
Enhanced Therapeutic Effect of Optimized Melittin-dKLA, a Peptide Agent Targeting M2-like Tumor-Associated Macrophages in Triple-Negative Breast Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
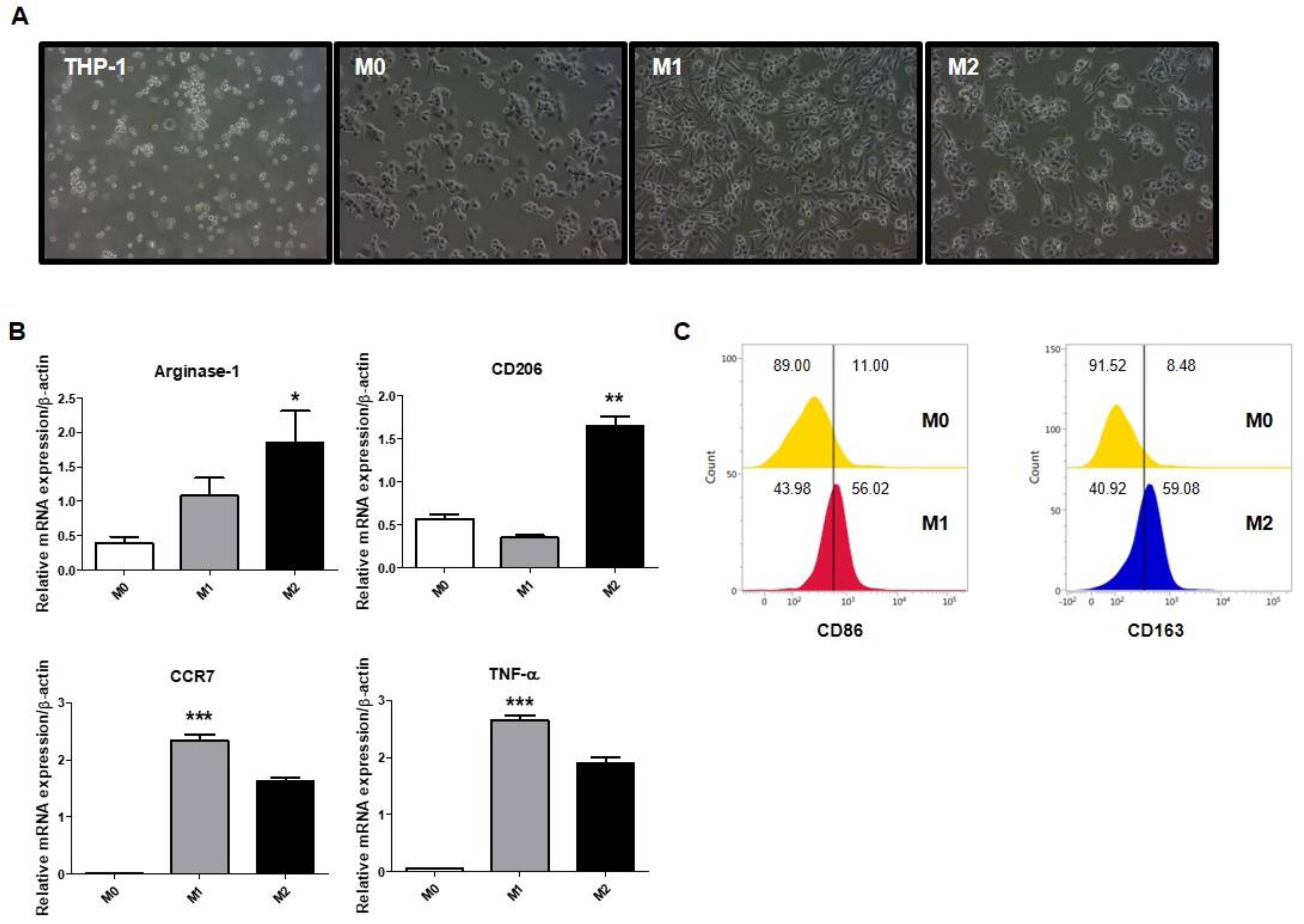
2.1. Polarization of Macrophages in THP-1-Derived Macrophages
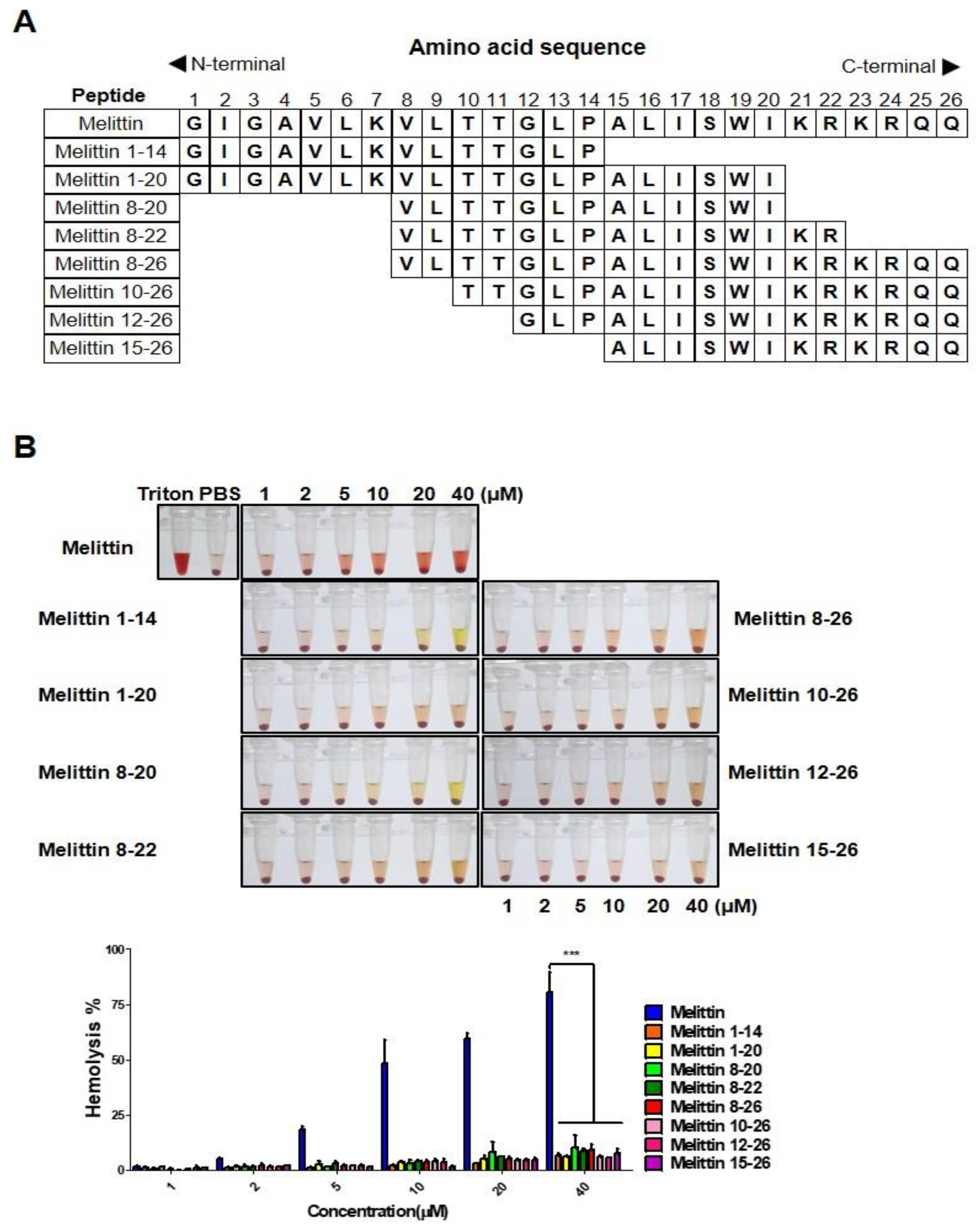
2.2. Melittin Fragments Has Fewer Hemolytic Effects than Melittin
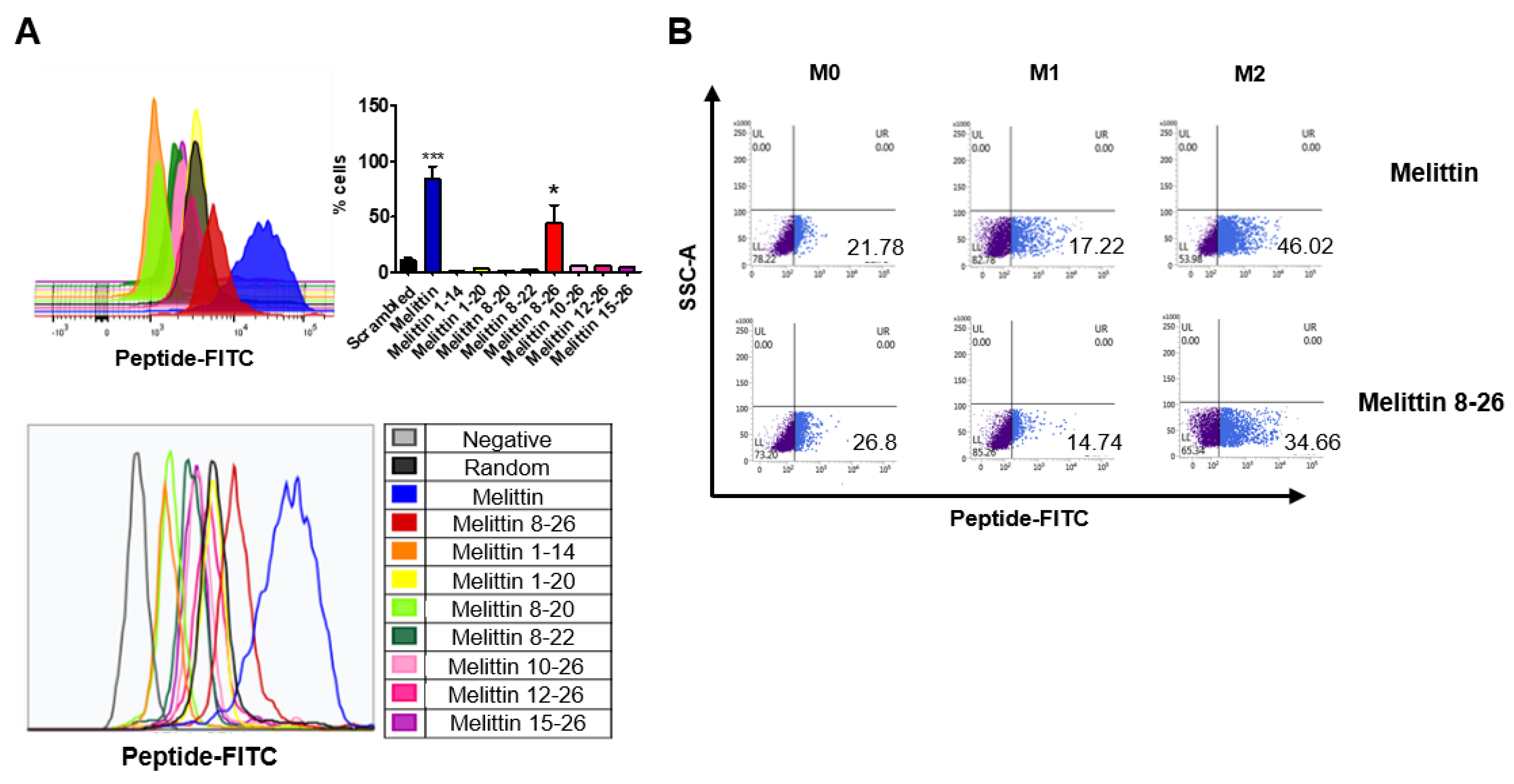
2.3. Binding of Melittin Fragments to the THP-1-Derived M2 Macrophages
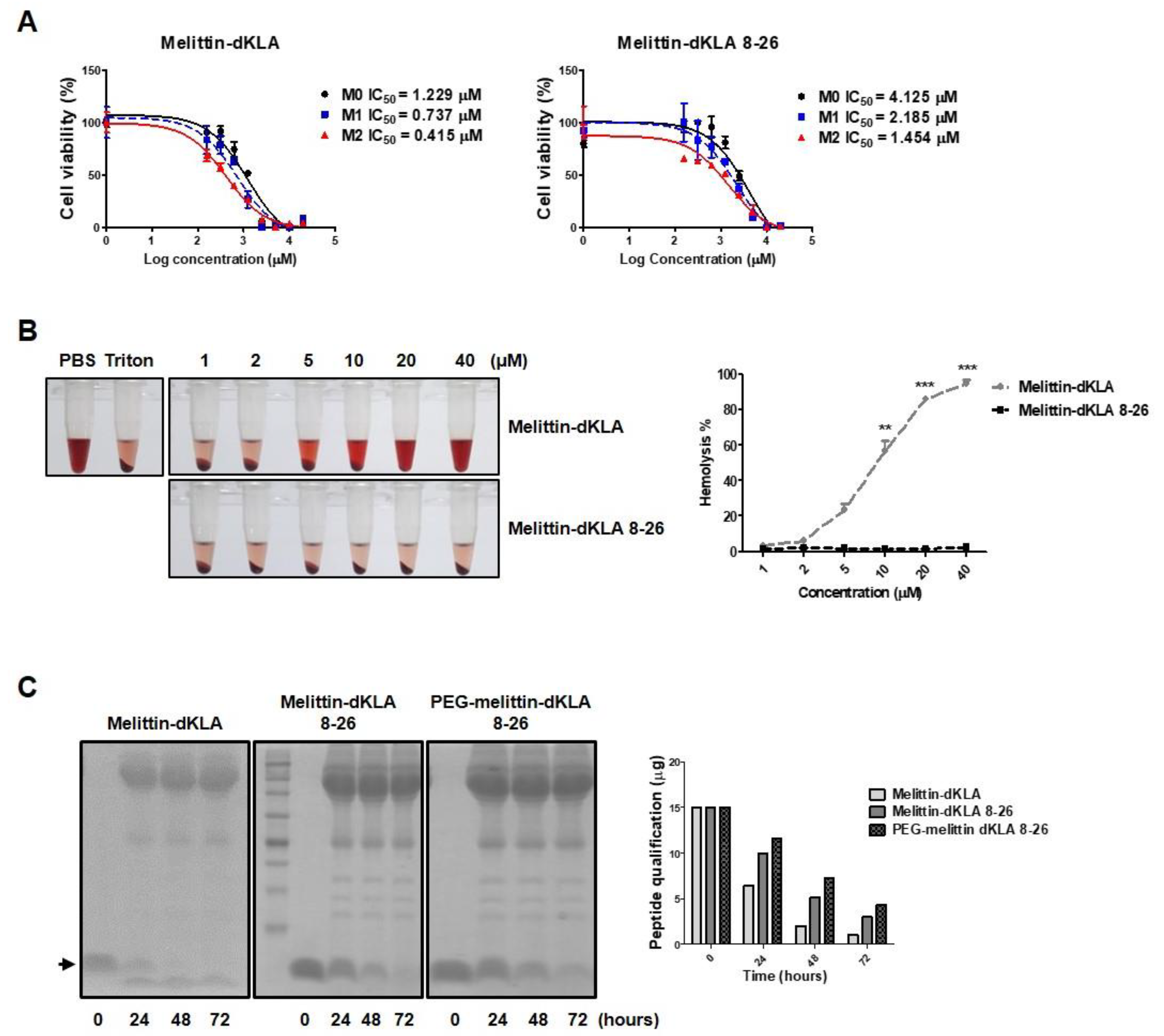
2.4. Characterization of the Optimized Hybrid Peptide Drug, PEG-Melittin-dKLA 8-26
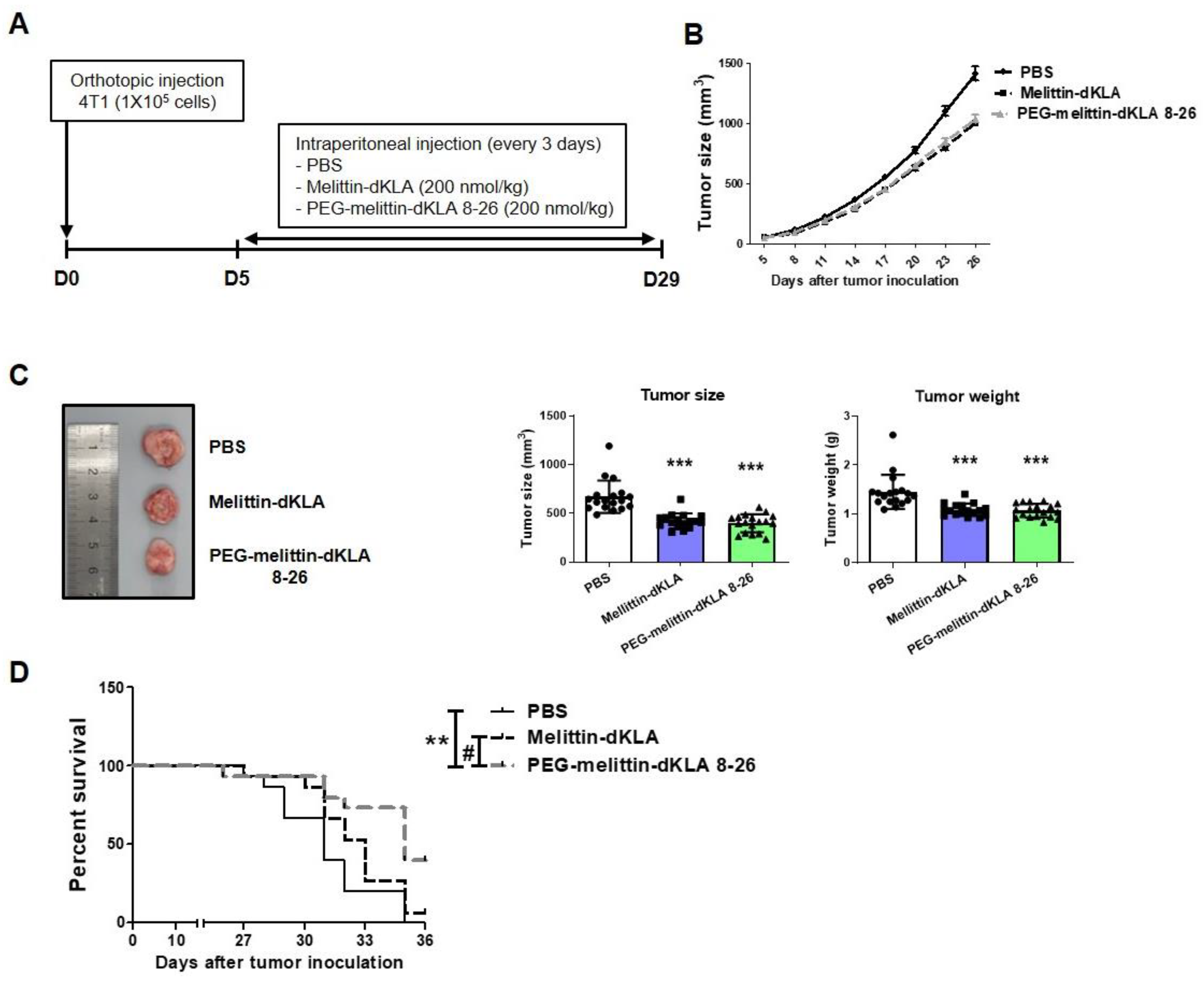
2.5. Inhibition of Tumor Growth and Increased Survival Rates by PEG-Melittin-dKLA 8-26 in 4T1 TNBC Mouse Model
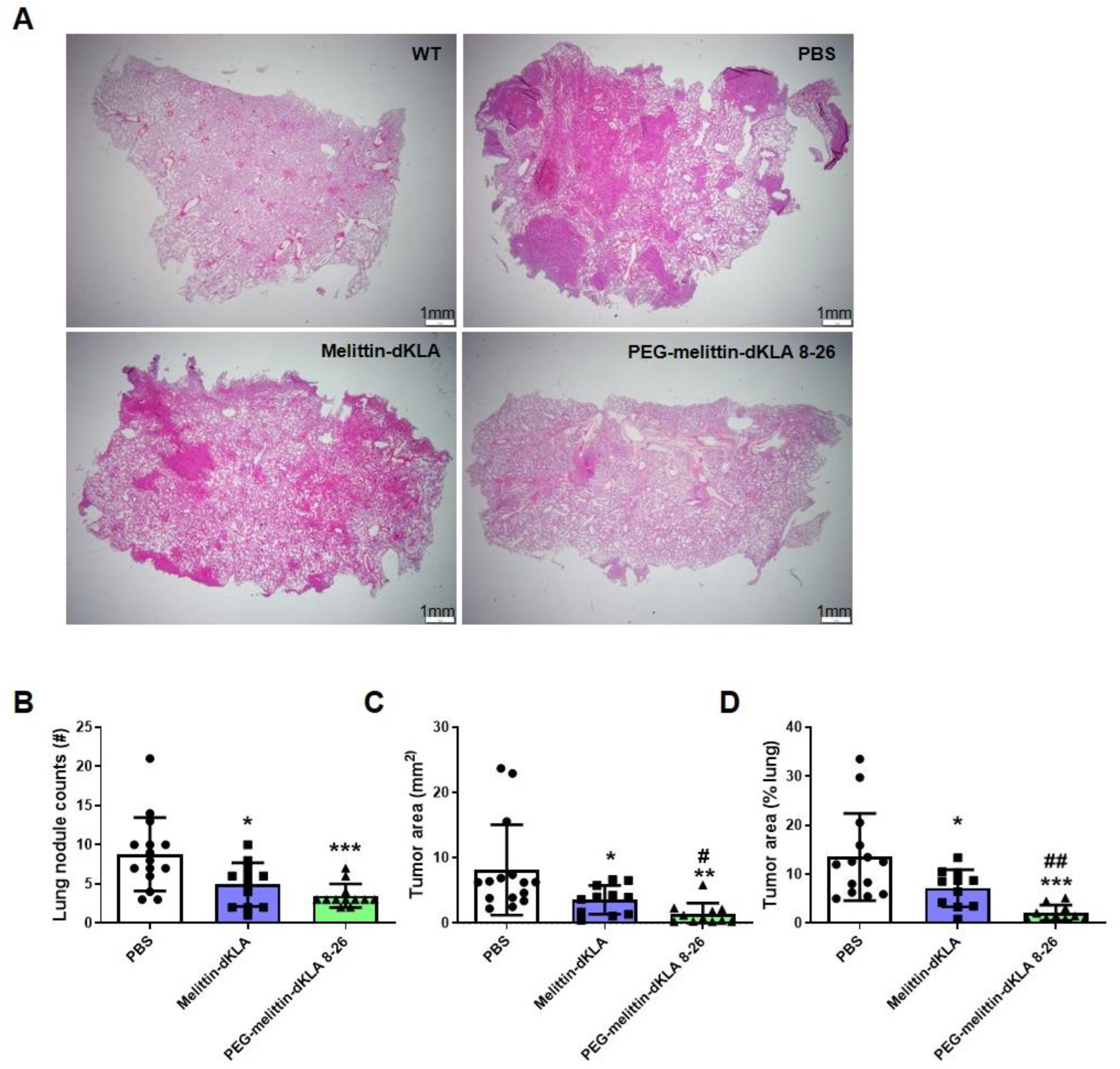
2.6. Enhanced Suppression of Lung Metastasis by PEG-Melittin-dKLA 8-26 in the TNBC Mouse Model
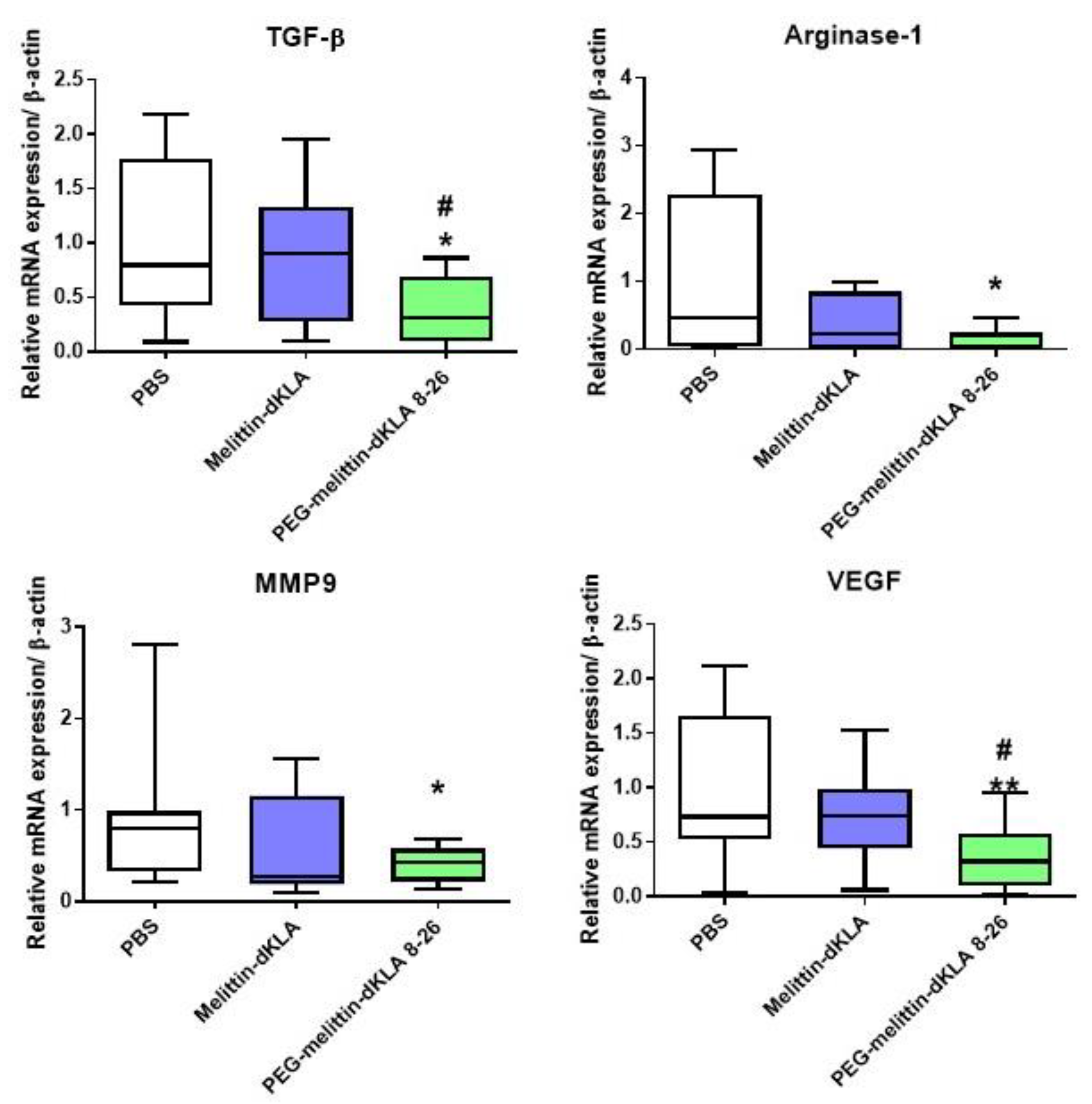
2.7. Reduction of M2-like TAMs by PEG-Melittin-dKLA 8-26 in the 4T1 TNBC Mouse Model
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. Cells and Mice
4.3. Macrophage Polarization
4.4. Flow Cytometry
4.5. MTS Assay
4.6. Stability Test
4.7. Hemolysis Assay
4.8. Animal Study
4.9. Quantitative Real-Time PCR
4.10. H&E Staining
4.11. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. BCR 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Won, K.A.; Spruck, C. Triplenegative breast cancer therapy: Current and future perspectives (Review). Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, D.; Sendi, M.A.; Kelly, C.M. Overview of recent advances in metastatic triple negative breast cancer. World J. Clin. Oncol. 2021, 12, 164–182. [Google Scholar] [CrossRef]
- Lv, Y.; Ma, X.; Du, Y.; Feng, J. Understanding Patterns of Brain Metastasis in Triple-Negative Breast Cancer and Exploring Potential Therapeutic Targets. OncoTargets Ther. 2021, 14, 589–607. [Google Scholar] [CrossRef]
