
Cost-Effective Mechanical Aggregation of Cardiac Progenitors and Encapsulation in Matrigel Support Self-Organization in a Dynamic Culture Environment
 ,
, 
 , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
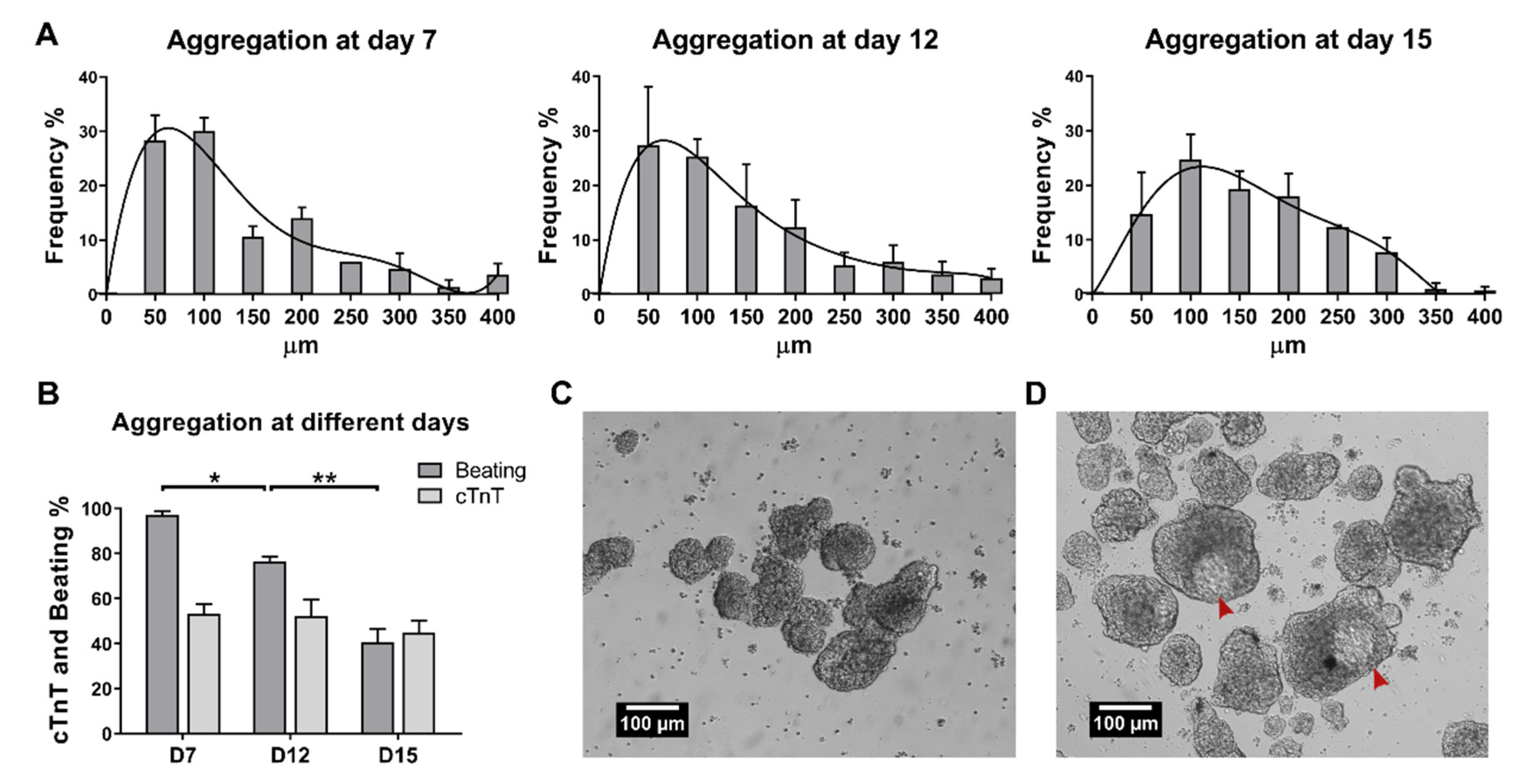
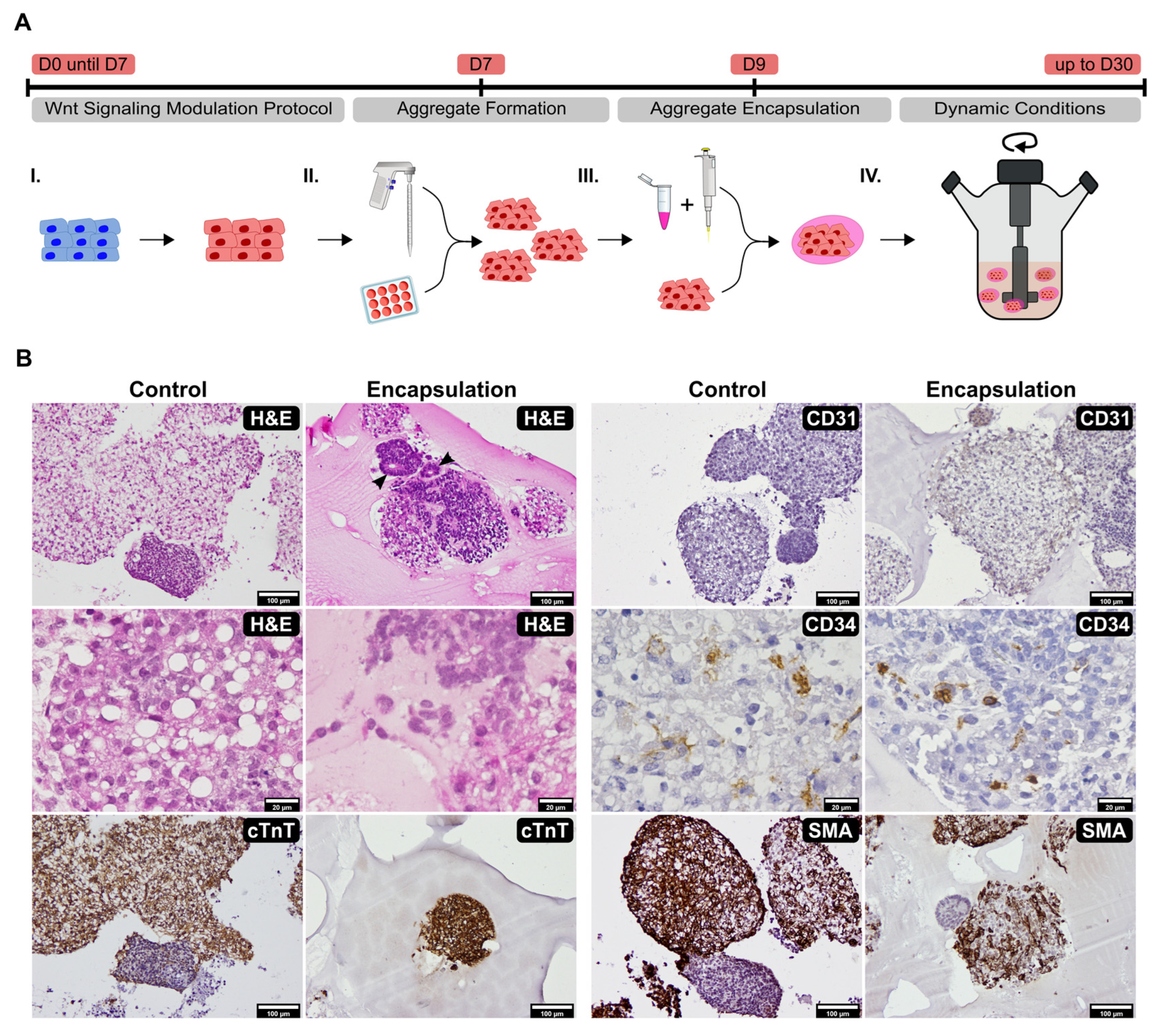
2.1. Establishment of Culture Conditions for the Generation of 3D Cardiac Microtissues from hiPSCs
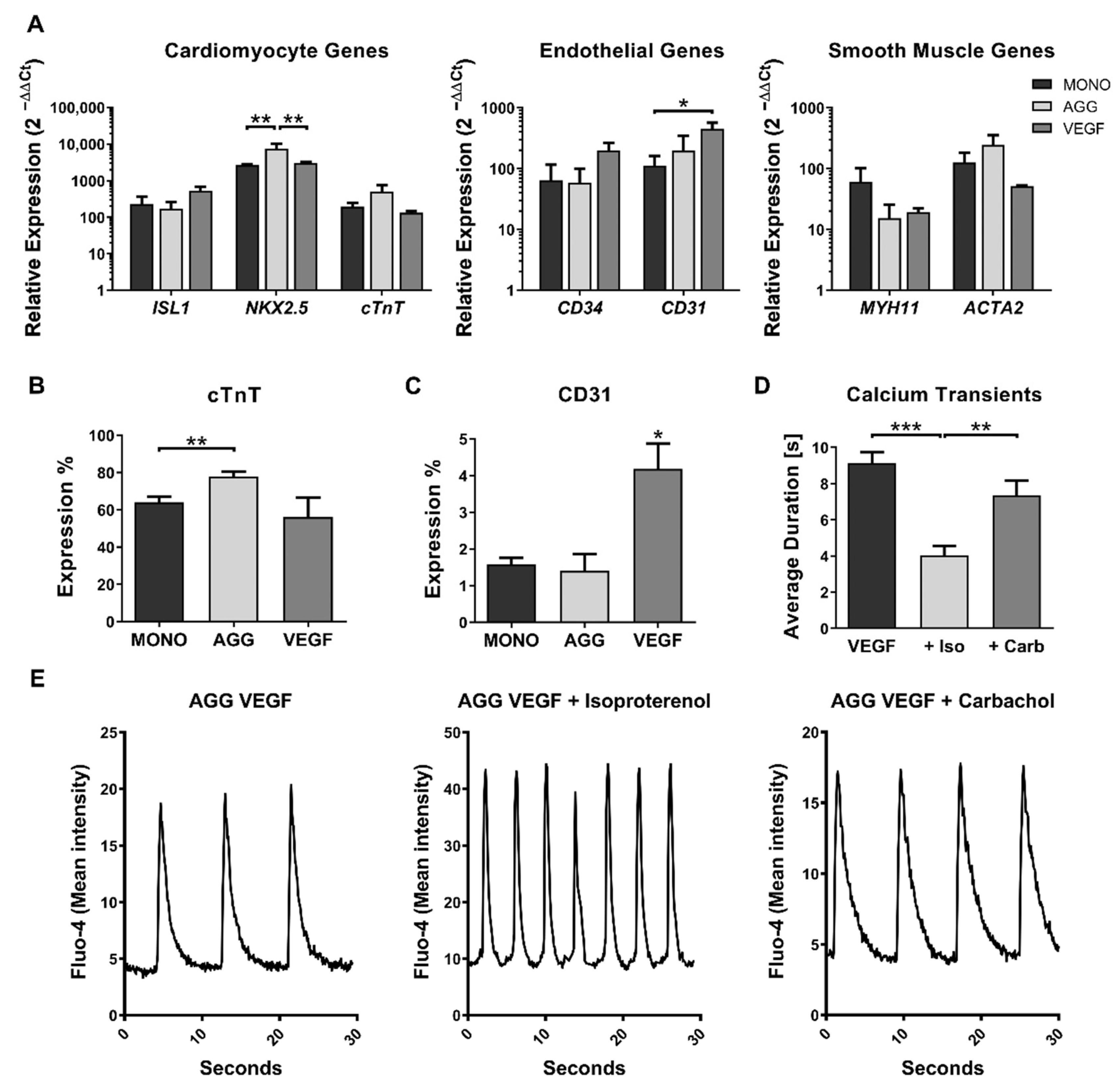
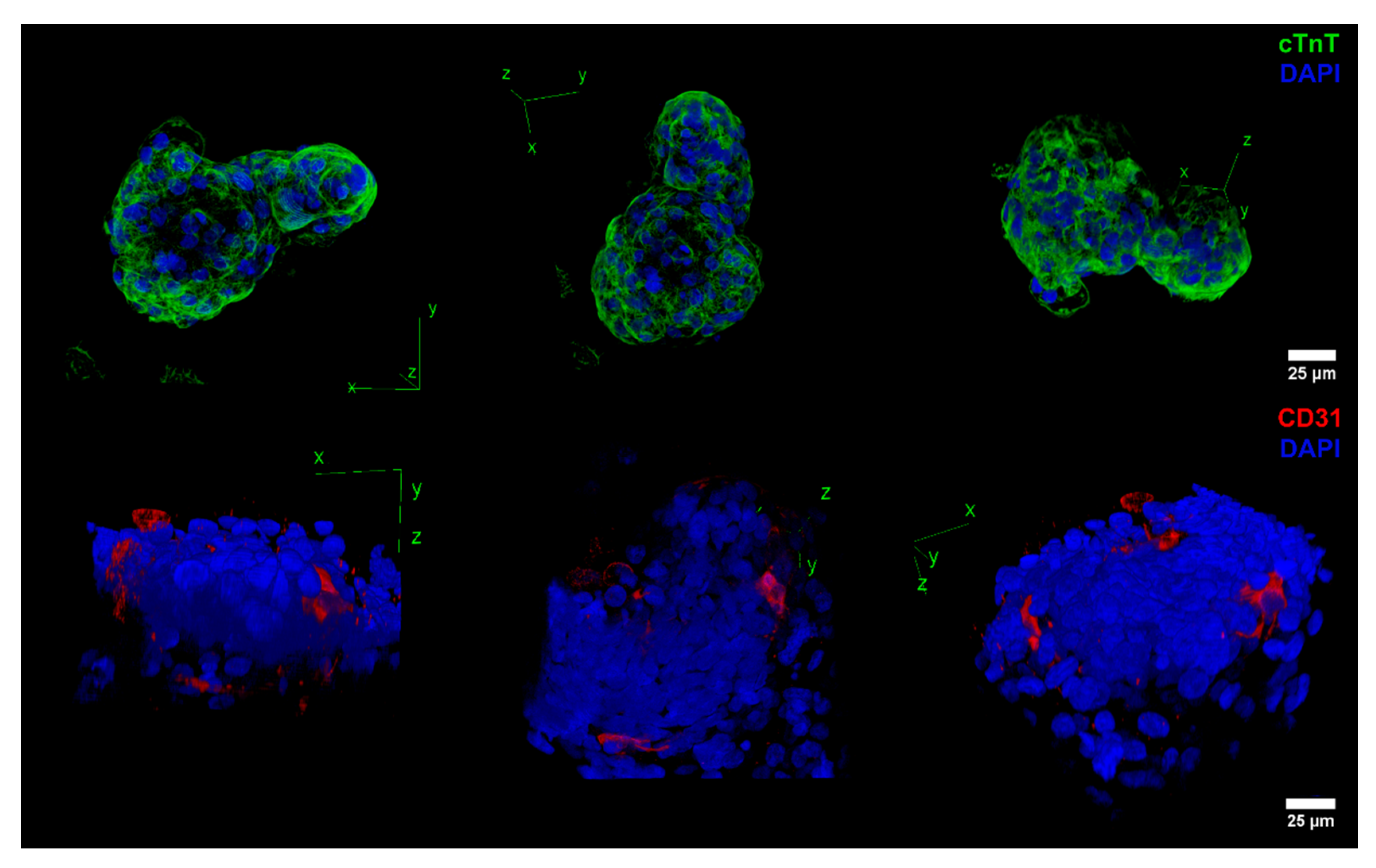
2.2. Impact of VEGF Supplementation and Cell Aggregation on the Development of Microvessel-like Structures and Cardiomyocyte Functionality of Cardiac Microtissues
3. Discussion
4. Materials and Methods
4.1. Human Induced Pluripotent Stem Cell Culture
4.2. Human iPSC Cardiomyocyte Differentiation, Aggregation, and Encapsulation
4.3. Flow Cytometry
4.4. Real-Time PCR
4.5. H&E Staining and Immunohistochemistry
4.6. Immunofluorescence Staining and Confocal Microscopy
4.7. Calcium Imaging
4.8. Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Force, T.; Kolaja, K.L. Cardiotoxicity of Kinase Inhibitors: The Prediction and Translation of Preclinical Models to Clinical Outcomes. Nat. Rev. Drug Discov. 2011, 10, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An Analysis of the Attrition of Drug Candidates from Four Major Pharmaceutical Companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.A.; Kavanagh, S.L.; Mellor, H.R.; Pollard, C.E.; Robinson, S.; Platz, S.J. Reducing Attrition in Drug Development: Smart Loading Preclinical Safety Assessment. Drug Discov. Today 2014, 19, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.; Knoblich, J. