
Single-Cell RNA Sequencing for the Detection of Clonotypic V(D)J Rearrangements in Multiple Myeloma

 ,
,  , ,
, ,  , ,
, ,  , and
, and
Abstract
1. Introduction
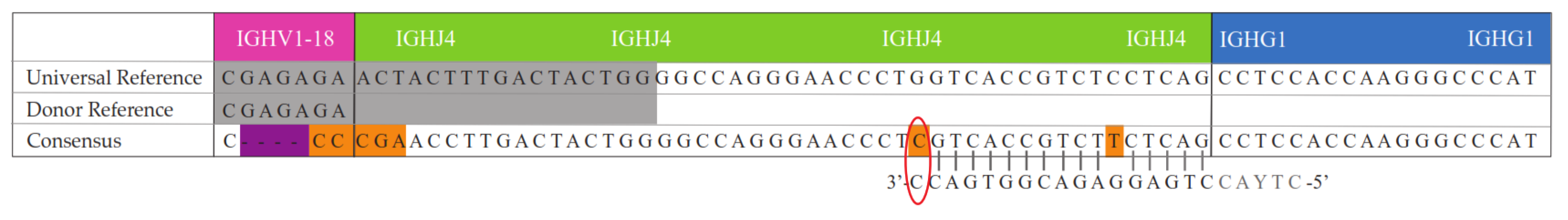
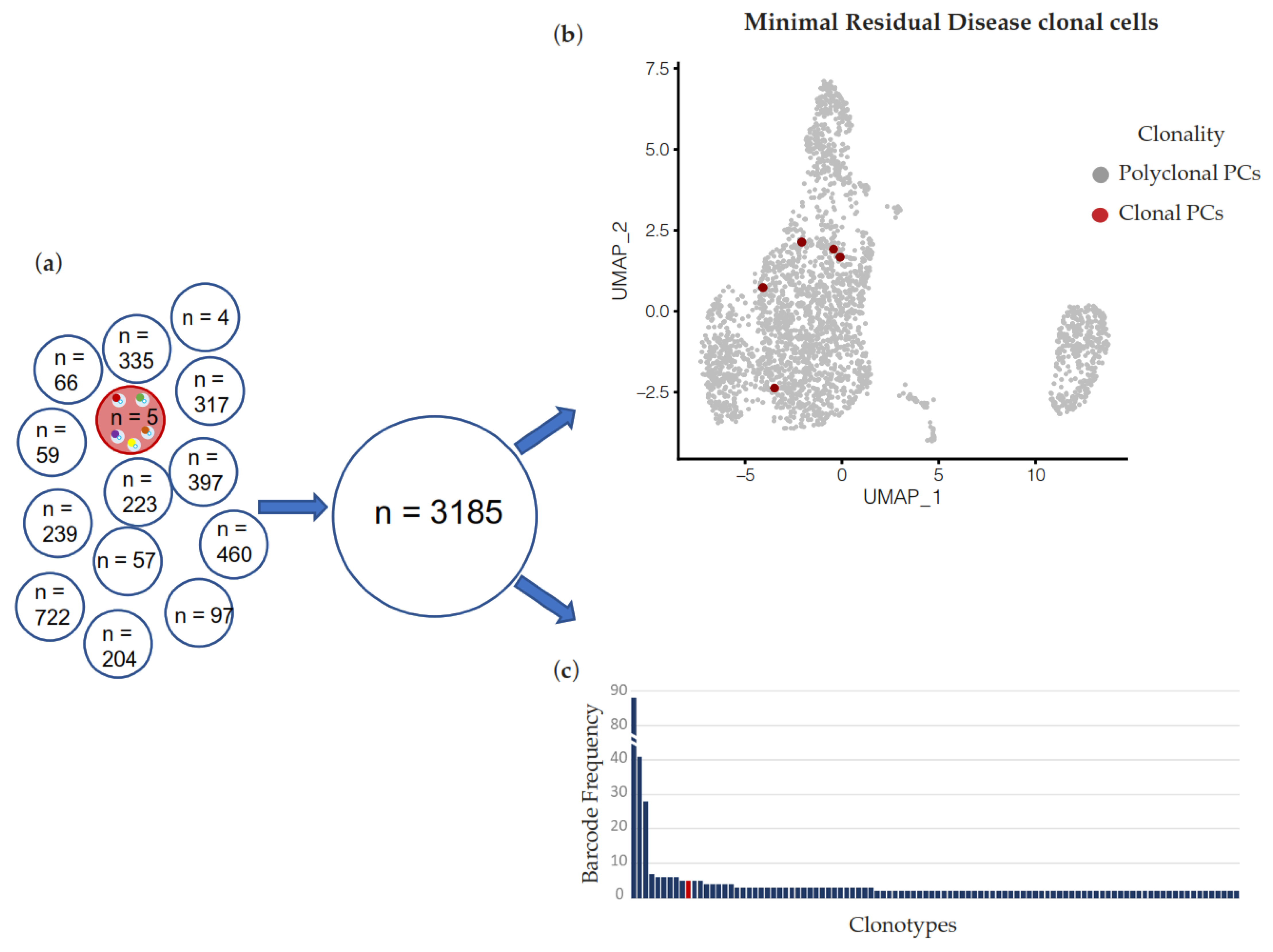
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA-Based Molecular Analysis of IGHV Rearrangement
4.3. Single-Cell V(D)J Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Bolli, N.; Biancon, G.; Moarii, M.; Gimondi, S.; Li, Y.; de Philippis, C.; Maura, F.; Sathiaseelan, V.; Tai, Y.T.; Mudie, L.; et al. Analysis of the Genomic Landscape of Multiple Myeloma Highlights Novel Prognostic Markers and Disease Subgroups. Leukemia 2018, 32, 2604–2616. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The Genetic Architecture of Multiple Myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic Patterns of Progression in Smoldering Multiple Myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef] [PubMed]
- Oben, B.; Froyen, G.; Maclachlan, K.H.; Leongamornlert, D.; Abascal, F.; Zheng-Lin, B.; Yellapantula, V.; Derkach, A.; Geerdens, E.; Diamond, B.T.; et al. Whole-Genome Sequencing Reveals Progressive versus Stable Myeloma Precursor Conditions as Two Distinct Entities. Nat. Commun. 2021, 12, 1861. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of Genomic Evolution and Mutational Profiles in Multiple Myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [PubMed]
- Misund, K.; Bruinink, D.H.O.; Coward, E.; Hoogenboezem, R.M.; Rustad, E.H.; Sanders, M.A.; Rye, M.; Sponaas, A.-M.; van der Holt, B.; Zweegman, S.; et al. Clonal Evolution after Treatment Pressure in Multiple Myeloma: Heterogenous Genomic Aberrations and Transcriptomic Convergence. Leukemia 2022, 36, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Ziccheddu, B.; Biancon, G.; Bagnoli, F.; De Philippis, C.; Maura, F.; Rustad, E.H.; Dugo, M.; Devecchi, A.; De Cecco, L.; Sensi, M.; et al. Integrative Analysis of the Genomic and Transcriptomic Landscape of Double-Refractory Multiple Myeloma. Blood Adv. 2020, 4, 830–844. [Google Scholar] [CrossRef]
- Rustad, E.H.; Misund, K.; Bernard, E.; Coward, E.; Yellapantula, V.D.; Hultcrantz, M.; Ho, C.; Kazandjian, D.; Korde, N.; Mailankody, S.; et al. Stability and Uniqueness of Clonal Immunoglobulin CDR3 Sequences for MRD Tracking in Multiple Myeloma. Am. J. Hematol. 2019, 94, 1364–1373. [Google Scholar] [CrossRef]
