
TDP-43 Proteinopathy and Tauopathy: Do They Have Pathomechanistic Links?
 ,
,  , and
, and
Abstract
1. Introduction
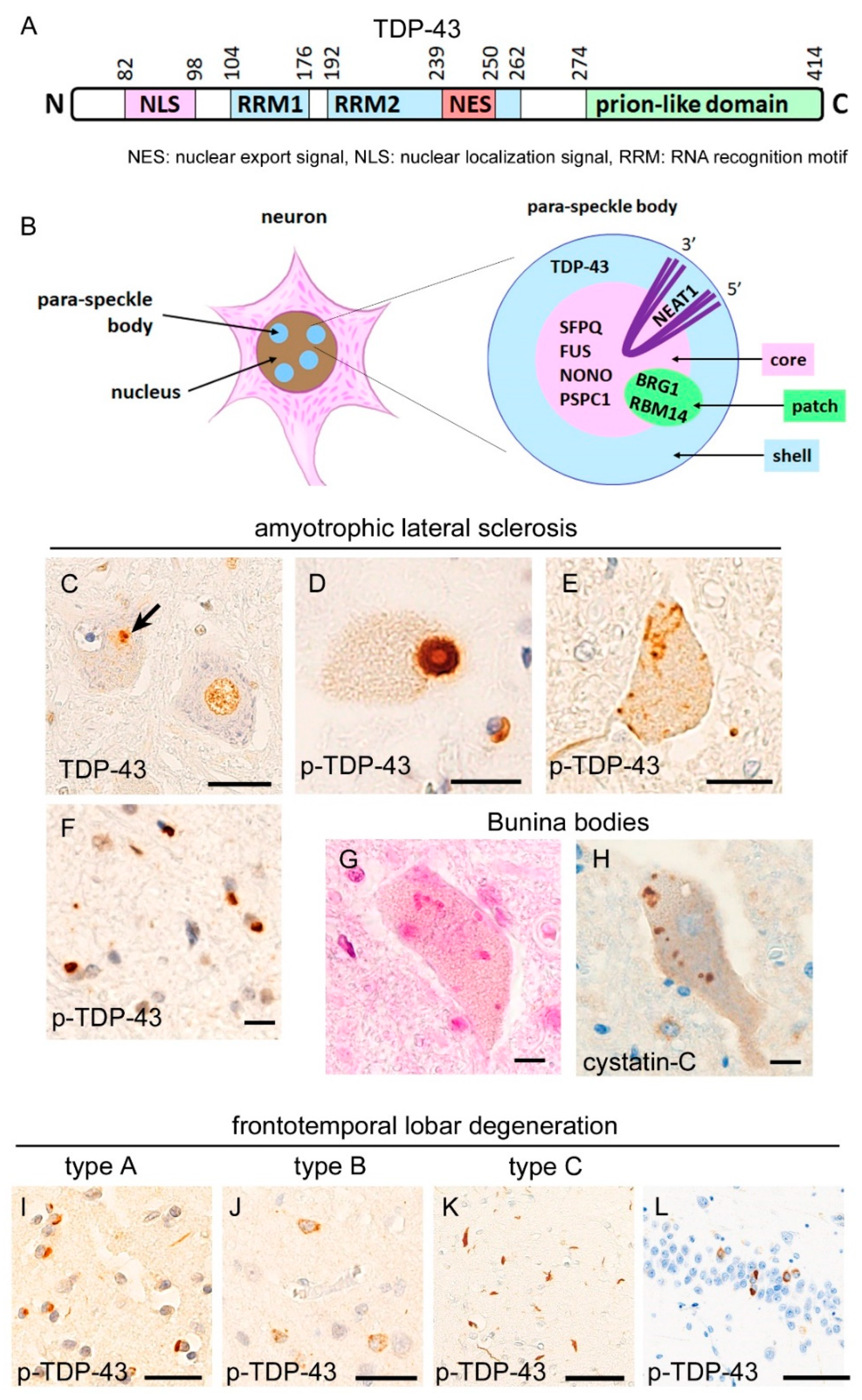
2. Neuropathology of TDP-43 Proteinopathies
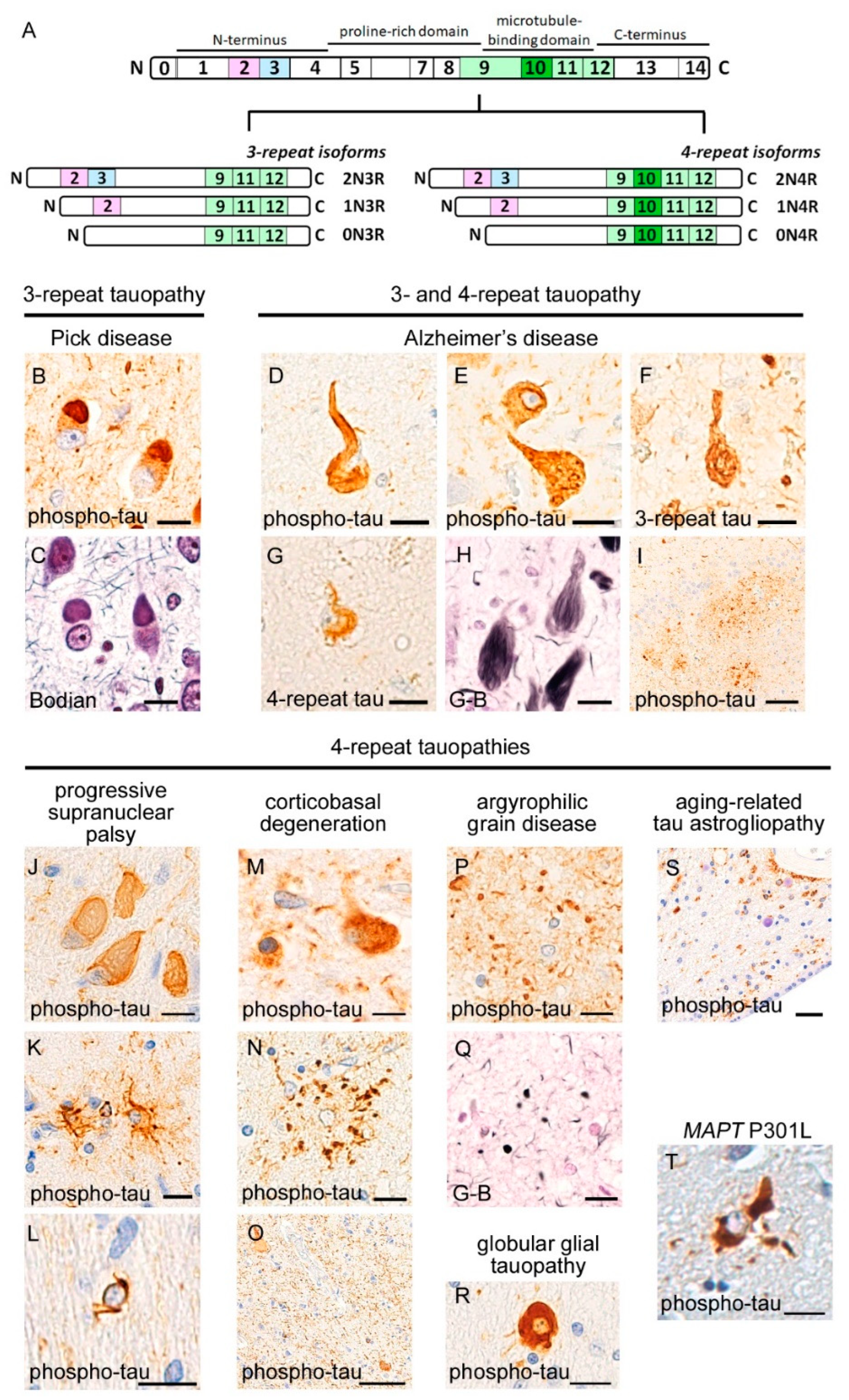
3. Neuropathology of Tauopathies