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N.; et al. Vascularized and Functional Human Liver from an IPSC-Derived Organ Bud Transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed Differentiation of Human Pluripotent Stem Cells into Intestinal Tissue in Vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Peerani, R.; Rao, B.M.; Bauwens, C.; Yin, T.; Wood, G.; Nagy, A.; Kumacheva, E.; Zandstra, P.W. Niche-Mediated Control of Human Embryonic Stem Cell Self-Renewal and Differentiation. EMBO J. 2007, 26, 4744–4755. [Google Scholar] [CrossRef]
- Rossi, G.; Broguiere, N.; Miyamoto, M.; Boni, A.; Guiet, R.; Girgin, M.; Kelly, R.G.; Kwon, C.; Lutolf, M.P. Capturing Cardiogenesis in Gastruloids. Cell Stem Cell 2021, 28, 230–240.e6. [Google Scholar] [CrossRef]
- Takebe, T.; Wells, J.M. Organoids by Design. Science 2019, 364, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, C.L.; Peerani, R.; Niebruegge, S.; Woodhouse, K.A.; Kumacheva, E.; Husain, M.; Zandstra, P.W. Control of Human Embryonic Stem Cell Colony and Aggregate Size Heterogeneity Influences Differentiation Trajectories. Stem Cells 2008, 26, 2300–2310. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, C.L.; Song, H.; Thavandiran, N.; Ungrin, M.; Massé, S.; Nanthakumar, K.; Seguin, C.; Zandstra, P.W. Geometric Control of Cardiomyogenic Induction in Human Pluripotent Stem Cells. Tissue Eng. Part A 2011, 17, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Niebruegge, S.; Bauwens, C.L.; Peerani, R.; Thavandiran, N.; Masse, S.; Sevaptisidis, E.; Nanthakumar, K.; Woodhouse, K.; Husain, M.; Kumacheva, E.; et al. Generation of Human Embryonic Stem Cell-Derived Mesoderm and Cardiac Cells Using Size-Specified Aggregates in an Oxygen-Controlled Bioreactor. Biotechnol. Bioeng. 