- Ledergor, G.; Weiner, A.; Zada, M.; Wang, S.Y.; Cohen, Y.C.; Gatt, M.E.; Snir, N.; Magen, H.; Koren-Michowitz, M.; Herzog-Tzarfati, K.; et al. Single Cell Dissection of Plasma Cell Heterogeneity in Symptomatic and Asymptomatic Myeloma. Nat. Med. 2018, 24, 1867–1876. [Google Scholar] [CrossRef]
- Liu, R.; Gao, Q.; Foltz, S.M.; Fowles, J.S.; Yao, L.; Wang, J.T.; Cao, S.; Sun, H.; Wendl, M.C.; Sethuraman, S.; et al. Co-Evolution of Tumor and Immune Cells during Progression of Multiple Myeloma. Nat. Commun. 2021, 12, 2559. [Google Scholar] [CrossRef] [PubMed]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-Cell RNA Sequencing Reveals Compromised Immune Microenvironment in Precursor Stages of Multiple Myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Frede, J.; Anand, P.; Sotudeh, N.; Pinto, R.A.; Nair, M.S.; Stuart, H.; Yee, A.J.; Vijaykumar, T.; Waldschmidt, J.M.; Potdar, S.; et al. Dynamic Transcriptional Reprogramming Leads to Immunotherapeutic Vulnerabilities in Myeloma. Nat. Cell Biol. 2021, 23, 1199–1211. [Google Scholar] [CrossRef]
- Rashid, N.U.; Sperling, A.S.; Bolli, N.; Wedge, D.C.; Van Loo, P.; Tai, Y.T.; Shammas, M.A.; Fulciniti, M.; Samur, M.K.; Richardson, P.G.; et al. Differential and Limited Expression of Mutant Alleles in Multiple Myeloma. Blood 2014, 124, 3110–3117. [Google Scholar] [CrossRef]
- Brüggemann, M.; Kotrova, M.; Knecht, H.; Bartram, J.; Boudjogrha, M.; Bystry, V.; Fazio, G.; Froňková, E.; Giraud, M.; Grioni, A.; et al. Standardized Next-Generation Sequencing of Immunoglobulin and T-Cell Receptor Gene Recombinations for MRD Marker Identification in Acute Lymphoblastic Leukaemia; a EuroClonality-NGS Validation Study. Leukemia 2019, 33, 2241–2253. [Google Scholar] [CrossRef]
- Duez, M.; Giraud, M.; Herbert, R.; Rocher, T.; Salson, M.; Thonier, F. Vidjil: A Web Platform for Analysis of High-Throughput Repertoire Sequencing. PLoS ONE 2016, 11, e0166126. [Google Scholar] [CrossRef] [PubMed]
- Bashford-Rogers, R.J.M.; Nicolaou, K.A.; Bartram, J.; Goulden, N.J.; Loizou, L.; Koumas, L.; Chi, J.; Hubank, M.; Kellam, P.; Costeas, P.A.; et al. Eye on the B-ALL: B-Cell Receptor Repertoires Reveal Persistence of Numerous B-Lymphoblastic Leukemia Subclones from Diagnosis to Relapse. Leukemia 2016, 30, 2312–2321. [Google Scholar] [CrossRef]