3.1. 3R-Tauopathy (PiD)
3.2. 4R-Tauopathies (PSP, CBD, AGD, and GGT)
3.3. 3R and 4R-Tauopathy (AD)
3.4. Aging-Related Tauopathies
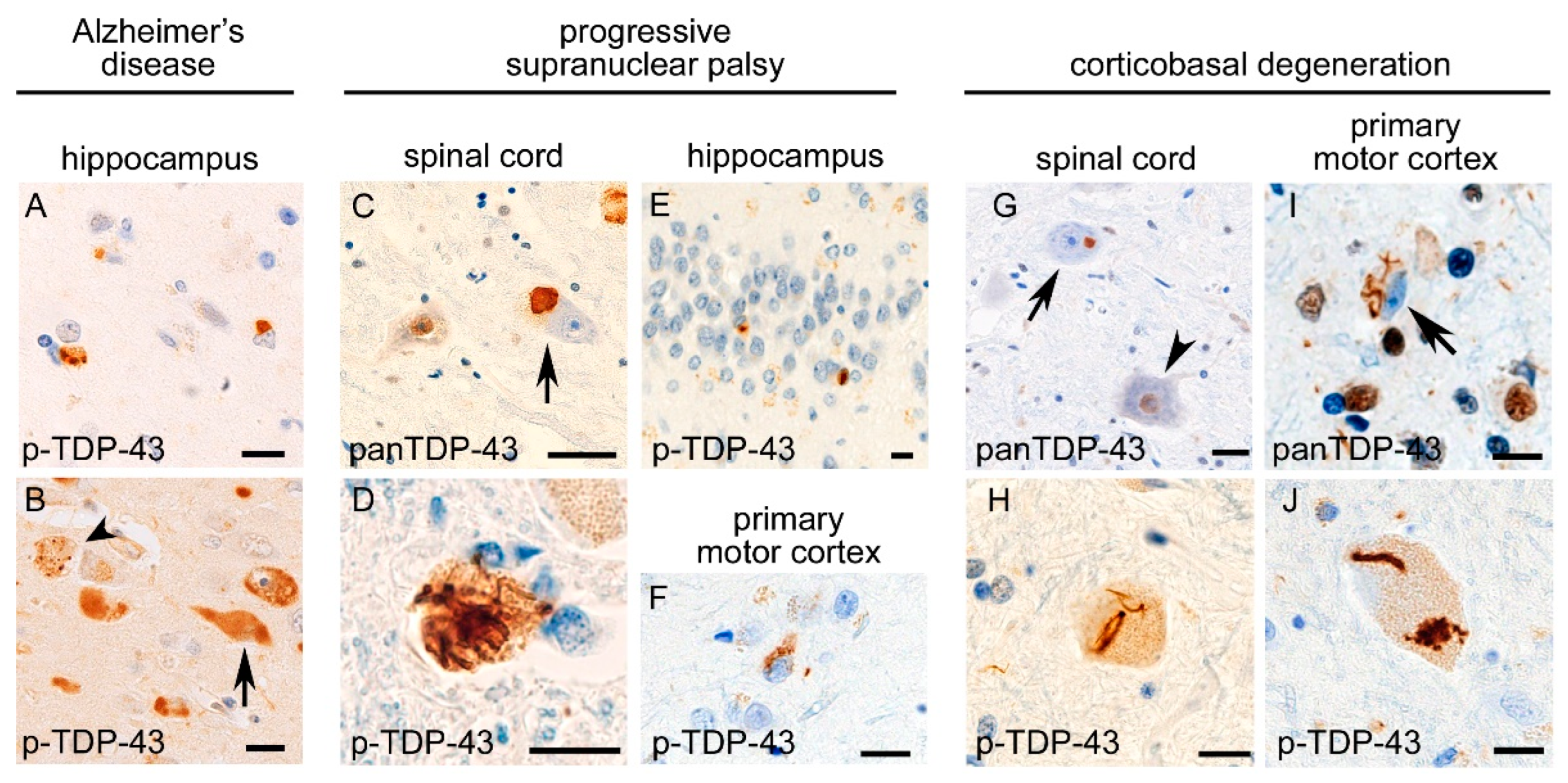
4. Comorbid TDP-43 Pathology in Tauopathies and Comorbid Tau Pathology in TDP-43 Proteinopathies

