate Dioxygenase by Analogues of the Herbicide Nitisinone as a Strategy to Decrease Homogentisic Acid Levels, the Causative Agent of Alkaptonuria. ChemMedChem 2016, 11, 674–678. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Milan, A.M.; Hughes, A.T.; Davison, A.S.; Norman, B.P.; Bou-Gharios, G.; Gallagher, J.A.; Imrich, R.; Arnoux, J.B.; Rudebeck, M.; et al. Determinants of Tyrosinaemia during Nitisinone Therapy in Alkaptonuria. Sci. Rep. 2022, 12, 16083. [Google Scholar] [CrossRef]
- Davison, A.S.; Strittmatter, N.; Sutherland, H.; Hughes, A.T.; Hughes, J.; Bou-Gharios, G.; Milan, A.M.; Goodwin, R.J.A.; Ranganath, L.R.; Gallagher, J.A. Assessing the Effect of Nitisinone Induced Hypertyrosinaemia on Monoamine Neurotransmitters in Brain Tissue from a Murine Model of Alkaptonuria Using Mass Spectrometry Imaging. Metabolomics 2019, 15, 68. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Khedr, M.; Milan, A.M.; Davison, A.S.; Hughes, A.T.; Usher, J.L.; Taylor, S.; Loftus, N.; Daroszewska, A.; West, E.; et al. Nitisinone Arrests Ochronosis and Decreases Rate of Progression of Alkaptonuria: Evaluation of the Effect of Nitisinone in the United Kingdom National Alkaptonuria Centre. Mol Genet Metab 2018, 125, 127–134. [Google Scholar] [CrossRef]
- White, A.; Tchan, M.C. Nitisinone-Induced Keratopathy in Alkaptonuria: A Challenging Diagnosis Despite Clinical Suspicion. JIMD Rep. 2018, 40, 7. [Google Scholar] [CrossRef]
- Zatkova, A.; Olsson, B.; Ranganath, L.R.; Imrich, R. Analysis of the Phenotype Differences in Siblings with Alkaptonuria. Metabolites 2022, 12, 990. [Google Scholar] [CrossRef]
- Eisner, R.; Stretch, C.; Eastman, T.; Xia, J.; Hau, D.; Damaraju, S.; Greiner, R.; Wishart, D.S.; Baracos, V.E. Learning to Predict Cancer-Associated Skeletal Muscle Wasting from 1H-NMR Profiles of Urinary Metabolites. Metabolomics 2011, 7, 25–34. [Google Scholar] [CrossRef]
- Saude, E.J.; Sykes, B.D. Urine Stability for Metabolomic Studies: Effects of Preparation and Storage. Metabolomics 2007, 3, 19–27. [Google Scholar] [CrossRef]
- Mindikoglu, A.L.; Opekun, A.R.; Putluri, N.; Devaraj, S.; Sheikh-Hamad, D.; Vierling, J.M.; Goss, J.A.; Rana, A.; Sood, G.K.; Jalal, P.K.; et al. Unique Metabolomic Signature Associated with Hepatorenal Dysfunction and Mortality in Cirrhosis. Transl. Res. 2018, 195, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Cao, S.; Shi, X.; Nie, X.; Zheng, J.; Deng, Y.; Ruan, L.; Peng, D.; Sun, M. Genetic and Biochemical Characterization of a Gene Operon for Trans-Aconitic Acid, a Novel Nematicide from Bacillus Thuringiensis. J. Biol. Chem. 2017, 292, 3517–3530. [Google Scholar] [CrossRef] [PubMed]
- Saffran, M.; Prado, J.L. Inhibition of aconitase by trans-aconitate. J. Biol. Chem. 1949, 180, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Villafranca, J.J. The Mechanism of Aconitase Action: Evidence for an enzyme isomerization by studies of inhibition by tricarboxylic acids. J. Biol. Chem. 1974, 249, 6149–6155. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, M.E.; Brosnan, J.T. Formate: The Neglected Member of One-Carbon Metabolism. Annu. Rev. Nutr. 2016, 36, 369–388. [Google Scholar] [CrossRef]
- Stretch, C.; Eastman, T.; Mandal, R.; Eisner, R.; Wishart, D.S.; Mourtzakis, M.; Prado, C.M.M.; Damaraju, S.; Ball, R.O.; Greiner, R.; et al. Prediction of Skeletal Muscle and Fat Mass in Patients with Advanced Cancer Using a Metabolomic Approach. J. Nutr. 2012, 142, 14–21. [Google Scholar] [CrossRef]