- Jin, L.; Han, B.; Siegel, E.; Cui, Y.; Giuliano, A.; Cui, X. Breast cancer lung metastasis: Molecular biology and therapeutic implications. Cancer Biol. Ther. 2018, 19, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Deepak, K.G.K.; Vempati, R.; Nagaraju, G.P.; Dasari, V.R.; Nagini, S.; Rao, D.N.; Malla, R.R. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 2020, 153, 104683. [Google Scholar] [CrossRef]
- Kuroda, H.; Jamiyan, T.; Yamaguchi, R.; Kakumoto, A.; Abe, A.; Harada, O.; Masunaga, A. Tumor microenvironment in triple-negative breast cancer: The correlation of tumor-associated macrophages and tumor-infiltrating lymphocytes. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2021, 23, 2513–2525. [Google Scholar] [CrossRef]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothe, F.; et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J. Natl. Cancer Inst. 2020, 112, 708–719. [Google Scholar] [CrossRef]
- Mehta, A.K.; Kadel, S.; Townsend, M.G.; Oliwa, M.; Guerriero, J.L. Macrophage Biology and Mechanisms of Immune Suppression in Breast Cancer. Front. Immunol. 2021, 12, 643771. [Google Scholar] [CrossRef]
- Jang, H.; Kim, E.H.; Chi, S.G.; Kim, S.H.; Yang, Y. Nanoparticles Targeting Innate Immune Cells in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 10009. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Cheng, T.; Zhan, J.; Peng, X.; Zhang, Y.; Wen, J.; Chen, X.; Ying, M. Targeting tumor-associated macrophages in the tumor microenvironment. Oncol. Lett. 2020, 20, 234. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Rebelo, S.P.; Pinto, C.; Martins, T.R.; Harrer, N.; Estrada, M.F.; Loza-Alvarez, P.; Cabecadas, J.; Alves, P.M.; Gualda, E.J.; Sommergruber, W.; et al. 3D-3-culture: A tool to unveil macrophage plasticity in the tumour microenvironment. Biomaterials 2018, 163, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, A.; Michel, J.; Schonhaar, K.; Goerdt, S.; Schledzewski, K. Differentiation and gene expression profile of tumor-associated macrophages. Semin. Cancer Biol. 2012, 22, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheng, S.; Zhang, M.; Zhen, L.; Pang, D.; Zhang, Q.; Li, Z. High-infiltration of tumor-associated macrophages predicts unfavorable clinical outcome for node-negative breast cancer. PLoS ONE 2013, 8, e76147. [Google Scholar] [CrossRef]
- Chen, X.; Yang, M.; Yin, J.; Li, P.; Zeng, S.; Zheng, G.; He, Z.; Liu, H.; Wang, Q.; Zhang, F.; et al. Tumor-associated macrophages promote epithelial-mesenchymal transition and the cancer stem cell properties in triple-negative breast cancer through CCL2/AKT/beta-catenin signaling. Cell Commun. Signal. CCS 2022, 20, 92. [Google Scholar] [CrossRef]
- Rady, I.; Siddiqui, I.A.; Rady, M.; Mukhtar, H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett. 2017, 402, 16–31. [Google Scholar] [CrossRef]
- Carpena, M.; Nunez-Estevez, B.; Soria-Lopez, A.; Simal-Gandara, J. Bee Venom: An Updating Review of Its Bioactive Molecules and Its Health Applications. Nutrients 2020, 12, 3360. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, S.; Li, J.; Yuan, B.; Yang, K.; Ma, Y. Molecular details on the intermediate states of melittin action on a cell membrane. Biochim. Et Biophys. Acta Biomembr. 2018, 1860, 2234–2241. [Google Scholar] [CrossRef]
- Pandidan, S.; Mechler, A. Nano-viscosimetry analysis of the membrane disrupting action of the bee venom peptide melittin. Sci. Rep. 2019, 9, 10841. [Google Scholar] [CrossRef]
- Soman, N.R.; Baldwin, S.L.; Hu, G.; Marsh, J.N.; Lanza, G.M.; Heuser, J.E.; Arbeit, J.M.; Wickline, S.A.; Schlesinger, P.H. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J. Clin. Investig. 2009, 119, 2830–2842. [Google Scholar] [CrossRef] [PubMed]