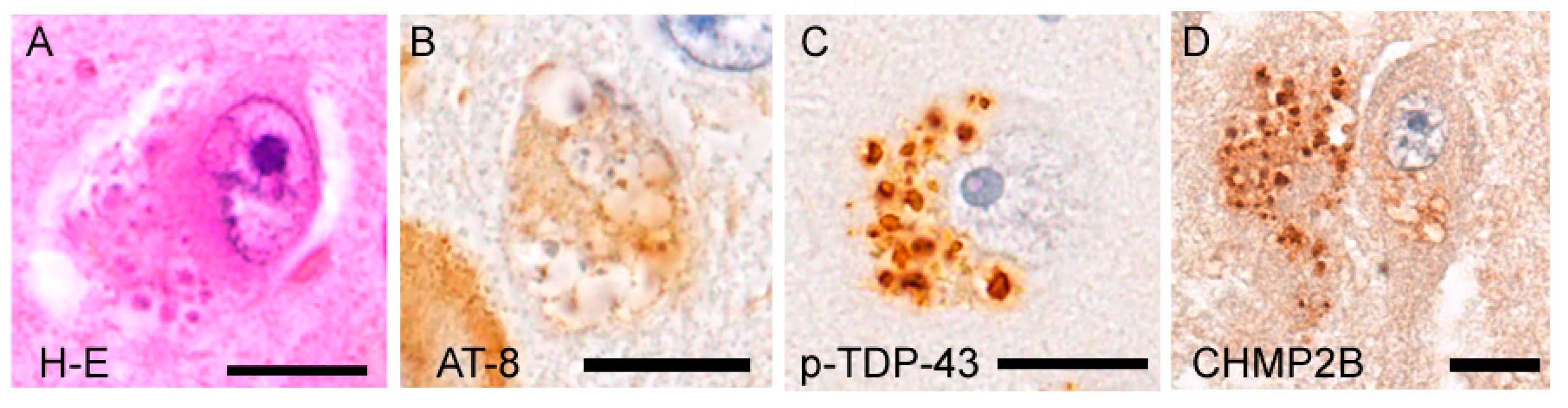
5. Granulovacuolar Degeneration: A Unique Pathology Involving TDP-43 and Tau
6. Do TDP-43 Proteinopathy and Tauopathy Have Mechanical Links?
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation List
| AD | Alzheimer’s disease |
| AGD | argyrophilic grain disease |
| ALS | amyotrophic lateral sclerosis |
| ARTAG | aging-related tau astrogliopathy |
| BRG1 | brahma-related gene 1 |
| bv-FTD | behavior-variant frontotemporal dementia |
| CBD | corticobasal degeneration |
| CHMP2B | charged multivesicular body protein 2B |
| FTD-MND | FTD with motor neuron disease |
| FTLD | frontotemporal lobar degeneration |
| FUS | fused-in-sarcoma |
| GGT | globular glial tauopathy |
| GVD | granulovacuolar degeneration |
| IBMPFD | inclusion body myopathy, Paget’s disease, and frontotemporal dementia |
| LATE | limbic-predominant age-related TDP-43 encephalopathy |
| MAPT | microtubule-associated protein tau |
| NFT | neurofibrillary tangle |
| NEAT1 | nuclear paraspeckle assembly transcript 1 |
| NONO | non-POU domain-containing octamer-binding protein |
| OPTN | Optineurin |
| PART | primary age-related tauopathy |
| PiD | Pick disease |
| PSP | progressive supranuclear palsy |
| PSPC1 | paraspeckle component 1 |
| RBM14 | RNA binding motif protein 14 |
| SD-NFT | senile dementia of the neurofibrillary tangle type |
| SFPQ | splicing factor proline/glutamine rich |
| TDP-43 | transactivation response DNA binding protein 43 kDa |
| TMEM106B | transmembrane protein 106B. |
References
- Buratti, E.; Dörk, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20, 1774–1784. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef]
- Mann, D.M.A.; Snowden, J.S. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 2017, 27, 723–736. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Majtenyi, K.; Spina, S.; Murrell, J.R.; Gelpi, E.; Hoftberger, R.; Fraser, G.; Crowther, R.A.; Goedert, M.; Budka, H.; et al. White matter tauopathy with globular glial inclusions: A distinct sporadic frontotemporal lobar degeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 963–975. [Google Scholar] [CrossRef]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Ferrer, I.; Grinberg, L.T.; Alafuzoff, I.; Attems, J.; Budka, H.; Cairns, N.J.; Crary, J.F.; Duyckaerts, C.; Ghetti, B.; et al. Aging-related tau astrogliopathy (ARTAG): Harmonized evaluation strategy. Acta Neuropathol. 2016, 131, 87–102. [Google Scholar] [CrossRef]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Tosakulwong, N.; Weigand, S.D.; Petrucelli, L.; Liesinger, A.M.; Petersen, R.C.; Parisi, J.E.; Dickson, D.W. Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol. 2016, 131, 571–585. [Google Scholar] [CrossRef]
- Josephs, K.A.; Martin, P.R.; Weigand, S.D.; Tosakulwong, N.; Buciuc, M.; Murray, M.E.; Petrucelli, L.; Senjem, M.L.; Spychalla, A.J.; Knopman, D.S.; et al. Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 2020, 143, 3463–3476. [Google Scholar] [CrossRef] [PubMed]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.H.; Ke, Y.D.; Ittner, L.M.; Halliday, G.M. ALS/FTLD: Experimental models and reality. Acta Neuropathol. 2017, 133, 177–196. [Google Scholar] [CrossRef] [PubMed]
- West, J.A.; Mito, M.; Kurosaka, S.; Takumi, T.; Tanegashima, C.; Chujo, T.; Yanaka, K.; Kingston, R.E.; Hirose, T.; Bond, C.; et al. Structural, super-resolution microscopy analysis of paraspeckle nuclear body organization. J. Cell Biol. 2016, 214, 817–830. [Google Scholar] [CrossRef]
- Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Co-localization of Bunina bodies and TDP-43 inclusions in lower motor neurons in amyotrophic lateral sclerosis. Neuropathology 2014, 34, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Seilhean, D.; Le Ber, I.; Millecamps, S.; Sazdovitch, V.; Kitagawa, K.; Uchihara, T.; Duyckaerts, C. Amygdala TDP-43 Pathology in Frontotemporal Lobar Degeneration and Motor Neuron Disease. J. Neuropathol. Exp. Neurol. 2017, 76, 800–812. [Google Scholar] [CrossRef]
- Riku, Y.; Watanabe, H.; Yoshida, M.; Mimuro, M.; Iwasaki, Y.; Masuda, M.; Ishigaki, S.; Katsuno, M.; Sobue, G. Marked Involvement of the Striatal Efferent System in TAR DNA-Binding Protein 43 kDa-Related Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2016, 75, 801–811. [Google Scholar] [CrossRef]
- Riku, Y.; Watanabe, H.; Yoshida, M.; Mimuro, M.; Iwasaki, Y.; Masuda, M.; Ishigaki, S.; Katsuno, M.; Sobue, G. Pathologic involvement of glutamatergic striatal inputs from the cortices in TAR DNA-binding protein 43 kDa-related frontotemporal lobar degeneration and amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2017, 76, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.; Lee, V.M. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin- containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef]
- Ando, T.; Nakamura, R.; Kuru, S.; Yokoi, D.; Atsuta, N.; Koike, H.; Suzuki, M.; Hara, K.; Iguchi, Y.; Harada, Y.; et al. The wide-ranging clinical and genetic features in Japanese families with valosin-containing protein proteinopathy. Neurobiol. Aging 2021, 100, 120.e1–120.e6. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Weng, S.M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Gijselinck, I.; van der Zee, J.; Engelborghs, S.; Wils, H.; Pirici, D.; Rademakers, R.; Vandenberghe, R.; Dermaut, B.; Martin, J.J.; et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 2006, 442, 920–924. [Google Scholar] [CrossRef]
