
Loss of Myostatin Alters Mitochondrial Oxidative Phosphorylation, TCA Cycle Activity, and ATP Production in Skeletal Muscle

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
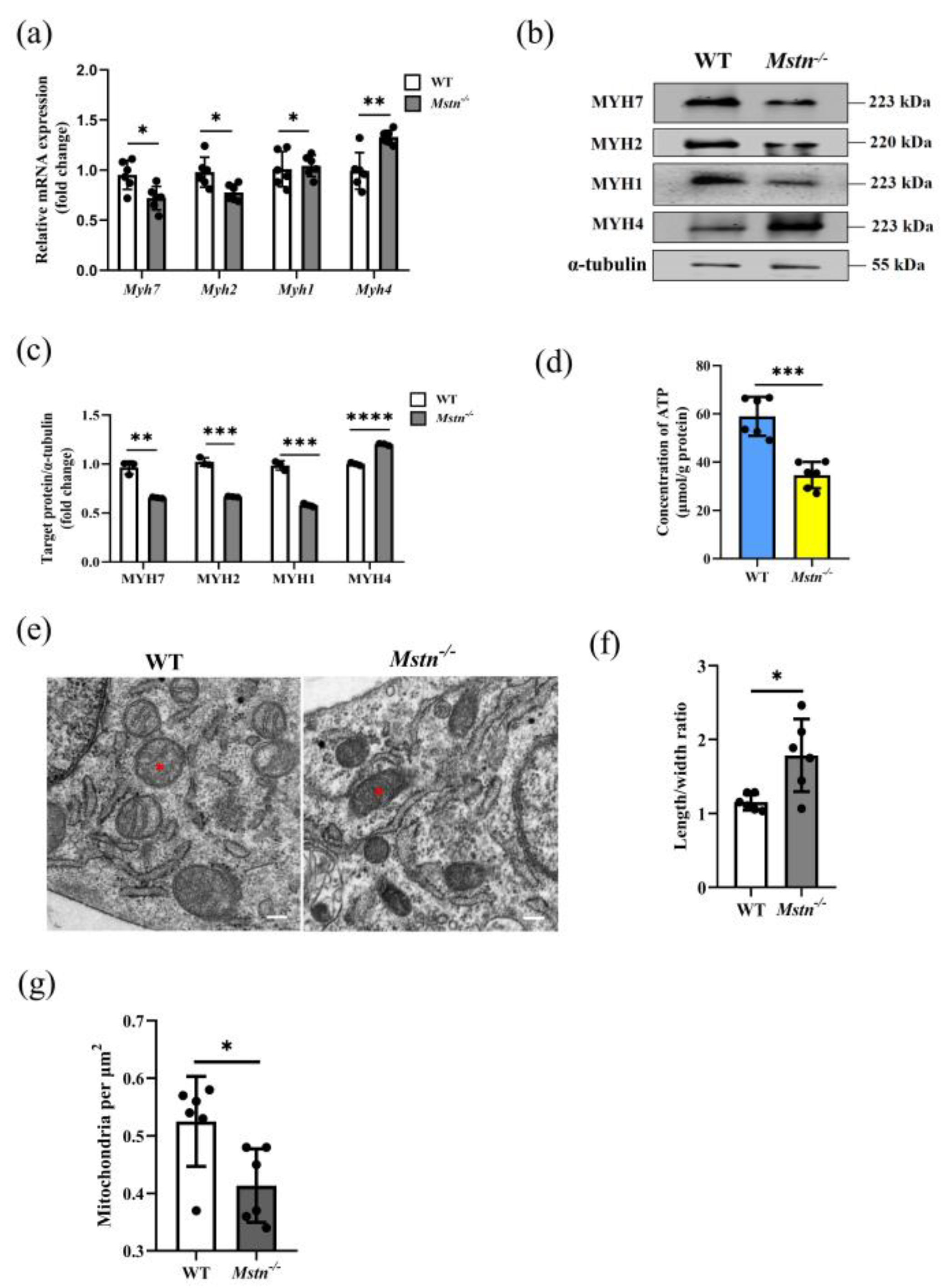
2.1. Mstn−/− Mice Developed a Typical Muscle Hypertrophy Phenotype
2.2. Mstn−/− Mice Significantly Reduced Muscle ATP Level
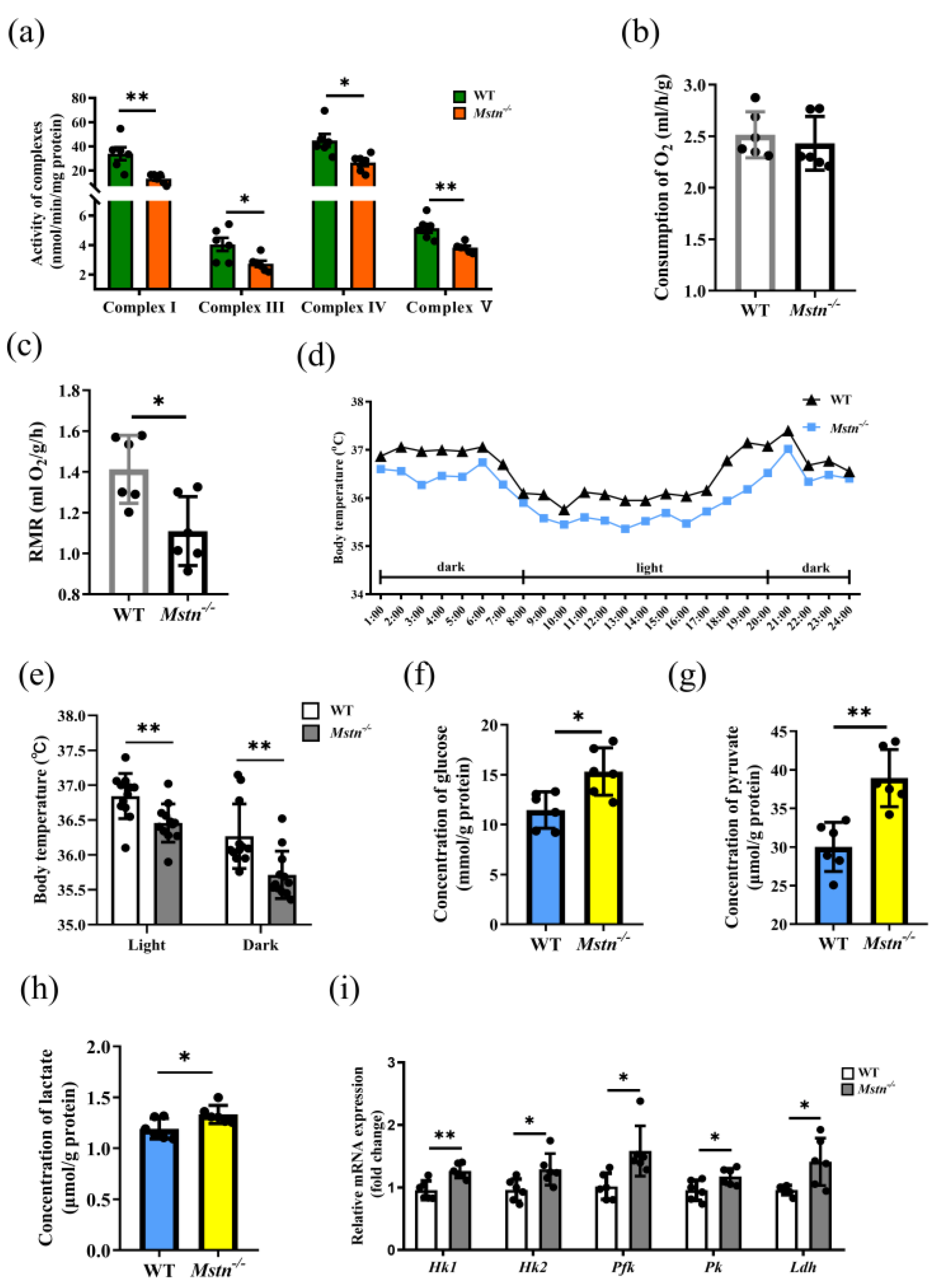
2.3. Reduced ATP Level of Mstn−/− Mice Resulted from Decreased OXPHOS Activity
2.4. The TCA Cycle Does Not Provide Sufficient Substrates for OXPHOS in Mstn−/− Muscles
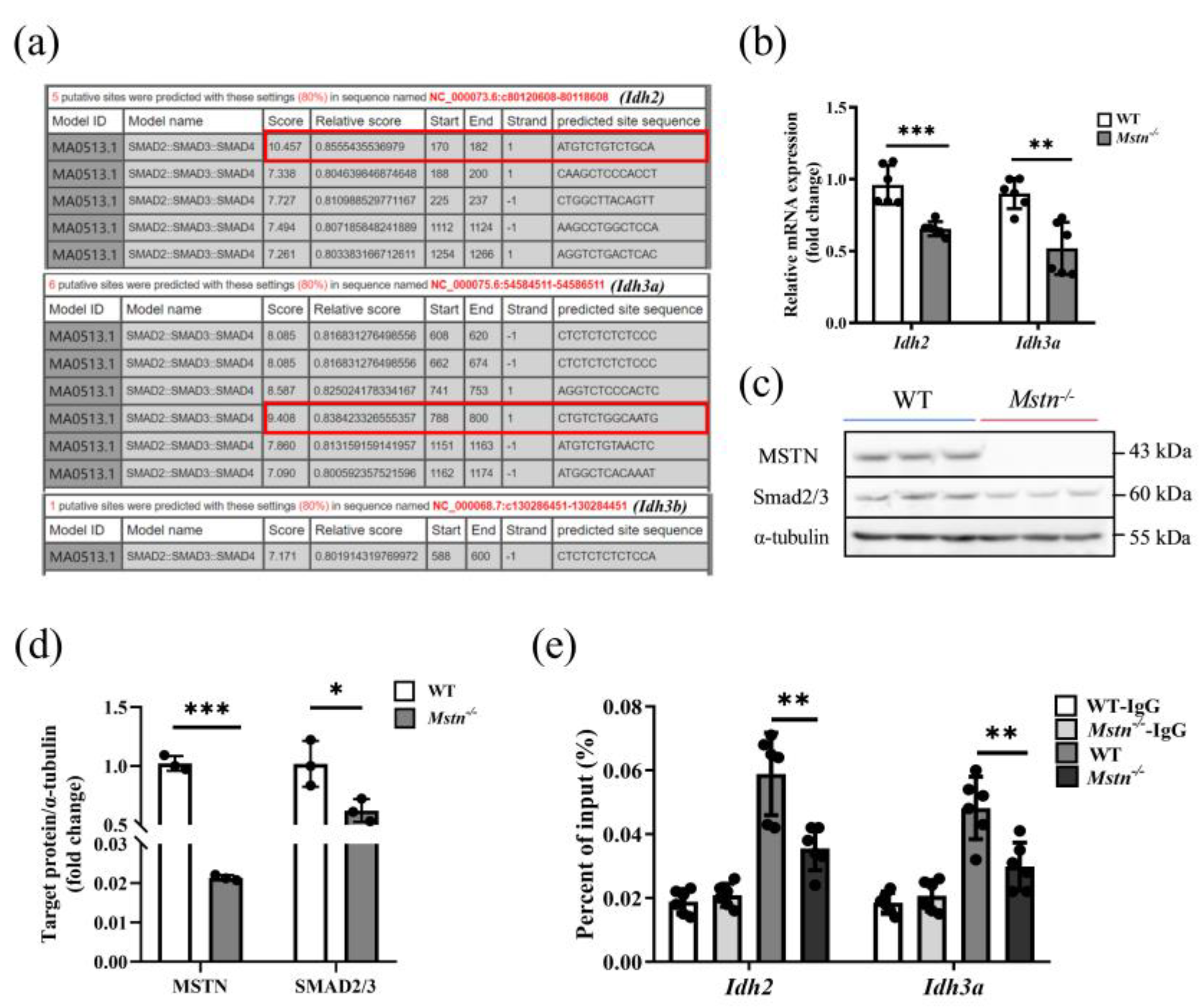
2.5. Loss of MSTN Disrupted Promotive Regulation of Idh2 and Idh3a through Smad2/3
3. Discussion
3.1. The Effect of Mstn−/− on Muscle Morphology
3.2. The Effect of Mstn−/− on OXPHOS
3.3. The Effect of Mstn−/− on Glycolysis
3.4. The Effect of Mstn−/− on the TCA Cycle
3.5. Mstn−/− Regulates Muscle Energy Metabolism through Smad2/3
4. Materials and Methods
4.1. Ethics Statement
4.2. Generation of Mstn−/− Mice
4.3. Separation of Single Muscle Fibers
4.4. RT-qPCR Assay
4.5. Western Blotting Analysis
4.6. Transmission Electron Microscopy (TEM)
4.7. Mice Resting Metabolic Rate (RMR) and Body Temperature Assays
4.8. Metabolic Substrates and Enzymes Assays
4.9. ChIP-qPCR
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aiello, D.; Patel, K.; Lasagna, E. The myostatin gene: An overview of mechanisms of action and its relevance to livestock animals. Anim. Genet. 2018, 49, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.C.; Cramer, M.S.; Mitchell, P.J.; Lucchesi, J.; Ortega, A.M.; Livingston, E.W.; Ballard, D.; Zhang, L.; Hanson, J.; Barton, K.; et al. Inhibition of myostatin prevents microgravity-induced loss of skeletal muscle mass and strength. PLoS ONE 2020, 15, e0230818. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, R.; Lachey, J.; Wallner, S.; Strand, J.; Liharska, K.; Watanabe, A.E.; Cannell, M.; Davies, M.V.; Sako, D.; Troy, M.E.; et al. Follistatin-288-Fc fusion protein promotes localized growth of skeletal muscle. J. Pharmacol. Exp. Ther. 2019, 368, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Long, K.K.; O’Shea, K.M.; Khairallah, R.J.; Howell, K.; Paushkin, S.; Chen, K.S.; Cote, S.M.; Webster, M.T.; Stains, J.P.; Treece, E.; et al. Specific inhibition of myostatin activation is beneficial in mouse models of SMA therapy. Hum. Mol. Genet. 2019, 28, 1076–1089. [Google Scholar] [CrossRef] [PubMed]