- McCann, M.R.; de la Rosa, M.V.G.; Rosania, G.R.; Stringer, K.A. L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine. Metabolites 2021, 11, 51. [Google Scholar] [CrossRef]
- Reuter, S.E.; Evans, A.M. Carnitine and Acylcarnitines. Clin. Pharmacokinet. 2012, 51, 553–572. [Google Scholar] [CrossRef]
- Evans, A.M.; Fornasini, G. Pharmacokinetics of L-Carnitine. Clin. Pharmacokinet. 2012, 42, 941–967. [Google Scholar] [CrossRef] [PubMed]
- Bruls, Y.M.; de Ligt, M.; Lindeboom, L.; Phielix, E.; Havekes, B.; Schaart, G.; Kornips, E.; Wildberger, J.E.; Hesselink, M.K.; Muoio, D.; et al. Carnitine Supplementation Improves Metabolic Flexibility and Skeletal Muscle Acetylcarnitine Formation in Volunteers with Impaired Glucose Tolerance: A Randomised Controlled Trial. EBioMedicine 2019, 49, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M. Metabolic Inflexibility: When Mitochondrial Indecision Leads to Metabolic Gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.A.; Atherton, H.J.; Dodd, M.S.; Lee, P.; Cochlin, L.E.; Radda, G.K.; Clarke, K.; Tyler, D.J. The Cycling of Acetyl-CoA through Acetylcarnitine Buffers Cardiac Substrate Supply: A Hyperpolarised 13C Magnetic Resonance Study. Circ Cardiovasc Imaging 2012, 5, 201. [Google Scholar] [CrossRef] [PubMed]
- Dall’Asta, M.; Calani, L.; Tedeschi, M.; Jechiu, L.; Brighenti, F.; del Rio, D. Identification of Microbial Metabolites Derived from in Vitro Fecal Fermentation of Different Polyphenolic Food Sources. Nutrition 2012, 28, 197–203. [Google Scholar] [CrossRef]
- Lehotay, D.C.; Clarke, J.T.R. Organic Acidurias and Related Abnormalities. Crit. Rev. Clin. Lab Sci. 1995, 32, 377–429. [Google Scholar] [CrossRef]
- Dzúrik, R.; Spustová, V.; Krivošíková, Z.; Gazíiková, K. Hippurate Participates in the Correction of Metabolic Acidosis. Kidney Int. 2001, 59, S278–S281. [Google Scholar] [CrossRef]
- Davison, A.S.; Milan, A.M.; Gallagher, J.A.; Ranganath, L.R. Acute Fatal Metabolic Complications in Alkaptonuria. J. Inherit. Metab. Dis. 2016, 39, 203–210. [Google Scholar] [CrossRef]
- Pallister, T.; Jackson, M.A.; Martin, T.C.; Zierer, J.; Jennings, A.; Mohney, R.P.; MacGregor, A.; Steves, C.J.; Cassidy, A.; Spector, T.D.; et al. Hippurate as a Metabolomic Marker of Gut Microbiome Diversity: Modulation by Diet and Relationship to Metabolic Syndrome. Sci. Rep. 2017, 7, 13670. [Google Scholar] [CrossRef]
- Joncquel-Chevalier Curt, M.; Voicu, P.M.; Fontaine, M.; Dessein, A.F.; Porchet, N.; Mention-Mulliez, K.; Dobbelaere, D.; Soto-Ares, G.; Cheillan, D.; Vamecq, J. Creatine Biosynthesis and Transport in Health and Disease. Biochimie 2015, 119, 146–165. [Google Scholar] [CrossRef]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and Creatinine Metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.L.; Wassif, W.S.; Bell, J.D.; Hurley, M.; Scott, D.L. Urinary Levels of Creatine and Other Metabolites in the Assessment of Polymyositis and Dermatomyositis. Rheumatology 2003, 42, 298–303. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, D.; Zhao, L.; Jiang, Y.; Li, L.; Guo, M.; Mu, Y.; Zhu, H. Integrated Analysis of Plasma and Urine Reveals Unique Metabolomic Profiles in Idiopathic Inflammatory Myopathies Subtypes. J. Cachexia Sarcopenia Muscle 2022, 13, 2456–2472. [Google Scholar] [CrossRef]