- van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Okamoto, K.; Fujita, Y.; Hoshino, E.; Tamura, Y.; Fukuda, T.; Hasegawa, M.; Takatama, M. An autopsy case of familial amyotrophic lateral sclerosis with a TARDBP Q343R mutation. Neuropathology 2015, 35, 462–468. [Google Scholar] [CrossRef]
- Geser, F.; Martinez-Lage, M.; Robinson, J.; Uryu, K.; Neumann, M.; Brandmeir, N.J.; Xie, S.X.; Kwong, L.K.; Elman, L.; McCluskey, L.; et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. 2009, 66, 180–189. [Google Scholar] [CrossRef]
- Riku, Y.; Watanabe, H.; Yoshida, M.; Tatsumi, S.; Mimuro, M.; Iwasaki, Y.; Katsuno, M.; Iguchi, Y.; Masuda, M.; Senda, J.; et al. Lower motor neuron involvement in TAR DNA-binding protein of 43 kDa-related frontotemporal lobar degeneration and amyotrophic lateral sclerosis. JAMA Neurol. 2014, 71, 172–179. [Google Scholar] [CrossRef]
- Nishihira, Y.; Tan, C.F.; Onodera, O.; Toyoshima, Y.; Yamada, M.; Morita, T.; Nishizawa, M.; Kakita, A.; Takahashi, H. Sporadic amyotrophic lateral sclerosis: Two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 2008, 116, 169–182. [Google Scholar] [CrossRef]
- Robinson, J.L.; Porta, S.; Garrett, F.G.; Zhang, P.; Xie, S.X.; Suh, E.; Van Deerlin, V.M.; Abner, E.L.; Jicha, G.A.; Barber, J.M.; et al. Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain 2020, 143, 2844–2857. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, D.S.; Salmon, D.P.; Galasko, D.; Edland, S.D.; Pizzo, D.P.; Goodwill, V.; Hiniker, A. TDP-43 Pathology Exacerbates Cognitive Decline in Primary Age-Related Tauopathy. Ann. Neurol. 2022, 92, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3858–3863. [Google Scholar] [CrossRef]
- Baskaran, P.; Shaw, C.; Guthrie, S. TDP-43 causes neurotoxicity and cytoskeletal dysfunction in primary cortical neurons. PLoS ONE 2018, 13, e0196528. [Google Scholar]
- Ebstein, S.Y.; Yagudayeva, I.; Shneider, N.A. Mutant TDP-43 Causes early-stage dose-dependent motor neuron degeneration in a TARDBP knockin mouse model of ALS. Cell Rep. 2019, 26, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 4220. [Google Scholar] [CrossRef]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis-a model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef]
- Yang, C.; Wang, H.; Qiao, T.; Yang, B.; Aliaga, L.; Qiu, L.; Tan, W.; Salameh, J.; McKenna-Yasek, D.M.; Smith, T.; et al. Partial loss of TDP-43 function causes phenotypes of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, E1121–E1129. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.S.; Cheng, W.C.; Chen, C.Y.; Wu, M.C.; Wang, Y.C.; Tseng, Y.H.; Chuang, T.J.; Shen, C.J. Transcriptomopathies of pre- and post-symptomatic frontotemporal dementia-like mice with TDP-43 depletion in forebrain neurons. Acta Neuropathol. Commun. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Donde, A.; Sun, M.; Ling, J.P.; Braunstein, K.E.; Pang, B.; Wen, X.; Cheng, X.; Chen, L.; Wong, P.C. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019, 138, 813–826. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Mariani, A.; Watkins, L.R.; Link, C.D. TDP-43 knockdown causes innate immune activation via protein kinase R in astrocytes. Neurobiol. Dis. 2019, 132, 104514. [Google Scholar] [CrossRef]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef]
- Boluda, S.; Iba, M.; Zhang, B.; Raible, K.M.; Lee, V.M.; Trojanowski, J.Q. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015, 129, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Hench, J.; Lavenir, I.; Schweighauser, G.; Frank, S.; Goedert, M.; Tolnay, M. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014, 127, 299–301. [Google Scholar] [CrossRef]
- Espino de la Fuente-Muñoz, C.; Rosas-Lemus, M.; Moreno-Castilla, P.; Bermúdez-Rattoni, F.; Uribe-Carvajal, S.; Arias, C. Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 8727. [Google Scholar] [CrossRef]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef]
- Hara, M.; Hirokawa, K.; Kamei, S.; Uchihara, T. Isoform transition from four-repeat to three-repeat tau underlies dendrosomatic and regional progression of neurofibrillary pathology. Acta Neuropathol. 2013, 125, 565–579. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Braak, H.; Brion, J.P.; Buée, L.; Del Tredici, K.; Goedert, M.; Halliday, G.; Neumann, M.; Spillantini, M.G.; Tolnay, M.; et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015, 129, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Abner, E.L.; Schmitt, F.A.; Kryscio, R.J.; Jicha, G.A.; Santacruz, K.; Smith, C.D.; Patel, E.; Markesbery, W.R. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M. Senile dementia of the neurofibrillary tangle type (tangle-only dementia): Neuropathological criteria and clinical guidelines for diagnosis. Neuropathology 2003, 23, 311–317. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Weigand, S.D.; Murray, M.E.; Tosakulwong, N.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Knopman, D.S.; Boeve, B.F.; et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014, 127, 811–824. [Google Scholar] [CrossRef] [PubMed]
- McAleese, K.E.; Walker, L.; Erskine, D.; Thomas, A.J.; McKeith, I.G.; Attems, J. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 2017, 27, 472–479. [Google Scholar] [CrossRef]
- Koga, S.; Sanchez-Contreras, M.; Josephs, K.A.; Uitti, R.J.; Graff-Radford, N.; van Gerpen, J.A.; Cheshire, W.P.; Wszolek, Z.K.; Rademakers, R.; Dickson, D.W. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov. Disord. 2017, 32, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Yokota, O.; Davidson, Y.; Bigio, E.H.; Ishizu, H.; Terada, S.; Arai, T.; Hasegawa, M.; Akiyama, H.; Sikkink, S.; Pickering-Brown, S.; et al. Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol. 2010, 120, 55–66. [Google Scholar] [CrossRef]