- Lamar, K.M.; Bogdanovich, S.; Gardner, B.B.; Gao, Q.Q.; Miller, T.; Earley, J.U.; Hadhazy, M.; Vo, A.H.; Wren, L.; Molkentin, J.D.; et al. Overexpression of latent TGFbeta binding protein 4 in muscle ameliorates muscular dystrophy through myostatin and TGFbeta. PLoS Genet. 2016, 12, e1006019. [Google Scholar] [CrossRef]
- Ran, N.; Gao, X.; Dong, X.; Li, J.; Lin, C.; Geng, M.; Yin, H. Effects of exosome-mediated delivery of myostatin propeptide on functional recovery of mdx mice. Biomaterials 2020, 236, 119826. [Google Scholar] [CrossRef]
- Hoogaars WM, H.; Jaspers, R.T. Past, present, and future perspective of targeting myostatin and related signaling pathways to counteract muscle atrophy. Adv. Exp. Med. Biol. 2018, 1088, 153–206. [Google Scholar] [CrossRef]
- Gu, H.; Cao, Y.; Qiu, B.; Zhou, Z.; Deng, R.; Chen, Z.; Li, R.; Li, X.; Wei, Q.; Xia, X.; et al. Establishment and phenotypic analysis of an Mstn knockout rat. Biochem. Biophys. Res. Commun. 2016, 477, 115–122. [Google Scholar] [CrossRef]
- Baltusnikas, J.; Kilikevicius, A.; Venckunas, T.; Fokin, A.; Bunger, L.; Lionikas, A.; Ratkevicius, A. Myostatin dysfunction impairs force generation in extensor digitorum longus muscle and increases exercise-induced protein efflux from extensor digitorum longus and soleus muscles. Appl. Physiol. Nutr. Metab. 2015, 40, 817–821. [Google Scholar] [CrossRef]
- Stantzou, A.; Ueberschlag-Pitiot, V.; Thomasson, R.; Furling, D.; Bonnieu, A.; Amthor, H.; Ferry, A. Effect of constitutive inactivation of the myostatin gene on the gain in muscle strength during postnatal growth in two murine models. Muscle Nerve 2017, 55, 254–261. [Google Scholar] [CrossRef]
- Amthor, H.; Macharia, R.; Navarrete, R.; Schuelke, M.; Brown, S.C.; Otto, A.; Voit, T.; Muntoni, F.; Vrbova, G.; Partridge, T.; et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Natl. Acad. Sci. USA 2007, 104, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.S.; Kim, D.H.; Park, H.B.; Han, S.H.; Hwang, S.; Cho, I.C.; Lee, J.W. Ectopic overexpression of porcine Myh1 increased in slow muscle fibers and enhanced endurance exercise in transgenic mice. Int. J. Mol. Sci. 2018, 19, 2959. [Google Scholar] [CrossRef]
- Ishihara, A.; Taguchi, S. Histochemical differentiation of fibers in the rat slow and fast twitch muscles. Jpn. J. Physiol. 1991, 41, 251–258. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Girgenrath, S.; Song, K.; Whittemore, L.A. Loss of myostatin expression alters fiber-type distribution and expression of myosin heavy chain isoforms in slow- and fast-type skeletal muscle. Muscle Nerve 2005, 31, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, C.; Huang, L.; Zeng, K.; Cao, X.; Gao, J. Inefficient ATP synthesis by inhibiting mitochondrial respiration causes lipids to decrease in MSTN-lacking muscles of loach Misgurnus anguillicaudatus. Funct. Integr. Genom. 