- Soliman, C.; Eastwood, S.; Truong, V.K.; Ramsland, P.A.; Elbourne, A. The membrane effects of melittin on gastric and colorectal cancer. PLoS ONE 2019, 14, e0224028. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Park, M.H.; Kollipara, P.S.; An, B.J.; Song, H.S.; Han, S.B.; Kim, J.H.; Song, M.J.; Hong, J.T. Anti-cancer effect of bee venom toxin and melittin in ovarian cancer cells through induction of death receptors and inhibition of JAK2/STAT3 pathway. Toxicol. Appl. Pharmacol. 2012, 258, 72–81. [Google Scholar] [CrossRef]
- Kokot, G.; Mally, M.; Svetina, S. The dynamics of melittin-induced membrane permeability. Eur. Biophys. J. EBJ 2012, 41, 461–474. [Google Scholar] [CrossRef]
- Van den Bogaart, G.; Guzman, J.V.; Mika, J.T.; Poolman, B. On the mechanism of pore formation by melittin. J. Biol. Chem. 2008, 283, 33854–33857. [Google Scholar] [CrossRef]
- Lee, C.; Bae, S.S.; Joo, H.; Bae, H. Melittin suppresses tumor progression by regulating tumor-associated macrophages in a Lewis lung carcinoma mouse model. Oncotarget 2017, 8, 54951–54965. [Google Scholar] [CrossRef]
- Javadpour, M.M.; Juban, M.M.; Lo, W.C.; Bishop, S.M.; Alberty, J.B.; Cowell, S.M.; Becker, C.L.; McLaughlin, M.L. De novo antimicrobial peptides with low mammalian cell toxicity. J. Med. Chem. 1996, 39, 3107–3113. [Google Scholar] [CrossRef] [PubMed]
- Ellerby, H.M.; Arap, W.; Ellerby, L.M.; Kain, R.; Andrusiak, R.; Rio, G.D.; Krajewski, S.; Lombardo, C.R.; Rao, R.; Ruoslahti, E.; et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat. Med. 1999, 5, 1032–1038. [Google Scholar] [CrossRef]
- Agemy, L.; Friedmann-Morvinski, D.; Kotamraju, V.R.; Roth, L.; Sugahara, K.N.; Girard, O.M.; Mattrey, R.F.; Verma, I.M.; Ruoslahti, E. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17450–17455. [Google Scholar] [CrossRef]
- Lee, C.; Jeong, H.; Bae, Y.; Shin, K.; Kang, S.; Kim, H.; Oh, J.; Bae, H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J. Immunother. Cancer 2019, 7, 147. [Google Scholar] [CrossRef] [PubMed]