- Koga, S.; Kouri, N.; Walton, R.L.; Ebbert, M.T.W.; Josephs, K.A.; Litvan, I.; Graff-Radford, N.; Ahlskog, J.E.; Uitti, R.J.; van Gerpen, J.A.; et al. Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: A distinct clinicopathologic subtype. Acta Neuropathol. 2018, 136, 389–404. [Google Scholar] [CrossRef]
- Uryu, K.; Nakashima-Yasuda, H.; Forman, M.S.; Kwong, L.K.; Clark, C.M.; Grossman, M.; Miller, B.L.; Kretzschmar, H.A.; Lee, V.M.; Trojanowski, J.Q.; et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 555–564. [Google Scholar] [CrossRef]
- Aoki, N.; Murray, M.E.; Ogaki, K.; Fujioka, S.; Rutherford, N.J.; Rademakers, R.; Ross, O.A.; Dickson, D.W. Hippocampal sclerosis in Lewy body disease is a TDP-43 proteinopathy similar to FTLD-TDP Type A. Acta Neuropathol. 2015, 129, 53–64. [Google Scholar] [CrossRef]
- Nakashima-Yasuda, H.; Uryu, K.; Robinson, J.; Xie, S.X.; Hurtig, H.; Duda, J.E.; Arnold, S.E.; Siderowf, A.; Grossman, M.; Leverenz, J.B.; et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007, 114, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Amador-Ortiz, C.; Lin, W.L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 2007, 61, 435–445. [Google Scholar] [CrossRef]
- Kenney, K.; Iacono, D.; Edlow, B.L.; Katz, D.I.; Diaz-Arrastia, R.; Dams-O’Connor, K.; Daneshvar, D.H.; Stevens, A.; Moreau, A.L.; Tirrell, L.S.; et al. Dementia After Moderate-Severe Traumatic Brain Injury: Coexistence of Multiple Proteinopathies. J. Neuropathol. Exp. Neurol. 2018, 77, 50–63. [Google Scholar] [CrossRef]
- Prabowo, A.S.; Iyer, A.M.; Veersema, T.J.; Anink, J.J.; Schouten-van Meeteren, A.Y.; Spliet, W.G.; van Rijen, P.C.; Ferrier, C.H.; Thom, M.; Aronica, E. Expression of neurodegenerative disease-related proteins and caspase-3 in glioneuronal tumours. Neuropathol. Appl. Neurobiol. 2015, 41, e1–e15. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Matsubara, T.; Miyamoto, R.; Sumikura, H.; Takeuchi, T.; Maruyama Saladini, K.; Kawarai, T.; Nodera, H.; Udaka, F.; Kume, K.; et al. Co-morbidity of progressive supranuclear palsy and amyotrophic lateral sclerosis: A clinical-pathological case report. BMC Neurol. 2019, 19, 168. [Google Scholar] [CrossRef]
- Koga, S.; Zhou, X.; Murakami, A.; Fernandez De Castro, C.; Baker, M.C.; Rademakers, R.; Dickson, D.W. Concurrent tau pathologies in frontotemporal lobar degeneration with TDP-43 pathology. Neuropathol. Appl. Neurobiol. 2022, 48, e12778. [Google Scholar] [CrossRef]
- Tomé, S.O.; Vandenberghe, R.; Ospitalieri, S.; Van Schoor, E.; Tousseyn, T.; Otto, M.; von Arnim, C.A.F.; Thal, D.R. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathol. Commun. 2020, 8, 61. [Google Scholar] [CrossRef]
- Robinson, J.L.; Yan, N.; Caswell, C.; Xie, S.X.; Suh, E.; Van Deerlin, V.M.; Gibbons, G.; Irwin, D.J.; Grossman, M.; Lee, E.B.; et al. Primary Tau Pathology, Not Copathology, Correlates With Clinical Symptoms in PSP and CBD. J. Neuropathol. Exp. Neurol. 2020, 79, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Riku, Y.; Iwasaki, Y.; Ishigaki, S.; Akagi, A.; Hasegawa, M.; Nishioka, K.; Li, Y.; Riku, M.; Ikeuchi, T.; Fujioka, Y.; et al. Motor neuron TDP-43 proteinopathy in progressive supranuclear palsy and corticobasal degeneration. Brain 2022, 145, 2769–2784. [Google Scholar] [CrossRef]
- Behrouzi, R.; Liu, X.; Wu, D.; Robinson, A.C.; Tanaguchi-Watanabe, S.; Rollinson, S.; Shi, J.; Tian, J.; Hamdalla, H.H.; Ealing, J.; et al. Pathological tau deposition in Motor Neurone Disease and frontotemporal lobar degeneration associated with TDP-43 proteinopathy. Acta Neuropathol. Commun. 2016, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Strong, M.J. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotroph. Lateral. Scler. 2012, 13, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.H.; Guthrie, N.J.; van Roijen, M.; Halliday, G.M.; Ooi, L. Increased Tau Phosphorylation in Motor Neurons from Clinically Pure Sporadic Amyotrophic Lateral Sclerosis Patients. J. Neuropathol. Exp. Neurol. 2019, 78, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Bieniek, K.F.; Murray, M.E.; Rutherford, N.J.; Castanedes-Casey, M.; DeJesus-Hernandez, M.; Liesinger, A.M.; Baker, M.C.; Boylan, K.B.; Rademakers, R.; Dickson, D.W. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. 2013, 125, 289–302. [Google Scholar] [CrossRef]
- Robinson, A.C.; Thompson, J.C.; Weedon, L.; Rollinson, S.; Pickering-Brown, S.; Snowden, J.S.; Davidson, Y.S.; Mann, D.M. No interaction between tau and TDP-43 pathologies in either frontotemporal lobar degeneration or motor neurone disease. Neuropathol. Appl. Neurobiol. 2014, 40, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.D.; Bachstetter, A.D.; Ighodaro, E.; Roberts, K.; Abner, E.L.; Fardo, D.W.; Nelson, P.T. Overlapping but distinct TDP-43 and tau pathologic patterns in aged hippocampi. Brain Pathol. 2018, 28, 264–273. [Google Scholar] [CrossRef]
- Köhler, C. Granulovacuolar degeneration: A neurodegenerative change that accompanies tau pathology. Acta Neuropathol. 2016, 132, 339–359. [Google Scholar] [CrossRef]
- Funk, K.E.; Mrak, R.E.; Kuret, J. Granulovacuolar degeneration (GVD) bodies of Alzheimer’s disease (AD) resemble late-stage autophagic organelles. Neuropathol. Appl. Neurobiol. 2011, 37, 295–306. [Google Scholar] [CrossRef]