2019, 19, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Ploquin, C.; Chabi, B.; Fouret, G.; Vernus, B.; Feillet-Coudray, C.; Coudray, C.; Bonnieu, A.; Ramonatxo, C. Lack of myostatin alters intermyofibrillar mitochondria activity, unbalances redox status, and impairs tolerance to chronic repetitive contractions in muscle. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1000–E1008. [Google Scholar] [CrossRef]
- Bechir, N.; Pecchi, E.; Relizani, K.; Vilmen, C.; Le Fur, Y.; Bernard, M.; Amthor, H.; Bendahan, D.; Giannesini, B. Mitochondrial impairment induced by postnatal ActRIIB blockade does not alter function and energy status in exercising mouse glycolytic muscle in vivo. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E539–E549. [Google Scholar] [CrossRef]
- Mouisel, E.; Relizani, K.; Mille-Hamard, L.; Denis, R.; Hourde, C.; Agbulut, O.; Patel, K.; Arandel, L.; Morales-Gonzalez, S.; Vignaud, A.; et al. Myostatin is a key mediator between energy metabolism and endurance capacity of skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R444–R454. [Google Scholar] [CrossRef]
- Zhao, B.; Li, E.J.; Wall, R.J.; Yang, J. Coordinated patterns of gene expressions for adult muscle build-up in transgenic mice expressing myostatin propeptide. BMC Genom. 2009, 10, 305. [Google Scholar] [CrossRef]
- Manfredi, L.H.; Ang, J.; Peker, N.; Dagda, R.K.; McFarlane, C. G protein-coupled receptor kinase 2 regulates mitochondrial bioenergetics and impairs myostatin-mediated autophagy in muscle cells. Am. J. Physiol. Cell Physiol. 2019, 317, C674–C686. [Google Scholar] [CrossRef]
- Singh, R.; Braga, M.; Pervin, S. Regulation of brown adipocyte metabolism by myostatin/follistatin signaling. Front. Cell Dev. Biol. 2014, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; McFarlane, C.; Kambadur, R.; Kukreti, H.; Bonala, S.; Srinivasan, S. Myostatin: Expanding horizons. IUBMB Life 2015, 67, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Colgan, T.D.; Walton, K.L.; Gregorevic, P.; Harrison, C.A. The TGF-beta signalling network in muscle development, adaptation and disease. Adv. Exp. Med. Biol. 2016, 900, 97–131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, X.; Wei, Z.; Yang, M.; Zhou, X.; Lei, J.; Bai, C.; Su, G.; Liu, X.; Yang, L.; et al. Myostatin deficiency enhances antioxidant capacity of bovine muscle via the SMAD-AMPK-G6PD pathway. Oxid. Med. Cell Longev. 