- Hultcrantz, M.; Rustad, E.H.; Yellapantula, V.; Arcila, M.; Ho, C.; Syed, M.H.; Papaemmanuil, E.; Zhang, Y.; Maura, F.; Landgren, O. Baseline VDJ Clonotype Detection Using a Targeted Sequencing NGS Assay: Allowing for Subsequent MRD Assessment. Blood Cancer J. 2020, 10, 76. [Google Scholar] [CrossRef]
- van der Velden, V.H.J.; Bastian, L.; Brüggemann, M.; Hartmann, A.M.; Darzentas, N. Immunoglobulin/T-Cell Receptor Gene Rearrangement Analysis Using RNA-Seq. Methods Mol. Biol. 2022, 2453, 61–77. [Google Scholar] [CrossRef]
- Bolli, N.; Genuardi, E.; Ziccheddu, B.; Martello, M.; Oliva, S.; Terragna, C. Next-Generation Sequencing for Clinical Management of Multiple Myeloma: Ready for Prime Time? Front. Oncol. 2020, 10, 189. [Google Scholar] [CrossRef]
- Yellapantula, V.; Hultcrantz, M.; Rustad, E.H.; Wasserman, E.; Londono, D.; Cimera, R.; Ciardiello, A.; Landau, H.; Akhlaghi, T.; Mailankody, S.; et al. Comprehensive Detection of Recurring Genomic Abnormalities: A Targeted Sequencing Approach for Multiple Myeloma. Blood Cancer J. 2019, 9, 101. [Google Scholar] [CrossRef]
- Maura, F.; Bolli, N.; Rustad, E.H.; Hultcrantz, M.; Munshi, N.; Landgren, O. Moving From Cancer Burden to Cancer Genomics for Smoldering Myeloma: A Review. JAMA Oncol. 2019, 6, 425. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality Reduction for Visualizing Single-Cell Data Using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | scRNA-Seq | Amplicon-Based NGS from Bulk DNA | Common CDR3 | ||||
|---|---|---|---|---|---|---|---|
| IGHV | IGHJ | Proportion | IGHV | IGHJ | Proportion | ||
| PLC-01 | IGHV3-48 | IGHJ4 | 92.90% | IGHV3-30*09 | IGHJ4*02 | 67.55% | CARDSYEDYVYW |
| PLC-02 | IGHV3-21 | IGHJ4 | 97.93% | IGHV3-21*01 | IGHJ4*02 | 74.39% | CARYQLDAVAGKWGHYFDYW |
| PLC-03 | IGHV3-43 | IGHJ5 | 97.77% | IGHV3-9*01 | IGHJ4*02 | 66.23% | CAKARLPLVGGLDSW |
| PLC-05 | IGHV3-43 | IGHJ6 | 49.79% | IGHV3-9*01 | IGHJ6*02 | 13.62% | CTRVIGSGASCYDCYYHGMDVW |
| PLC-06 | IGHV3-53 | IGHJ5 | 87.91% | IGHV3-53*01 | IGHJ4*02 | 57.58% | CARGLTAPGFPLDSW |
| PLC-07 | IGHV3-23 | IGHJ6 | 65.26% | IGHV3-23*01 | IGHJ6*01 | 56.92% | CAKGRADCTDGVCYRRYGMDVW |
| PLC-08 | IGHV3-43 | IGHJ4 | 99.44% | IGHV3-43*01 | IGHJ4*02 | 81.44% | CVKGQGGYTYGGFDCW |
| PLC-10 | IGHV5-51 | IGHJ4 | 77.63% | IGHV5-51*01 | IGHJ4*02 | 57.39% | CARTNWPYYFDHW |