2009, 102, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Branco, M.A.; Cotovio, J.P.; Rodrigues, C.A.V.; Vaz, S.H.; Fernandes, T.G.; Moreira, L.M.; Cabral, J.M.S.; Diogo, M.M. Transcriptomic Analysis of 3D Cardiac Differentiation of Human Induced Pluripotent Stem Cells Reveals Faster Cardiomyocyte Maturation Compared to 2D Culture. Sci. Rep. 2019, 9, 9229. [Google Scholar] [CrossRef] [PubMed]
- Thavandiran, N.; Dubois, N.; Mikryukov, A.; Masse, S.; Beca, B.; Simmons, C.A.; Deshpande, V.S.; McGarry, J.P.; Chen, C.S.; Nanthakumar, K.; et al. Design and Formulation of Functional Pluripotent Stem Cell-Derived Cardiac Microtissues. Proc. Natl. Acad. Sci. USA 2013, 110, E4698–E4707. [Google Scholar] [CrossRef]
- Nguyen, D.C.; Hookway, T.A.; Wu, Q.; Jha, R.; Preininger, M.K.; Chen, X.; Easley, C.A.; Spearman, P.; Deshpande, S.R.; Maher, K.; et al. Microscale Generation of Cardiospheres Promotes Robust Enrichment of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2014, 3, 260–268. [Google Scholar] [CrossRef]
- Voges, H.K.; Mills, R.J.; Elliott, D.A.; Parton, R.G.; Porrello, E.R.; Hudson, J.E. Development of a Human Cardiac Organoid Injury Model Reveals Innate Regenerative Potential. Development 2017, 144, 1118–1127. [Google Scholar] [CrossRef]
- Mills, R.J.; Parker, B.L.; Quaife-Ryan, G.A.; Voges, H.K.; Needham, E.J.; Bornot, A.; Ding, M.; Andersson, H.; Polla, M.; Elliott, D.A.; et al. Drug Screening in Human PSC-Cardiac Organoids Identifies Pro-Proliferative Compounds Acting via the Mevalonate Pathway. Cell Stem Cell 2019, 24, 895–907.e6. [Google Scholar] [CrossRef]