2022, 2022, 3497644. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Zhou, X.; Zhu, L.; Gao, Y.; Gao, L.; Bai, C.; Yang, L.; Li, G. Myostatin mutation promotes glycolysis by increasing phosphorylation of phosphofructokinase via activation of PDE5A-cGMP-PKG in cattle heart. Front. Cell Dev. Biol. 2021, 9, 774185. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, Y.Q.; Yuan, Y.X.; Xu, P.W.; Zhang, C.; Li, F.; Wang, L.N.; Yin, C.; Zhang, L.; Cai, X.C.; et al. Succinate induces skeletal muscle fiber remodeling via SUNCR1 signaling. EMBO Rep. 2019, 20, e47892. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Fania, C.; Torretta, E.; Vigano, A.; Moriggi, M.; Bravata, V.; Caretti, A.; Levett DZ, H.; Grocott MP, W.; Samaja, M.; et al. TCA cycle rewiring fosters metabolic adaptation to oxygen restriction in skeletal muscle from rodents and humans. Sci. Rep. 2017, 7, 9723. [Google Scholar] [CrossRef]
- Xing, X.X.; Xuan, M.F.; Jin, L.; Guo, Q.; Luo, Z.B.; Wang, J.X.; Luo, Q.R.; Zhang, G.L.; Cui, C.D.; Cui, Z.Y.; et al. Fiber-type distribution and expression of myosin heavy chain isoforms in newborn heterozygous myostatin-knockout pigs. Biotechnol. Lett. 2017, 39, 1811–1819. [Google Scholar] [CrossRef]
- Hennebry, A.; Berry, C.; Siriett, V.; O’Callaghan, P.; Chau, L.; Watson, T.; Sharma, M.; Kambadur, R. Myostatin regulates fiber-type composition of skeletal muscle by regulating MEF2 and MyoD gene expression. Am. J. Physiol. Cell Physiol. 2009, 296, C525–C534. [Google Scholar] [CrossRef]
- Charles, J.P.; Cappellari, O.; Spence, A.J.; Hutchinson, J.R.; Wells, D.J. Musculoskeletal geometry, muscle architecture and functional specialisations of the mouse hindlimb. PLoS ONE 2016, 11, e0147669. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Humphries, K.M. Selective inhibition of deactivated mitochondrial complex I by biguanides. Biochemistry 2015, 54, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Conlon, I.; Raff, M. Size control in animal development. Cell 1999, 96, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Gabillard, J.C.; Biga, P.R.; Rescan, P.Y.; Seiliez, I. Revisiting the paradigm of myostatin in vertebrates: Insights from fishes. Gen. Comp. Endocrinol. 2013, 194, 45–54. [Google Scholar] [CrossRef]
- Jia, H.; Zhao, Y.; Li, T.; Zhang, Y.; Zhu, D. miR-30e is negatively regulated by myostatin in skeletal muscle and is functionally related to fiber-type composition. Acta Biochim. Biophys. Sin. 2017, 49, 392–399. [Google Scholar] [CrossRef]
