
Celiac Disease and Neurological Manifestations: From Gluten to Neuroinflammation

 ,
,  ,
,  ,
,  , and
, and
Abstract
1. Introduction
1.1. The History of Celiac Disease
1.2. Pathophysiology of Celiac Disease
2. Celiac Disease and the Brain
2.1. Overview
2.2. Focus on Gluten Ataxia
3. Celiac Disease, Gut Microbiota, and Inflammation
3.1. Gut Microbiota and Gluten Digestion
3.2. Changes in Gut Microbiota in Celiac Disease
4. Biomarkers for Neurological Manifestation of Celiac Disease
5. Neurophysiological Findings in Celiac Disease
5.1. Electroencephalography
5.2. Multimodal Evoked Potentials
5.3. Transcranial Magnetic Simulation
6. Neuroimaging Findings in Celiac Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pennisi, M.; Bramanti, A.; Cantone, M.; Pennisi, G.; Bella, R.; Lanza, G. Neurophysiology of the ‘celiac brain’: Disentangling gut-brain connections. Front. Neurosci. 2017, 11, 498. [Google Scholar] [CrossRef] [PubMed]
- Morello, F.; Ronzani, G.; Cappellari, F. Migraine, cortical blindness, multiple cerebral infarctions and hypocoagulopathy in celiac disease. Neurol. Sci. 2003, 24, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Bingley, P.J.; Norcross, A.J.; Lock, R.J.; Ness, A.; Jones, R.W. Undiagnosed coeliac disease at age seven: Population based prospective birth cohort study. BMJ 2004, 328, 322–323. [Google Scholar] [CrossRef] [PubMed]
- Dicke, W.K.; Weijers, H.A.; Kamer, J.H.V.D. Coeliac Disease the Presence in Wheat of a Factor Having a Deleterious Effect in Cases of Coeliac Disease. Acta Paediatr. 1953, 42, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Paulley, J.W. Observations on the Aetiology of Idiopathic Steatorrhoea. BMJ 1954, 2, 1318–1321. [Google Scholar] [CrossRef]
- Taylor, K.B.; Thomson, D.L.; Truelove, S.C.; Wright, R. An Immunological Study of Coeliac Disease and Idiopathic Steatorrhoea. Serological reactions to gluten and milk proteins. BMJ 1961, 2, 1727–1731. [Google Scholar] [CrossRef]
- Marks, J.; Birkett, D.; Shuster, S.; Roberts, D.F. Small intestinal mucosal abnormalities in relatives of patients with dermatitis herpetiformis. Gut 1970, 11, 493–497. [Google Scholar] [CrossRef][Green Version]
- Hadjivassiliou, M.; Gibson, A.; Davies-Jones, G.; Lobo, A.; Stephenson, T.; Milford-Ward, A. Does cryptic gluten sensitivity play a part in neurological illness? Lancet 1996, 347, 369–371. [Google Scholar] [CrossRef]
- Schuppan, D.; Junker, Y.; Barisani, D. Celiac Disease: From Pathogenesis to Novel Therapies. Gastroenterology 2009, 137, 1912–1933. [Google Scholar] [CrossRef]
- Patel, S.C.; Shreya, D.; Zamora, D.I.; Patel, G.S.; Grossmann, I.; Rodriguez, K.; Soni, M.; Joshi, P.K.; Sange, I. Celiac Disease, Beyond the Bowel: A Review of Its Neurological Manifestations. Cureus 2021, 13, e20112. [Google Scholar] [CrossRef]
- Verdu, E.F.; Schuppan, D. Co-factors, Microbes, and Immunogenetics in Celiac Disease to Guide Novel Approaches for Diagnosis and Treatment. Gastroenterology 2021, 161, 1395–1411.e4. [Google Scholar] [CrossRef]