| PLC-12 | IGHV3-43 | IGHJ4 | 93.73% | IGHV3-9*01 | IGHJ4*02 | 62.86% | CARDRYQLIIYYFDRW |
| PLC-15 | IGHV3-43 | IGHJ4 | 98.97% | IGHV3-9*01 | IGHJ4*02 | 32.77% | CAKDVRYGYGSTQSAGFDYW |
| PLC-17 | IGHV4-39 | IGHJ4 | 95.53% | IGHV4-39*07 | IGHJ4*02 | 22.10% | CARDKTTMTFSSPIFDYW |
| PLC-18 | IGHV2-5 | IGHJ1 | 95.78% | IGHV2-5*02 | IGHJ1*01 | 56.58% | CAHSGSMWSGYAGTEYFQHW |
| PLC-19 | IGHV4-59 | IGHJ4 | 67.50% | IGHV4-59*01 | IGHJ4*02 | 21.08% | CARAGDYDLLLLDYW |
| PLC-21 | IGHV5-51 | IGHJ6 | 80.02% | IGHV5-51*03 | IGHJ6*03 | 56.06% | CARLPQGGYYYMDVW |
| PLC-22 | IGHV3-53 | IGHJ5 | 85.10% | IGHV3-53*01 | IGHJ4*02 | 77.01% | CARGLTAPGFPLDSW |
| PLC-23 | IGHV3-33 | IGHJ1 | 56.11% | IGHV3-30-3*02 | IGHJ1*01 | 61.13% | CAFAIGADGEYFQHW |
| PLC-24 | IGHV2-70 | IGHJ4 | 69.68% | IGHV2-70*01 | IGHJ4*02 | 40.67% | CARGASETQVAMSTAELYFFDSW |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matera, A.; Marella, A.; Maeda, A.; Da Vià, M.C.; Lazzaroni, F.; Fabris, S.; Pioggia, S.; Porretti, L.; Colombo, F.; Torricelli, F.; et al. Single-Cell RNA Sequencing for the Detection of Clonotypic V(D)J Rearrangements in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 15691. https://doi.org/10.3390/ijms232415691
Matera A, Marella A, Maeda A, Da Vià MC, Lazzaroni F, Fabris S, Pioggia S, Porretti L, Colombo F, Torricelli F, et al. Single-Cell RNA Sequencing for the Detection of Clonotypic V(D)J Rearrangements in Multiple Myeloma. International Journal of Molecular Sciences. 2022; 23(24):15691. https://doi.org/10.3390/ijms232415691
Chicago/Turabian StyleMatera, Antonio, Alessio Marella, Akihiro Maeda, Matteo C. Da Vià, Francesca Lazzaroni, Sonia Fabris, Stefania Pioggia, Laura Porretti, Federico Colombo, Federica Torricelli, and et al. 2022. "Single-Cell RNA Sequencing for the Detection of Clonotypic V(D)J Rearrangements in Multiple Myeloma" International Journal of Molecular Sciences 23, no. 24: 15691. https://doi.org/10.3390/ijms232415691
APA StyleMatera, A., Marella, A., Maeda, A., Da Vià, M. C., Lazzaroni, F., Fabris, S., Pioggia, S., Porretti, L., Colombo, F., Torricelli, F., Neri, A., Taiana, E., Fabbiano, G., Traini, V., Genuardi, E., Drandi, D., Bolli, N., & Lionetti, M. (2022). Single-Cell RNA Sequencing for the Detection of Clonotypic V(D)J Rearrangements in Multiple Myeloma. International Journal of Molecular Sciences, 23(24), 15691. https://doi.org/10.3390/ijms232415691