- Silva, A.C.; Matthys, O.B.; Joy, D.A.; Kauss, M.A.; Natarajan, V.; Lai, M.H.; Turaga, D.; Blair, A.P.; Alexanian, M.; Bruneau, B.G.; et al. Co-Emergence of Cardiac and Gut Tissues Promotes Cardiomyocyte Maturation within Human IPSC-Derived Organoids. Cell Stem Cell 2021, 28, 2137–2152.e6. [Google Scholar] [CrossRef]
- Drakhlis, L.; Biswanath, S.; Farr, C.M.; Lupanow, V.; Teske, J.; Ritzenhoff, K.; Franke, A.; Manstein, F.; Bolesani, E.; Kempf, H.; et al. Human Heart-Forming Organoids Recapitulate Early Heart and Foregut Development. Nat. Biotechnol. 2021, 39, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Israeli, Y.R.; Wasserman, A.H.; Gabalski, M.A.; Volmert, B.D.; Ming, Y.; Ball, K.A.; Yang, W.; Zou, J.; Ni, G.; Pajares, N.; et al. Self-Assembling Human Heart Organoids for the Modeling of Cardiac Development and Congenital Heart Disease. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, P.; Jahnel, S.M.; Papai, N.; Giesshammer, M.; Deyett, A.; Schmidt, C.; Penc, M.; Tavernini, K.; Grdseloff, N.; Meledeth, C.; et al. Cardioids Reveal Self-Organizing Principles of Human Cardiogenesis. Cell 2021, 184, 3299–3317.e22. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.P.; Sousa-Luís, R.; Fernandes, T.G.; Bekman, E.P.; Rodrigues, C.A.V.; Vaz, S.H.; Moreira, L.M.; Hashimura, Y.; Jung, S.; Lee, B.; et al. Transcriptome Profiling of Human Pluripotent Stem Cell-derived Cerebellar Organoids Reveals Faster Commitment under Dynamic Conditions. Biotechnol. Bioeng. 2021, 118, 2781–2803. [Google Scholar] [CrossRef]
- Nogueira, D.E.S.; Rodrigues, C.A.V.; Carvalho, M.S.; Miranda, C.C.; Hashimura, Y.; Jung, S.; Lee, B.; Cabral, J.M.S. Strategies for the Expansion of Human Induced Pluripotent Stem Cells as Aggregates in Single-Use Vertical-WheelTM Bioreactors. J. Biol. Eng. 2019, 13, 74. [Google Scholar] [CrossRef]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust Cardiomyocyte Differentiation from Human Pluripotent Stem Cells via Temporal Modulation of Canonical Wnt Signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef]