- Hunter, S.; Minett, T.; Polvikoski, T.; Mukaetova-Ladinska, E.; Brayne, C.; Cambridge City over-75s Cohort Collaboration. Re-examining tau-immunoreactive pathology in the population: Granulovacuolar degeneration and neurofibrillary tangles. Alzheimers Res. Ther. 2015, 7, 57. [Google Scholar] [CrossRef]
- Thal, D.R.; Del Tredici, K.; Ludolph, A.C.; Hoozemans, J.J.; Rozemuller, A.J.; Braak, H.; Knippschild, U. Stages of granulovacuolar degeneration: Their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol. 2011, 122, 577–589. [Google Scholar] [CrossRef]
- Riku, Y.; Duyckaerts, C.; Boluda, S.; Plu, I.; Le Ber, I.; Millecamps, S.; Salachas, F.; Brainbank NeuroCEB Neuropathology Network; Yoshida, M.; Ando, T.; et al. Increased prevalence of granulovacuolar degeneration in C9orf72 mutation. Acta Neuropathol. 2019, 138, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.H.; Hung, S.T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- Roos, P.; Johannsen, P.; Lindquist, S.G.; Brown, J.M.; Waldemar, G.; Duno, M.; Nielsen, T.T.; Budtz-Jørgensen, E.; Gydesen, S.; Holm, I.E.; et al. Six generations of CHMP2B-mediated Frontotemporal Dementia: Clinical features, predictive testing, progression, and survival. Acta Neurol. Scand. 2022, 145, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Van Schoor, E.; Koper, M.J.; Ospitalieri, S.; Dedeene, L.; Tomé, S.O.; Vandenberghe, R.; Brenner, D.; Otto, M.; Weishaupt, J.; Ludolph, A.C.; et al. Necrosome-positive granulovacuolar degeneration is associated with TDP-43 pathological lesions in the hippocampus of ALS/FTLD cases. Neuropathol. Appl. Neurobiol. 2021, 47, 328–345. [Google Scholar] [CrossRef]
- Kierdaszuk, B.; Berdynski, M.; Palczewski, P.; Golebiowski, M.; Zekanowski, C.; Kaminska, A.M. Sporadic inclusion body myositis: Clinical, pathological, and genetic analysis of eight Polish patients. Folia Neuropathol. 2015, 53, 355–366. [Google Scholar] [CrossRef]
- Ling, H.; Kara, E.; Revesz, T.; Lees, A.J.; Plant, G.T.; Martino, D.; Houlden, H.; Hardy, J.; Holton, J.L. Concomitant progressive supranuclear palsy and chronic traumatic encephalopathy in a boxer. Acta Neuropathol. Commun. 2014, 2, 24. [Google Scholar] [CrossRef]
- Ferrer, I.; Legati, A.; García-Monco, J.C.; Gomez-Beldarrain, M.; Carmona, M.; Blanco, R.; Seeley, W.W.; Coppola, G. Familial behavioral variant frontotemporal dementia associated with astrocyte-predominant tauopathy. J. Neuropathol. Exp. Neurol. 2015, 74, 370–379. [Google Scholar] [CrossRef]
- St-Amour, I.; Turgeon, A.; Goupil, C.; Planel, E.; Hébert, S.S. Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease. Acta Neuropathol. 2018, 135, 249–265. [Google Scholar] [CrossRef]
- Murata-Shinozaki, Y.; Takahashi, T.; Matsubara, T.; Maruyama, H.; Izumi, Y.; Matsumoto, M. The origins of rimmed vacuoles and granulovacuolar degeneration bodies are associated with the Wnt signaling pathway. Neurosci. Lett. 2017, 638, 55–59. [Google Scholar] [CrossRef]
- Weihl, C.C.; Temiz, P.; Miller, S.E.; Watts, G.; Smith, C.; Forman, M.; Hanson, P.I.; Kimonis, V.; Pestronk, A. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1186–1189. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, E.; Höftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Högl, B.; Schmutzhard, E.; et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016, 132, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Götzl, J.K.; Mori, K.; Damme, M.; Fellerer, K.; Tahirovic, S.; Kleinberger, G.; Janssens, J.; van der Zee, J.; Lang, C.M.; Kremmer, E.; et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014, 127, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Koga, S.; Lin, W.L.; Kasanuki, K.; Castanedes-Casey, M.; Wszolek, Z.K.; Oh, S.J.; Tsuboi, Y.; Dickson, D.W. Perry Syn-drome: A Distinctive Type of TDP-43 Proteinopathy. J. Neuropathol. Exp. Neurol. 2017, 76, 676–682. [Google Scholar] [CrossRef]
- Haraguchi, T.; Terada, S.; Ishizu, H.; Yokota, O.; Yoshida, H.; Takeda, N.; Kishimoto, Y.; Katayama, N.; Takata, H.; Akagi, M.; et al. Coexistence of TDP-43 and tau pathology in neurodegeneration with brain iron accumulation type 1 (NBIA-1, formerly Hallervorden-Spatz syndrome). Neuropathology 2011, 31, 531–539. [Google Scholar] [CrossRef]
- Ayaki, T.; Ito, H.; Komure, O.; Kamada, M.; Nakamura, M.; Wate, R.; Kusaka, H.; Yamaguchi, Y.; Li, F.; Kawakami, H.; et al. Multiple Proteinopathies in Familial ALS Cases With Optineurin Mutations. J. Neuropathol. Exp. Neurol. 2018, 77, 128–138. [Google Scholar] [CrossRef]
- Honda, H.; Sasagasako, N.; Shen, C.; Shijo, M.; Hamasaki, H.; Suzuki, S.O.; Tsuboi, Y.; Fujii, N.; Iwaki, T. DCTN1 F52L mutation case of Perry syndrome with progressive supranuclear palsy-like tauopathy. Parkinsonism. Relat. Disord. 2018, 51, 105–110. [Google Scholar] [CrossRef]
- Wisniewski, K.; Jervis, G.A.; Moretz, R.C.; Wisniewski, H.M. Alzheimer neurofibrillary tangles in diseases other than senile and presenile dementia. Ann. Neurol. 1979, 5, 288–294. [Google Scholar] [CrossRef]
- Huin, V.; Barbier, M.; Bottani, A.; Lobrinus, J.A.; Clot, F.; Lamari, F.; Chat, L.; Rucheton, B.; Fluchère, F.; Auvin, S.; et al. Homozygous GRN mutations: New phenotypes and new insights into pathological and molecular mechanisms. Brain 2020, 143, 303–319. [Google Scholar] [CrossRef]
- Schwab, C.; Arai, T.; Hasegawa, M.; Yu, S.; McGeer, P.L. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1159–1165. [Google Scholar] [CrossRef]