- Han, I.H.; Jeong, C.; Yang, J.; Park, S.H.; Hwang, D.S.; Bae, H. Therapeutic Effect of Melittin-dKLA Targeting Tumor-Associated Macrophages in Melanoma. Int. J. Mol. Sci. 2022, 23, 3094. [Google Scholar] [CrossRef] [PubMed]
- Lyu, C.; Fang, F.; Li, B. Anti-Tumor Effects of Melittin and Its Potential Applications in Clinic. Curr. Protein Pept. Sci. 2019, 20, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Tender, T.; Rahangdale, R.R.; Balireddy, S.; Nampoothiri, M.; Sharma, K.K.; Raghu Chandrashekar, H. Melittin, a honeybee venom derived peptide for the treatment of chemotherapy-induced peripheral neuropathy. Med. Oncol. 2021, 38, 52. [Google Scholar] [CrossRef]
- Sessa, G.; Freer, J.H.; Colacicco, G.; Weissmann, G. Interaction of alytic polypeptide, melittin, with lipid membrane systems. J. Biol. Chem. 1969, 244, 3575–3582. [Google Scholar] [CrossRef]
- Gevod, V.S.; Birdi, K.S. Melittin and the 8-26 fragment. Differences in ionophoric properties as measured by monolayer method. Biophys. J. 1984, 45, 1079–1083. [Google Scholar] [CrossRef]
- Gagliardi, M.; Pitner, M.K.; Park, J.; Xie, X.; Saso, H.; Larson, R.A.; Sammons, R.M.; Chen, H.; Wei, C.; Masuda, H.; et al. Differential functions of ERK1 and ERK2 in lung metastasis processes in triple-negative breast cancer. Sci. Rep. 2020, 10, 8537. [Google Scholar] [CrossRef]
- Yi, H.; Li, Y.; Tan, Y.; Fu, S.; Tang, F.; Deng, X. Immune Checkpoint Inhibition for Triple-Negative Breast Cancer: Current Landscape and Future Perspectives. Front. Oncol. 2021, 11, 648139. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schroder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Aharinejad, S.; Paulus, P.; Sioud, M.; Hofmann, M.; Zins, K.; Schafer, R.; Stanley, E.R.; Abraham, D. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res. 2004, 64, 5378–5384. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Steenbrugge, J.; Breyne, K.; Demeyere, K.; De Wever, O.; Sanders, N.N.; Van Den Broeck, W.; Colpaert, C.; Vermeulen, P.; Van Laere, S.; Meyer, E. Anti-inflammatory signaling by mammary tumor cells mediates prometastatic macrophage polarization in an innovative intraductal mouse model for triple-negative breast cancer. J. Exp. Clin. Cancer Res. CR 2018, 37, 191. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, J.; Chen, S.; Lu, M.; Luo, X.; Yao, S.; Liu, S.; Qin, Y.; Chen, H. Tumor-associated macrophages provide a suitable microenvironment for non-small lung cancer invasion and progression. Lung Cancer 2011, 74, 188–196. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef]
- Liu, C.; Chikina, M.; Deshpande, R.; Menk, A.V.; Wang, T.; Tabib, T.; Brunazzi, E.A.; Vignali, K.M.; Sun, M.; Stolz, D.B.; et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8(+) T Cell-Derived Interferon-gamma. Immunity 2019, 51, 381–397.e386. [Google Scholar] [CrossRef]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Li, B.; Ren, M.; Zhou, X.; Han, Q.; Cheng, L. Targeting tumor-associated macrophages in head and neck squamous cell carcinoma. Oral Oncol. 2020, 106, 104723. [Google Scholar] [CrossRef]
- Lee, C.; Kim, S.; Jeong, C.; Cho, I.; Jo, J.; Han, I.H.; Bae, H. TAMpepK Suppresses Metastasis through the Elimination of M2-Like Tumor-Associated Macrophages in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 2157. [Google Scholar] [CrossRef]
- Pan, H.; Soman, N.R.; Schlesinger, P.H.; Lanza, G.M.; Wickline, S.A. Cytolytic peptide nanoparticles (‘NanoBees’) for cancer therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 318–327. [Google Scholar] [CrossRef] [PubMed]