- Al-Ajlouni, J.M.; Alisi, M.S.; Yasin, M.S.; Khanfar, A.; Hamdan, M.; Halaweh, A.A.; al Hawamdeh, H.; Elessi, K.; Alsbou, M.S. Long-Term Outcomes of the Knee and Hip Arthroplasties in Patients with Alkaptonuria. Arthroplast Today 2020, 6, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, L.R.; Jarvis, J.C.; Gallagher, J.A. Recent Advances in Management of Alkaptonuria (Invited Review; Best Practice Article). J. Clin. Pathol. 2013, 66, 367–373. [Google Scholar] [CrossRef]
- Brier, M.E.; Bowsher, R.R.; Mayer, P.R.; Henry, D.P. Conversion of P-Tyrosine to p-Tyramine in the Isolated Perfused Rat Kidney: Modulation by Perfusate Concentrations of p-Tyrosine. Life Sci. 1991, 48, 901–907. [Google Scholar] [CrossRef]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace Amines and Their Receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef]
- Berry, M.D. Mammalian Central Nervous System Trace Amines. Pharmacologic Amphetamines, Physiologic Neuromodulators. J. Neurochem. 2004, 90, 257–271. [Google Scholar] [CrossRef]
- Hoag, G.N.; Hill, A.; Zaleski, W. Urinary P-Tyramine in Hereditary Tyrosinemia: II. Origin of Urinary p-Tyramine. Clin. Biochem. 1977, 10, 26–28. [Google Scholar] [CrossRef]
- Berry, M.D.; Juorio, A.V.; Li, X.M.; Boulton, A.A. Aromatic L-Amino Acid Decarboxylase: A Neglected and Misunderstood Enzyme. Neurochem. Res. 1996, 21, 1075–1087. [Google Scholar] [CrossRef]
- Duchemin, A.M.; Neff, N.H.; Hadjiconstantinou, M. Aromatic L-Amino Acid Decarboxylase Phosphorylation and Activation by PKGIαin Vitro. J. Neurochem. 2010, 114, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Duchemin, A.M.; Berry, M.D.; Neff, N.H.; Hadjiconstantinou, M. Phosphorylation and Activation of Brain Aromatic L-Amino Acid Decarboxylase by Cyclic AMP-Dependent Protein Kinase. J. Neurochem. 2000, 75, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, L.R.; Milan, A.M.; Hughes, A.T.; Khedr, M.; Davison, A.S.; Shweihdi, E.; Norman, B.P.; Hughes, J.H.; Bygott, H.; Luangrath, E.; et al. Homogentisic Acid Is Not Only Eliminated by Glomerular Filtration and Tubular Secretion but Also Produced in the Kidney in Alkaptonuria. J. Inherit. Metab. Dis. 2020, 43, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Pillozzi, S.; Tazza, I.; Bertelli, M.; Campanacci, D.A.; Palchetti, I.; Bernini, A. An Improved NMR Approach for Metabolomics of Intact Serum Samples. Anal. Biochem. 2022, 654, 114826. [Google Scholar] [CrossRef]
- Ranganath, L.R.; Milan, A.M.; Hughes, A.T.; Davison, A.S.; Khedr, M.; Norman, B.P.; Bou-Gharios, G.; Gallagher, J.A.; Gornall, M.; Jackson, R.; et al. Characterization of Changes in the Tyrosine Pathway by 24-h Profiling during Nitisinone Treatment in Alkaptonuria. Mol. Genet. Metab. Rep. 2022, 30, 100846. [Google Scholar] [CrossRef]
- le Guennec, A.; Tayyari, F.; Edison, A.S. Alternatives to Nuclear Overhauser Enhancement Spectroscopy Presat and Carr−Purcell−Meiboom−Gill Presat for NMR-Based Metabolomics. Anal. Chem. 2017, 89, 52. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the Gap between Raw Spectra and Functional Insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Code | CHGA (mM) | S.D. | Patient Code | CHGA (mM) | S.D. |
|---|---|---|---|---|---|
| AKU.303 | 11.33 | 0.04 | AKU.413 | 21.36 | 0.17 |
| AKU.304 | 14.07 | 0.08 | AKU.414 | 52.98 | 0.35 |