- Forrest, S.L.; Shepherd, C.E.; McCann, H.; Kwok, J.B.; Halliday, G.M.; Kril, J.J. Globular glial tauopathy with a mutation in MAPT and unusual TDP-43 proteinopathy in a patient with behavioural-variant frontotemporal dementia. Acta Neuropathol. 2021, 141, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Bieniek, K.F.; Dickson, D.W. Concurrent neurodegenerative pathologies in periventricular nodular heterotopia. Acta Neuropathol. 2015, 130, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Steele, J.C.; Yu, S.; McCall, S.; Sandberg, G.; McGeer, E.G.; McGeer, P.L. Enduring involvement of tau, beta-amyloid, alpha-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam (ALS/PDC). Acta Neuropathol. 2008, 116, 625–637. [Google Scholar] [CrossRef]
- Mimuro, M.; Yoshida, M.; Kuzuhara, S.; Kokubo, Y. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: A multiple proteinopathy? Neuropathology 2018, 38, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Rogaeva, E. Motor neuron disease and frontotemporal dementia: Sometimes related, sometimes not. Exp. Neurol. 2014, 262, 75–83. [Google Scholar] [CrossRef]
- Guo, A.; Tapia, L.; Bamji, S.X.; Cynader, M.S.; Jia, W. Progranulin deficiency leads to enhanced cell vulnerability and TDP-43 translocation in primary neuronal cultures. Brain Res. 2010, 1366, 1–8. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, N.; Li, X.; Zheng, X.; Zhao, J.; Xia, H.; Mao, Q. Suppression of Progranulin Expression Leads to Formation of Intranuclear TDP-43 Inclusions In Vitro: A Cell Model of Frontotemporal Lobar Degeneration. J. Neuropathol. Exp. Neurol. 2019, 78, 1124–1129. [Google Scholar] [CrossRef]
- Hosokawa, M.; Arai, T.; Masuda-Suzukake, M.; Kondo, H.; Matsuwaki, T.; Nishihara, M.; Hasegawa, M.; Akiyama, H. Progranulin reduction is associated with increased tau phosphorylation in P301L tau transgenic mice. J. Neuropathol. Exp. Neurol. 2015, 74, 158–165. [Google Scholar] [CrossRef]
- Werner, G.; Damme, M.; Schludi, M.; Gnörich, J.; Wind, K.; Fellerer, K.; Wefers, B.; Wurst, W.; Edbauer, D.; Brendel, M.; et al. Loss of TMEM106B potentiates lysosomal and FTLD-like pathology in progranulin-deficient mice. EMBO Rep. 2020, 21, e50241. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Bereczki, E.; Zhang, H.; Wang, S.; Li, C.; Ji, X.; Branca, R.M.; Lehtiö, J.; Guan, Z.; Filipcik, P.; et al. Mammalian target of rapamycin (mTor) mediates tau protein dyshomeostasis: Implication for Alzheimer disease. J. Biol. Chem. 2013, 288, 15556–15570. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.F.; Guo, B.S.; Liu, Y.C.; Wu, C.C.; Yang, C.H.; Tsai, K.J.; Shen, C.K. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef]
- Llibre-Guerra, J.J.; Lee, S.E.; Suemoto, C.K.; Ehrenberg, A.J.; Kovacs, G.G.; Karydas, A.; Staffaroni, A.; Franca Resende, E.P.; Kim, E.J.; Hwang, J.H.; et al. A novel temporal-predominant neuro-astroglial tauopathy associated with TMEM106B gene polymorphism in FTLD/ALS-TDP. Brain Pathol. 2021, 31, 267–282. [Google Scholar] [CrossRef]
- White, M.A.; Kim, E.; Duffy, A.; Adalbert, R.; Phillips, B.U.; Peters, O.M.; Stephenson, J.; Yang, S.; Massenzio, F.; Lin, Z.; et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 2018, 21, 552–563. [Google Scholar] [CrossRef]
- Moszczynski, A.J.; Harvey, M.; Fulcher, N.; de Oliveira, C.; McCunn, P.; Donison, N.; Bartha, R.; Schmid, S.; Strong, M.J.; Volkening, K. Synergistic toxicity in an in vivo model of neurodegeneration through the co-expression of human TDP-43M337V and tauT175D protein. Acta Neuropathol. Commun. 2019, 7, 170. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Yokoyama, J.S.; Karch, C.M.; Fan, C.C.; Bonham, L.W.; Kouri, N.; Ross, O.A.; Rademakers, R.; Kim, J.; Wang, Y.; Höglinger, G.U.; et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017, 133, 825–837. [Google Scholar] [CrossRef]
- Karch, C.M.; Wen, N.; Fan, C.C.; Yokoyama, J.S.; Kouri, N.; Ross, O.A.; Höglinger, G.; Müller, U.; Ferrari, R.; Hardy, J.; et al. Selective Genetic Overlap Between Amyotrophic Lateral Sclerosis and Diseases of the Frontotemporal Dementia Spectrum. JAMA Neurol. 2018, 75, 860–875. [Google Scholar] [CrossRef]
- Cali, C.P.; Patino, M.; Tai, Y.K.; Ho, W.Y.; McLean, C.A.; Morris, C.M.; Seeley, W.W.; Miller, B.L.; Gaig, C.; Vonsattel, J.P.G.; et al. C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol. 2019, 138, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Fujioka, Y.; Okada, Y.; Riku, Y.; Udagawa, T.; Honda, D.; Yokoi, S.; Endo, K.; Ikenaka, K.; Takagi, S.; et al. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Rep. 2017, 18, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Luisier, R.; Tyzack, G.E.; Hall, C.E.; Mitchell, J.S.; Devine, H.; Taha, D.M.; Malik, B.; Meyer, I.; Greensmith, L.; Newcombe, J.; et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat. Commun. 2018, 9, 2010. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, G.E.; Neeves, J.; Crerar, H.; Klein, P.; Ziff, O.; Taha, D.M.; Luisier, R.; Luscombe, N.M.; Patani, R. Aberrant cytoplasmic intron retention is a blueprint for RNA binding protein mislocalization in amyotrophic lateral sclerosis. Brain 2021, 144, 1985–1993. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Riku, Y.; Fujioka, Y.; Endo, K.; Iwade, N.; Kawai, K.; Ishibashi, M.; Yokoi, S.; Katsuno, M.; Watanabe, H.; et al. Aberrant interaction between FUS and SFPQ in neurons in a wide range of FTLD spectrum diseases. Brain 2020, 143, 2398–2405. [Google Scholar] [CrossRef]