- Mohr, J.C.; Zhang, J.; Azarin, S.M.; Soerens, A.G.; de Pablo, J.J.; Thomson, J.A.; Lyons, G.E.; Palecek, S.P.; Kamp, T.J. The Microwell Control of Embryoid Body Size in Order to Regulate Cardiac Differentiation of Human Embryonic Stem Cells. Biomaterials 2010, 31, 1885–1893. [Google Scholar] [CrossRef]
- Branco, M.A.; Dias, T.P.; Cotovio, J.P.; Rodrigues, C.A.V.; Fernandes, T.G.; Cabral, J.M.S.; Diogo, M.M. 3D Microwell Platform for Cardiomyocyte Differentiation of Human Pluripotent Stem Cells. In Methods in Molecular Biology; Humana: New York, NY, USA, 2020. [Google Scholar] [CrossRef]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve Stem Cells Drive Self-Renewal in the Stomach and Build Long-Lived Gastric Units In Vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures in Vitro without a Mesenchymal Niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Stange, D.E.; Koo, B.-K.; Huch, M.; Sibbel, G.; Basak, O.; Lyubimova, A.; Kujala, P.; Bartfeld, S.; Koster, J.; Geahlen, J.H.; et al. Differentiated Troy+ Chief Cells Act as Reserve Stem Cells to Generate All Lineages of the Stomach Epithelium. Cell 2013, 155, 357–368. [Google Scholar] [CrossRef]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.W.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In Vitro Expansion of Single Lgr5+ Liver Stem Cells Induced by Wnt-Driven Regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-Organizing Optic-Cup Morphogenesis in Three-Dimensional Culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.