- Chen, X.; Luo, X.; Chen, D.; Yu, B.; He, J.; Huang, Z. Arginine promotes porcine type I muscle fibres formation through improvement of mitochondrial biogenesis. Br. J. Nutr. 2020, 123, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Sanchez, B.; Li, J.; Bragos, R.; Rutkove, S.B. Differentiation of the intracellular structure of slow- versus fast-twitch muscle fibers through evaluation of the dielectric properties of tissue. Phys. Med. Biol. 2014, 59, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef]
- Giannesini, B.; Vilmen, C.; Amthor, H.; Bernard, M.; Bendahan, D. Lack of myostatin impairs mechanical performance and ATP cost of contraction in exercising mouse gastrocnemius muscle in vivo. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E33–E40. [Google Scholar] [CrossRef]
- Nassar, R.; Vernus, B.; Carnac, G.; Fouret, G.; Goustard, B.; Casas, F.; Tintignac, L.; Cassar-Malek, I.; Picard, B.; Seiliez, I.; et al. Myostatin gene inactivation increases post-mortem calpain-dependent muscle proteolysis in mice. Meat Sci. 2022, 185, 108726. [Google Scholar] [CrossRef]
- Tajsharghi, H. Thick and thin filament gene mutations in striated muscle diseases. Int. J. Mol. Sci. 2008, 9, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Nicolae, A.; Wahrheit, J.; Nonnenmacher, Y.; Weyler, C.; Heinzle, E. Identification of active elementary flux modes in mitochondria using selectively permeabilized CHO cells. Metab. Eng. 2015, 32, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.F.; Porter, R.K.; Katz, L.M.; Hill, E.W. Skeletal muscle mitochondrial bioenergetics and associations with myostatin genotypes in the Thoroughbred horse. PLoS ONE 2017, 12, e0186247. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Streeper, R.S.; Farese, R.V.; Yamamoto, K.R., Jr. Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects. Proc. Natl. Acad. Sci. USA 2006, 103, 15675–15680. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef] [PubMed]
- Savabi, F. Interaction of creatine kinase and adenylate kinase systems in muscle cells. Mol. Cell Biochem. 1994, 133–134, 145–152. [Google Scholar] [CrossRef]
- Zeleznikar, R.J.; Heyman, R.A.; Graeff, R.M.; Walseth, T.F.; Dawis, S.M.; Butz, E.A.; Goldberg, N.D. Evidence for compartmentalized adenylate kinase catalysis serving a high energy phosphoryl transfer function in rat skeletal muscle. J. Biol. Chem. 1990, 265, 300–311. [Google Scholar] [CrossRef]