- DeGrado, W.F.; Musso, G.F.; Lieber, M.; Kaiser, E.T.; Kezdy, F.J. Kinetics and mechanism of hemolysis induced by melittin and by a synthetic melittin analogue. Biophys. J. 1982, 37, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, C.E. The actions of melittin on membranes. Biochim. Et Biophys. Acta 1990, 1031, 143–161. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, J.; Zhao, M.; Tang, S.; Cheng, X.; Zhang, W.; Li, W.; Liu, X.; Peng, H.; Wang, Q. Effects of polyethylene glycol on the surface of nanoparticles for targeted drug delivery. Nanoscale 2021, 13, 10748–10764. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Deluca, P.P.; Lee, K.C. Emerging PEGylated drugs. Expert Opin. Emerg. Drugs 2009, 14, 363–380. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Bae, H.D.; Pyun, H.; Kim, B.G.; Lee, S.I.; Song, J.S.; Lee, K. PEGylation improves the therapeutic potential of dimerized translationally controlled tumor protein blocking peptide in ovalbumin-induced mouse model of airway inflammation. Drug Deliv. 2022, 29, 2320–2329. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Gupta, V.; Bhavanasi, S.; Quadir, M.; Singh, K.; Ghosh, G.; Vasamreddy, K.; Ghosh, A.; Siahaan, T.J.; Banerjee, S.; Banerjee, S.K. Protein PEGylation for cancer therapy: Bench to bedside. J. Cell Commun. Signal. 2019, 13, 319–330. [Google Scholar] [CrossRef]
- Swierczewska, M.; Lee, K.C.; Lee, S. What is the future of PEGylated therapies? Expert Opin. Emerg. Drugs 2015, 20, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Dozier, J.K.; Distefano, M.D. Site-Specific PEGylation of Therapeutic Proteins. Int. J. Mol. Sci. 2015, 16, 25831–25864. [Google Scholar] [CrossRef] [PubMed]
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Li, Y.; Wang, H.; Qin, T.; Yin, X.; Ma, X. Therapeutic progress and challenges for triple negative breast cancer: Targeted therapy and immunotherapy. Mol. Biomed. 2022, 3, 8. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jeon, M.; Lee, J.E.; Nam, S.J. Zerumbone suppresses the motility and tumorigenecity of triple negative breast cancer cells via the inhibition of TGF-beta1 signaling pathway. Oncotarget 2016, 7, 1544–1558. [Google Scholar] [CrossRef]