- Tyzack, G.E.; Manferrari, G.; Newcombe, J.; Luscombe, N.M.; Luisier, R.; Patani, R. Paraspeckle components NONO and PSPC1 are not mislocalized from motor neuron nuclei in sporadic ALS. Brain 2020, 143, e66. [Google Scholar] [CrossRef]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef]
- Nelson, P.T.; Brayne, C.; Flanagan, M.E.; Abner, E.L.; Agrawal, S.; Attems, J.; Castellani, R.J.; Corrada, M.M.; Cykowski, M.D.; Di, J.; et al. Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: Combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol. 2022, 144, 27–44. [Google Scholar] [CrossRef]
- Wennberg, A.M.; Tosakulwong, N.; Lesnick, T.G.; Murray, M.E.; Whitwell, J.L.; Liesinger, A.M.; Petrucelli, L.; Boeve, B.F.; Parisi, J.E.; Knopman, D.S.; et al. Association of Apolipoprotein E ε4 With Transactive Response DNA-Binding Protein 43. JAMA Neurol. 2018, 75, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Vardarajan, B.N.; Reyes-Dumeyer, D.; Piriz, A.L.; Lantigua, R.A.; Medrano, M.; Rivera, D.; Jiménez-Velázquez, I.Z.; Martin, E.; Pericak-Vance, M.A.; Bush, W.; et al. Progranulin mutations in clinical and neuropathological Alzheimer’s disease. Alzheimers Dement. 2022. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorders | Pathogenesis/Key Molecule | Primary Aggregation | Description of Tau Pathology | Description of TDP-43 Pathology | Note, References |
|---|---|---|---|---|---|
| Aged people | PART/AD-NC | LATE | [4,7,8] | ||
| ALS/FTLD-TDP (sporadic or GRN, c9orf72, and CHMP2B mutations) | TDP-43 | mostly PART/AD-NC, but unclassifiable in some cases | ALS/FTLD-TDP | [81,82,83,84,94] | |
| ALS with OPTN E478G mutation | pathologic OPTN mutation | TDP-43 | neuronal and glial 3R/4R-tau inclusions | ALS/FTLD-TDP | [106] |
| FTD with FUS Q140H mutation | FUS mutation with unclear significance | 3R and 4R-tau | neuronal and glial 3R/4R-tau inclusions | granular neuronal inclusions in frontotemporal cortices | FUS pathology is absent [98] |
| ALS/FTLD-TDP with TMEM106B rs1990622 A/A | risk allele for TDP-43 pathology | TDP-43 | neuronal and astrocytic 4R-tau aggregates | ALS/FTLD-TDP | [125] |
| Perry syndrome | pathogenic DCTN1 mutation | TDP-43 | PSP-like (tau isoforms are not described) | FTD-MND-like, with prominent involvement of substantia nigra | [104,107] |
| PSP | 4R-tau | PSP-NC | prominent in hippocampus, ALS-like in motor neuron | [66,67,79,80] | |
| CBD | 4R-au | CBD-NC | prominent in midbrain, ALS-like in motor neuron | [68,69,79,80] | |
| FTD with MAPT IVS10 + 16 mutation | pathogenic MAPT mutation | 4R-tau | GGT | FTLD-TDP type A or B-like, densely spread | [111] |
| AD | 3R and 4R-tau, Aβ-amyloid | AD-NC | LATE or FTLD-TDP type A like | [10,11,64,65] | |
| Lewy body disease | α-synuclein | PART/AD-NC or colocalized with Lewy bodies | FTLD-TDP type A-like | [70,71,72] | |
| Guam ALS/PDC | tau, TDP-43, α-synuclein | NFT, granular or thorn-shaped astrocytic aggregates | ALS-TDP-like | [113] | |
| Kii peninsula ALS/PDC | tau, TDP-43, α-synuclein | NFT prominent in presubiculum | ALS-TDP-like | [114] | |
| Neuronal ceroid lipofuscinosis, including homozygous GRN mutation | lysosomal storage disease | mostly PART/AD-NC, but unclassifiable in some cases | cytoplasm of ballooned neurons | [103,108,109] | |
| Huntington disease | Huntingtin CAG repeat | CAG repeat | PART/AD-NC | colocalized with cytoplasmic/neuritic inclusions of huntingtin | [99,110] |
| NBIA type 1 | pathogenic PANK2 mutation | broadly extended NFT | neuronal and glial inclusions | [105] | |
| periventricular nodular heterotopia | focal NFT | neuronal cytoplasmic and neuritic inclusions, thorn-shaped astrocytic aggregates, and a perivascular inclusion body | immunoreactivities were presented within heterotopia [112] | ||
| glioneuronal tumors | activated mTOR signaling | diffuse cytoplasmic inclusions or neuropil threads in tumor | GVD-like | [75] | |
| chronic traumatic encephalopathy | 3R and 4R-tau | neuronal and glial inclusions prominent nearby traumatic sites and periventricular regions | sparse neuronal inclusions out of traumatic sites | [74,97] | |
| anti-IgLON5-related tauopathy | anti-IgLON5 antibody | 3R and 4R-tau | 3R/4R-tau pathology in hypothalamus and brainstem tegmentum | microglial and neuronal TDP-43 pathology in basal ganglia and midbrain | [102] |
| IBM (sporadic or VCP mutations) | muscle inclusion bodies | muscle inclusion bodies | GVD-related epitopes are also present [96,100,101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riku, Y.; Yoshida, M.; Iwasaki, Y.; Sobue, G.; Katsuno, M.; Ishigaki, S. TDP-43 Proteinopathy and Tauopathy: Do They Have Pathomechanistic Links? Int. J. Mol. Sci. 2022, 23, 15755. https://doi.org/10.3390/ijms232415755
Riku Y, Yoshida M, Iwasaki Y, Sobue G, Katsuno M, Ishigaki S. TDP-43 Proteinopathy and Tauopathy: Do They Have Pathomechanistic Links? International Journal of Molecular Sciences. 2022; 23(24):15755. https://doi.org/10.3390/ijms232415755
Chicago/Turabian StyleRiku, Yuichi, Mari Yoshida, Yasushi Iwasaki, Gen Sobue, Masahisa Katsuno, and Shinsuke Ishigaki. 2022. "TDP-43 Proteinopathy and Tauopathy: Do They Have Pathomechanistic Links?" International Journal of Molecular Sciences 23, no. 24: 15755. https://doi.org/10.3390/ijms232415755
APA StyleRiku, Y., Yoshida, M., Iwasaki, Y., Sobue, G., Katsuno, M., & Ishigaki, S. (2022). TDP-43 Proteinopathy and Tauopathy: Do They Have Pathomechanistic Links? International Journal of Molecular Sciences, 23(24), 15755. https://doi.org/10.3390/ijms232415755