- Xin, X.B.; Yang, S.P.; Li, X.; Liu, X.F.; Zhang, L.L.; Ding, X.B.; Zhang, S.; Li, G.P.; Guo, H. Proteomics insights into the effects of MSTN on muscle glucose and lipid metabolism in genetically edited cattle. Gen. Comp. Endocrinol. 2020, 291, 113237. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lee, S.J. Suppression of body fat accumulation in myostatin-deficient mice. J. Clin. Investig. 2002, 109, 595–601. [Google Scholar] [CrossRef]
- Lee, S.J.; Lehar, A.; Liu, Y.; Ly, C.H.; Pham, Q.M.; Michaud, M.; Rydzik, R.; Youngstrom, D.W.; Shen, M.M.; Kaartinen, V.; et al. Functional redundancy of type I and type II receptors in the regulation of skeletal muscle growth by myostatin and activin A. Proc. Natl. Acad. Sci. USA 2020, 117, 30907–30917. [Google Scholar] [CrossRef]
- Xie, L.; Wang, D.I. Energy metabolism and ATP balance in animal cell cultivation using a stoichiometrically based reaction network. Biotechnol. Bioeng. 1996, 52, 591–601. [Google Scholar] [CrossRef]
- Jarmuszkiewicz, W.; Swida, A.; Czarna, M.; Antos, N.; Sluse-Goffart, C.M.; Sluse, F.E. In phosphorylating Acanthamoeba castellanii mitochondria the sensitivity of uncoupling protein activity to GTP depends on the redox state of quinone. J. Bioenerg. Biomembr. 2005, 37, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Takehiro, M.; Fujimoto, S.; Shimodahira, M.; Shimono, D.; Mukai, E.; Nabe, K.; Radu, R.G.; Kominato, R.; Aramaki, Y.; Seino, Y.; et al. Chronic exposure to beta-hydroxybutyrate inhibits glucose-induced insulin release from pancreatic islets by decreasing NADH contents. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E372–E380. [Google Scholar] [CrossRef]
- Birrell, J.A.; Hirst, J. Investigation of NADH binding, hydride transfer, and NAD (+) dissociation during NADH oxidation by mitochondrial complex I using modified nicotinamide nucleotides. Biochemistry 2013, 52, 4048–4055. [Google Scholar] [CrossRef]
- Patterson, G.H.; Knobel, S.M.; Arkhammar, P.; Thastrup, O.; Piston, D.W. Separation of the glucose-stimulated cytoplasmic and mitochondrial NAD(P)H responses in pancreatic islet beta cells. Proc. Natl. Acad. Sci. USA 2000, 97, 5203–5207. [Google Scholar] [CrossRef] [PubMed]
- Leung, K. 18-[18F] Fluoro-4-thia-oleate. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar] [PubMed]
- Ma, Z.; Qin, X.; Zhong, X.; Liao, Y.; Su, Y.; Liu, X.; Liu, P.; Lu, J.; Zhou, S. Flavine adenine dinucleotide inhibits pathological cardiac hypertrophy and fibrosis through activating short chain acyl-CoA dehydrogenase. Biochem. Pharmacol. 2020, 178, 114100. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.D.; Bostrom, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. beta-Aminoisobutyric acid induces browning of white fat and hepatic beta-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar] [CrossRef]