- Green, P.H.; Cellier, C. Celiac Disease. N. Engl. J. Med. 2007, 357, 1731–1743. [Google Scholar] [CrossRef]
- Durazzo, M.; Ferro, A.; Brascugli, I.; Mattivi, S.; Fagoonee, S.; Pellicano, R. Extra-Intestinal Manifestations of Celiac Disease: What Should We Know in 2022? J. Clin. Med. 2022, 11, 258. [Google Scholar] [CrossRef]
- Kociszewska, D.; Vlajkovic, S.M. The Association of Inflammatory Gut Diseases with Neuroinflammatory and Auditory Disorders. Front. Biosci. 2022, 14, 8. [Google Scholar] [CrossRef]
- Mohan, M.; Okeoma, C.M.; Sestak, K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5407. [Google Scholar] [CrossRef]
- Philip, A.; White, N.D. Gluten, Inflammation, and Neurodegeneration. Am. J. Lifestyle Med. 2022, 16, 32–35. [Google Scholar] [CrossRef]
- Obrenovich, M.E.M. Leaky Gut, Leaky Brain? Microorganisms 2018, 6, 107. [Google Scholar] [CrossRef]
- Mann, E.R.; Li, X. Intestinal antigen-presenting cells in mucosal immune homeostasis: Crosstalk between dendritic cells, macrophages and B-cells. World J. Gastroenterol. 2014, 20, 9653–9664. [Google Scholar] [CrossRef]
- Mohan, M.; Chow, C.-E.T.; Ryan, C.N.; Chan, L.S.; Dufour, J.; Aye, P.P.; Blanchard, J.; Moehs, C.P.; Sestak, K. Dietary Gluten-Induced Gut Dysbiosis Is Accompanied by Selective Upregulation of microRNAs with Intestinal Tight Junction and Bacteria-Binding Motifs in Rhesus Macaque Model of Celiac Disease. Nutrients 2016, 8, 684. [Google Scholar] [CrossRef]
- Philips, C.A.; Rajesh, S.; Nair, D.C.; Ahamed, R.; Abduljaleel, J.K.; Augustine, P. Hepatocellular Carcinoma in 2021: An Exhaustive Update. Cureus 2021, 13, e19274. [Google Scholar] [CrossRef]
- Ford, R.P.K. The gluten syndrome: A neurological disease. Med. Hypotheses 2009, 73, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, L.; Beteta-Gorriti, V.; Alvarez, N.; de Castro, C.G.; de Dios, A.; Palacios, L.; Santos-Juanes, J. Cutaneous and Mucosal Manifestations Associated with Celiac Disease. Nutrients 2018, 10, 800. [Google Scholar] [CrossRef] [PubMed]
- Meresse, B.; Malamut, G.; Cerf-Bensussan, N. Celiac Disease: An Immunological Jigsaw. Immunity 2012, 36, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, V.; Ravich, W.J. Other Neurological Disorders Associated With Gastrointestinal, Liver, or Pancreatic Diseases. In Neurology and General Medicine: Expert Consult; Elsevier Inc.: Amsterdam, The Netherlands, 2008; pp. 281–292. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Sanders, D.S.; Grünewald, R.A.; Woodroofe, N.; Boscolo, S.; Aeschlimann, D. Gluten sensitivity: From gut to brain. Lancet Neurol. 2010, 9, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Sanders, D.D.; Aeschlimann, D.P. Gluten-Related Disorders: Gluten Ataxia. Dig. Dis. 2015, 33, 264–268. [Google Scholar] [CrossRef]
- Chin, R.L.; Sander, H.; Brannagan, T.H.; Green, P.H.; Hays, A.P.; Alaedini, A.; Latov, N. Celiac neuropathy. Neurology 2003, 60, 1581–1585. [Google Scholar] [CrossRef]
- Chin, R.L.; Latov, N. Peripheral neuropathy and celiac disease. Curr. Treat. Options Neurol. 2005, 7, 43–48. [Google Scholar] [CrossRef]