-H.; Ryeom, S.; Kim, C.F. Lung Stem Cell Differentiation in Mice Directed by Endothelial Cells via a BMP4-NFATc1-Thrombospondin-1 Axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Greggio, C.; De Franceschi, F.; Figueiredo-Larsen, M.; Gobaa, S.; Ranga, A.; Semb, H.; Lutolf, M.; Grapin-Botton, A. Artificial Three-Dimensional Niches Deconstruct Pancreas Development in Vitro. Development 2013, 140, 4452–4462. [Google Scholar] [CrossRef] [PubMed]
- Badenes, S.M.; Fernandes, T.G.; Miranda, C.C.; Pusch-Klein, A.; Haupt, S.; Rodrigues, C.A.V.; Diogo, M.M.; Brüstle, O.; Cabral, J.M.S. Long-Term Expansion of Human Induced Pluripotent Stem Cells in a Microcarrier-Based Dynamic System. J. Chem. Technol. Biotechnol. 2017, 92, 492–503. [Google Scholar] [CrossRef]
- Dias, T.P.; Pinto, S.N.; Santos, J.I.; Fernandes, T.G.; Fernandes, F.; Diogo, M.M.; Prieto, M.; Cabral, J.M.S. Biophysical Study of Human Induced Pluripotent Stem Cell-Derived Cardiomyocyte Structural Maturation during Long-Term Culture. Biochem. Biophys. Res. Commun. 2018, 499, 611–617. [Google Scholar] [CrossRef]
- Burridge, P.W.; Thompson, S.; Millrod, M.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A Universal System for Highly Efficient Cardiac Differentiation of Human Induced Pluripotent Stem Cells That Eliminates Interline Variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef]