- Ge, X.; Vajjala, A.; McFarlane, C.; Wahli, W.; Sharma, M.; Kambadur, R. Lack of Smad3 signaling leads to impaired skeletal muscle regeneration. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E90–E102. [Google Scholar] [CrossRef]
- Gao, L.; Yang, M.; Wei, Z.; Gu, M.; Yang, L.; Bai, C.; Wu, Y.; Li, G. MSTN mutant promotes myogenic differentiation by increasing demethylase TET1 expression via the SMAD2/SMAD3 pathway. Int. J. Biol. Sci. 2020, 16, 1324–1334. [Google Scholar] [CrossRef]
- Paek, H.J.; Luo, Z.B.; Choe, H.M.; Quan, B.H.; Gao, K.; Han, S.Z.; Li, Z.Y.; Kang, J.D.; Yin, X.J. Association of myostatin deficiency with collagen related disease-umbilical hernia and tippy toe standing in pigs. Transgenic Res. 2021, 30, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Go, Y.; Kim, D.Y.; Lee, S.H.; Kim, O.H.; Jeon, Y.H.; Kwon, T.K.; Bae, J.H.; Song, D.K.; Rhyu, I.J.; et al. Isocitrate dehydrogenase 2 protects mice from high-fat diet-induced metabolic stress by limiting oxidative damage to the mitochondria from brown adipose tissue. Exp. Mol. Med. 2020, 52, 238–252. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.L.; Corridon, P.R.; Zhang, S.; Xu, W.; Witzmann, F.A.; Collett, J.A.; Rhodes, G.J.; Winfree, S.; Bready, D.; Pfeffenberger, Z.J.; et al. Exogenous gene transmission of isocitrate dehydrogenase 2 mimics ischemic preconditioning protection. J. Am. Soc. Nephrol. 2018, 29, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Conte, E.; Pannunzio, A.; Imbrici, P.; Camerino, G.M.; Maggi, L.; Mora, M.; Gibertini, S.; Cappellari, O.; De Luca, A.; Coluccia, M.; et al. Gain-of-function STIM1 L96V mutation causes myogenesis alteration in muscle cells from a patient affected by tubular aggregate myopathy. Front. Cell Dev. Biol. 2021, 9, 635063. [Google Scholar] [CrossRef] [PubMed]
- Findlay, A.S.; Carter, R.N.; Starbuck, B.; McKie, L.; Novakova, K.; Budd, P.S.; Keighren, M.A.; Marsh, J.A.; Cross, S.H.; Simon, M.M.; et al. Mouse Idh3a mutations cause retinal degeneration and reduced mitochondrial function. Dis. Models Mech. 2018, 11, dmm036426. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Yang, L.; Song, L.S.; Liu, X.F.; Xia, Q.; Bai, L.G.; Gao, L.; Gao, G.Q.; Wang, Y.; Wei, Z.Y.; Bai, C.L.; et al. The maternal effect genes UTX and JMJD3 play contrasting roles in mus musculus preimplantation embryo development. Sci. Rep. 2016, 6, 26711. [Google Scholar] [CrossRef]