| AKU.305 | 11.22 | 0.04 | AKU.418 | 15.80 | 0.03 |
| AKU.338 | 14.89 | 0.03 | AKU.420 | 14.49 | 0.13 |
| AKU.350 | 37.25 | 0.12 | AKU.423 | 11.48 | 0.08 |
| AKU.356 | 2.77 | 0.02 | AKU.425 | 33.69 | 0.29 |
| AKU.364 | 17.12 | 0.14 | AKU.426 | 38.77 | 0.38 |
| AKU.367 | 16.17 | 0.13 | AKU.427 | 24.42 | 0.26 |
| AKU.369 | 24.22 | 0.26 | AKU.430 | 10.78 | 0.02 |
| AKU.370 | 11.19 | 0.08 | AKU.433 | 13.38 | 0.02 |
| AKU.371 | 35.77 | 0.37 | AKU.434 | 14.04 | 0.02 |
| AKU.379 | 21.36 | 0.15 | AKU.435 | 17.11 | 0.02 |
| AKU.381 | 16.12 | 0.12 | AKU.436 | 15.51 | 0.02 |
| AKU.385 | 12.38 | 0.11 | AKU.443 | 64.62 | 0.08 |
| AKU.390 | 23.01 | 0.17 | AKU.447 | 34.41 | 0.06 |
| AKU.410 | 12.19 | 0.08 | AKU.449 | 15.88 | 0.03 |
| AKU.411 | 23.39 | 0.03 | AKU.450 | 15.28 | 0.02 |
| Metabolite | Log2(FC) | Up/Down | Pathway | |
|---|---|---|---|---|
| Homogentisate a | 12.04 | p < 10−13 | ↑ | Phenylalanine and tyrosine metabolism |
| trans-Aconitate | 2.11 | p < 0.00001 | ↑ | TCA cycle |
| Trigonelline | 1.78 | p < 0.00001 | ↑ | SGOC b metabolic unit |
| Tyramine | 1.34 | p < 0.0001 | ↑ | Phenylalanine and tyrosine metabolism |
| O-Acetylcarnitine | 1.18 | p < 0.01 | ↑ | Acetyl-CoA conversion |
| Guanidinoacetate | 1.13 | p < 0.0001 | ↑ | SGOC metabolic unit |
| Tyrosine | 1.12 | p < 0.001 | ↑ | Phenylalanine and tyrosine metabolism |
| Hippurate | 1.08 | p < 0.0001 | ↑ | Modulator of metabolic acidosis (MAC) |
| Creatine | 0.99 | p < 0.001 | ↑ | SGOC metabolic unit |
| 2-Furoylglycine | 0.80 | p < 0.01 | ↑ | Minor fatty acid metabolite |
| 4-Hydroxyphenylacetate | 0.76 | p < 0.01 | ↑ | Phenylalanine and tyrosine metabolism |
| Glycine | 0.76 | p < 0.05 | ↑ | SGOC metabolic unit |
| Tryptophan | 0.71 | p < 0.05 | ↑ | Tryptophan metabolism |
| Taurine | 0.69 | p < 0.05 | ↑ | Taurine metabolism |
| Acetone | 0.62 | p < 0.01 | ↑ | Acetyl-CoA conversion |
| 3-Hydroxyisovalerate | 0.61 | p < 0.05 | ↑ | Leucine degradation |
| Formate | 0.44 | p < 0.05 | ↑ | SGOC metabolic unit |
| cis-Aconitate | −0.70 | p < 0.0001 | ↓ | TCA cycle |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grasso, D.; Geminiani, M.; Galderisi, S.; Iacomelli, G.; Peruzzi, L.; Marzocchi, B.; Santucci, A.; Bernini, A. Untargeted NMR Metabolomics Reveals Alternative Biomarkers and Pathways in Alkaptonuria. Int. J. Mol. Sci. 2022, 23, 15805. https://doi.org/10.3390/ijms232415805
Grasso D, Geminiani M, Galderisi S, Iacomelli G, Peruzzi L, Marzocchi B, Santucci A, Bernini A. Untargeted NMR Metabolomics Reveals Alternative Biomarkers and Pathways in Alkaptonuria. International Journal of Molecular Sciences. 2022; 23(24):15805. https://doi.org/10.3390/ijms232415805
Chicago/Turabian StyleGrasso, Daniela, Michela Geminiani, Silvia Galderisi, Gabriella Iacomelli, Luana Peruzzi, Barbara Marzocchi, Annalisa Santucci, and Andrea Bernini. 2022. "Untargeted NMR Metabolomics Reveals Alternative Biomarkers and Pathways in Alkaptonuria" International Journal of Molecular Sciences 23, no. 24: 15805. https://doi.org/10.3390/ijms232415805
APA StyleGrasso, D., Geminiani, M., Galderisi, S., Iacomelli, G., Peruzzi, L., Marzocchi, B., Santucci, A., & Bernini, A. (2022). Untargeted NMR Metabolomics Reveals Alternative Biomarkers and Pathways in Alkaptonuria. International Journal of Molecular Sciences, 23(24), 15805. https://doi.org/10.3390/ijms232415805