- Kempf, H.; Olmer, R.; Kropp, C.; Rückert, M.; Jara-Avaca, M.; Robles-Diaz, D.; Franke, A.; Elliott, D.A.; Wojciechowski, D.; Fischer, M.; et al. Controlling Expansion and Cardiomyogenic Differentiation of Human Pluripotent Stem Cells in Scalable Suspension Culture. Stem Cell Rep. 2014, 3, 1132–1146. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why Do Cancers Have High Aerobic Glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Tristan, C.A.; Ormanoglu, P.; Slamecka, J.; Malley, C.; Chu, P.-H.; Jovanovic, V.M.; Gedik, Y.; Jethmalani, Y.; Bonney, C.; Barnaeva, E.; et al. Robotic High-Throughput Biomanufacturing and Functional Differentiation of Human Pluripotent Stem Cells. Stem Cell Rep. 2021, 16, 3076–3092. [Google Scholar] [CrossRef]
- Zluhan, E.; Kelly, K.; LeClair, N.; Wortel, D.; Moody, K. Automating HESC Differentiation with 3D Printing and Legacy Liquid Handling Solutions. MethodsX 2016, 3, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Buikema, J.W.; Lee, S.; Goodyer, W.R.; Maas, R.G.; Chirikian, O.; Li, G.; Miao, Y.; Paige, S.L.; Lee, D.; Wu, H.; et al. Wnt Activation and Reduced Cell-Cell Contact Synergistically Induce Massive Expansion of Functional Human IPSC-Derived Cardiomyocytes. Cell Stem Cell 2020, 27, 50–63.e5. [Google Scholar] [CrossRef] [PubMed]
- Schade, D.; Drowley, L.; Wang, Q.-D.; Plowright, A.T.; Greber, B. Phenotypic Screen Identifies FOXO Inhibitor to Counteract Maturation and Promote Expansion of Human IPS Cell-Derived Cardiomyocytes. Bioorg. Med. Chem. 2022, 65, 116782. [Google Scholar] [CrossRef] [PubMed]
- Wesseler, F.; Riege, D.; Puthanveedu, M.; Halver, J.; Müller, E.; Bertrand, J.; Antonchick, A.P.; Sievers, S.; Waldmann, H.; Schade, D. Probing Embryonic Development Enables the Discovery of Unique Small-Molecule Bone Morphogenetic Protein Potentiators. J. Med. Chem. 2022, 65, 3978–3990. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed Cardiomyocyte Differentiation from Human Pluripotent Stem Cells by Modulating Wnt/β-Catenin Signaling under Fully Defined Conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias, T.P.; Pinto, S.N.; Carvalho, S.; Fernandes, T.G.; Fernandes, F.; Diogo, M.M.; Peleteiro, M.C.; Prieto, M.; Cabral, J.M.S. Cost-Effective Mechanical Aggregation of Cardiac Progenitors and Encapsulation in Matrigel Support Self-Organization in a Dynamic Culture Environment. Int. J. Mol. Sci. 2022, 23, 15785. https://doi.org/10.3390/ijms232415785
Dias TP, Pinto SN, Carvalho S, Fernandes TG, Fernandes F, Diogo MM, Peleteiro MC, Prieto M, Cabral JMS. Cost-Effective Mechanical Aggregation of Cardiac Progenitors and Encapsulation in Matrigel Support Self-Organization in a Dynamic Culture Environment. International Journal of Molecular Sciences. 2022; 23(24):15785. https://doi.org/10.3390/ijms232415785
Chicago/Turabian StyleDias, Tiago P., Sandra N. Pinto, Sandra Carvalho, Tiago G. Fernandes, Fábio Fernandes, Maria Margarida Diogo, Maria C. Peleteiro, Manuel Prieto, and Joaquim M. S. Cabral. 2022. "Cost-Effective Mechanical Aggregation of Cardiac Progenitors and Encapsulation in Matrigel Support Self-Organization in a Dynamic Culture Environment" International Journal of Molecular Sciences 23, no. 24: 15785. https://doi.org/10.3390/ijms232415785
APA StyleDias, T. P., Pinto, S. N., Carvalho, S., Fernandes, T. G., Fernandes, F., Diogo, M. M., Peleteiro, M. C., Prieto, M., & Cabral, J. M. S. (2022). Cost-Effective Mechanical Aggregation of Cardiac Progenitors and Encapsulation in Matrigel Support Self-Organization in a Dynamic Culture Environment. International Journal of Molecular Sciences, 23(24), 15785. https://doi.org/10.3390/ijms232415785