- Chen, C.; Zhu, L.; Zhou, X.Y.; Bai, C.L.; Zheng, Z.; Wei, Z.Y.; Li, G.P. The effect of mice (Mus musculus) Myostatin Y309 and C310 deletions on protein structure. J. Agric. Biotechnol. 2019, 27, 1051–1061. [Google Scholar]
- Pasut, A.; Jones, A.E.; Rudnicki, M.A. Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J. Vis. Exp. 2013, 73, e50074. [Google Scholar] [CrossRef]
- Thomas, K.C.; Zheng, X.F.; Garces Suarez, F.; Raftery, J.M.; Quinlan, K.G.; Yang, N.; North, K.N.; Houweling, P.J. Evidence based selection of commonly used RT-qPCR reference genes for the analysis of mouse skeletal muscle. PLoS ONE 2014, 9, e88653. [Google Scholar] [CrossRef]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.S.; Dirksen, R.T.; Protasi, F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell 2009, 20, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.C.; Vaanholt, L.M.; Sinclair, R.E.; Gamo, Y.; Speakman, J.R. Limits to sustained energy intake XII: Is the poor relation between resting metabolic rate and reproductive performance because resting metabolism is not a repeatable trait? J. Exp. Biol. 2010, 213, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Asp, P. How to Combine ChIP with qPCR. Methods Mol. Biol. 2018, 1689, 29–42. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wei, Z.; Gu, M.; Zhu, L.; Hai, C.; Di, A.; Wu, D.; Bai, C.; Su, G.; Liu, X.; et al. Loss of Myostatin Alters Mitochondrial Oxidative Phosphorylation, TCA Cycle Activity, and ATP Production in Skeletal Muscle. Int. J. Mol. Sci. 2022, 23, 15707. https://doi.org/10.3390/ijms232415707
Wang X, Wei Z, Gu M, Zhu L, Hai C, Di A, Wu D, Bai C, Su G, Liu X, et al. Loss of Myostatin Alters Mitochondrial Oxidative Phosphorylation, TCA Cycle Activity, and ATP Production in Skeletal Muscle. International Journal of Molecular Sciences. 2022; 23(24):15707. https://doi.org/10.3390/ijms232415707
Chicago/Turabian StyleWang, Xueqiao, Zhuying Wei, Mingjuan Gu, Lin Zhu, Chao Hai, Anqi Di, Di Wu, Chunling Bai, Guanghua Su, Xuefei Liu, and et al. 2022. "Loss of Myostatin Alters Mitochondrial Oxidative Phosphorylation, TCA Cycle Activity, and ATP Production in Skeletal Muscle" International Journal of Molecular Sciences 23, no. 24: 15707. https://doi.org/10.3390/ijms232415707
APA StyleWang, X., Wei, Z., Gu, M., Zhu, L., Hai, C., Di, A., Wu, D., Bai, C., Su, G., Liu, X., Yang, L., & Li, G. (2022). Loss of Myostatin Alters Mitochondrial Oxidative Phosphorylation, TCA Cycle Activity, and ATP Production in Skeletal Muscle. International Journal of Molecular Sciences, 23(24), 15707. https://doi.org/10.3390/ijms232415707