- Hew, K.E.; Miller, P.C.; El-Ashry, D.; Sun, J.; Besser, A.H.; Ince, T.A.; Gu, M.; Wei, Z.; Zhang, G.; Brafford, P.; et al. MAPK Activation Predicts Poor Outcome and the MEK Inhibitor, Selumetinib, Reverses Antiestrogen Resistance in ER-Positive High-Grade Serous Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 935–947. [Google Scholar] [CrossRef]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Lee, C.; Lee, H.; Cho, H.; Kim, S.; Choi, I.; Hwang, Y.S.; Jeong, H.; Jang, H.; Pak, S.; Hwang, D.S.; et al. Combination of anti-PD-L1 antibody with peptide MEL-dKLA targeting M2 tumor-associated macrophages suppresses breast cancer metastasis. Cancer Commun. 2022, 42, 345–349. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Choi, I.; Han, I.-H.; Bae, H. Enhanced Therapeutic Effect of Optimized Melittin-dKLA, a Peptide Agent Targeting M2-like Tumor-Associated Macrophages in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 15751. https://doi.org/10.3390/ijms232415751
Kim S, Choi I, Han I-H, Bae H. Enhanced Therapeutic Effect of Optimized Melittin-dKLA, a Peptide Agent Targeting M2-like Tumor-Associated Macrophages in Triple-Negative Breast Cancer. International Journal of Molecular Sciences. 2022; 23(24):15751. https://doi.org/10.3390/ijms232415751
Chicago/Turabian StyleKim, Soyoung, Ilseob Choi, Ik-Hwan Han, and Hyunsu Bae. 2022. "Enhanced Therapeutic Effect of Optimized Melittin-dKLA, a Peptide Agent Targeting M2-like Tumor-Associated Macrophages in Triple-Negative Breast Cancer" International Journal of Molecular Sciences 23, no. 24: 15751. https://doi.org/10.3390/ijms232415751
APA StyleKim, S., Choi, I., Han, I.-H., & Bae, H. (2022). Enhanced Therapeutic Effect of Optimized Melittin-dKLA, a Peptide Agent Targeting M2-like Tumor-Associated Macrophages in Triple-Negative Breast Cancer. International Journal of Molecular Sciences, 23(24), 15751. https://doi.org/10.3390/ijms232415751