- Mearns, E.S.; Taylor, A.; Thomas Craig, K.J.; Puglielli, S.; Leffler, D.A.; Sanders, D.S.; Lebwohl, B.; Hadjivassiliou, M. Neurological Manifestations of Neuropathy and Ataxia in Celiac Disease: A Systematic Review. Nutrients 2019, 11, 380. [Google Scholar] [CrossRef]
- Parisi, P.; Pietropaoli, N.; Ferretti, A.; Nenna, R.; Mastrogiorgio, G.; Del Pozzo, M.; Principessa, L.; Bonamico, M.; Villa, M. Role of the gluten-free diet on neurological-EEG findings and sleep disordered breathing in children with celiac disease. Seizure 2015, 25, 181–183. [Google Scholar] [CrossRef]
- Ludvigsson, J.F.; Zingone, F.; Tomson, T.; Ekbom, A.; Ciacci, C. Increased risk of epilepsy in biopsy-verified celiac disease: A population-based cohort study. Neurology 2012, 78, 1401–1407. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Grunewald, R.; Lawden, M.; Davies-Jones, G.; Powell, T.; Smith, C. Headache and CNS white matter abnormalities associated with gluten sensitivity. Neurology 2001, 56, 385–388. [Google Scholar] [CrossRef]
- Lichtwark, I.T.; Newnham, E.D.; Robinson, S.R.; Shepherd, S.J.; Hosking, P.; Gibson, P.R.; Yelland, G.W. Cognitive impairment in coeliac disease improves on a gluten-free diet and correlates with histological and serological indices of disease severity. Aliment. Pharmacol. Ther. 2014, 40, 160–170. [Google Scholar] [CrossRef]
- Yelland, G.W. Gluten-induced cognitive impairment (“brain fog”) in coeliac disease. J. Gastroenterol. Hepatol. 2017, 32, 90–93. [Google Scholar] [CrossRef]
- Carta, M.G.; Hardoy, M.C.; Usai, P.; Carpiniello, B.; Angst, J. Recurrent brief depression in celiac disease. J. Psychosom. Res. 2003, 55, 573–574. [Google Scholar] [CrossRef]
- Bushara, K.O. Neurologic presentation of celiac disease. Gastroenterology 2005, 128, S92–S97. [Google Scholar] [CrossRef]
- Barcia, G.; Posar, A.; Santucci, M.; Parmeggiani, A. Autism and Coeliac Disease. J. Autism Dev. Disord. 2007, 38, 407–408. [Google Scholar] [CrossRef]
- Karwautz, A.; Wagner, G.; Berger, G.; Sinnreich, U.; Grylli, V.; Huber, W.-D. Eating Pathology in Adolescents With Celiac Disease. J. Psychosom. Res. 2008, 49, 399–406. [Google Scholar] [CrossRef]
- Addolorato, G.; Capristo, E.; Ghittoni, G.; Valeri, C.; Mascianà, R.; Ancona, C.; Gasbarrini, G. Anxiety But Not Depression Decreases in Coeliac Patients After One-Year Gluten-free Diet: A Longitudinal Study. Scand. J. Gastroenterol. 2001, 36, 502–506. [Google Scholar] [CrossRef]
- Kumar, R.; Lumsden, A.; Ciclitira, P.J.; Ellis, H.; Laurie, G. Human genome search in celiac disease using gliadin cDNA as probe. J. Mol. Biol. 2000, 300, 1155–1167. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Aeschlimann, P.; Sanders, D.S.; Mäki, M.; Kaukinen, K.; Grunewald, R.A.; Bandmann, O.; Woodroofe, N.; Haddock, G.; Aeschlimann, D. Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology 2013, 80, 1740–1745. [Google Scholar] [CrossRef]
- Pinzon, N.E.; Sanz-Morello, B.; Brevé, J.J.P.; Bol, J.G.J.M.; Drukarch, B.; Bauer, J.; Baron, W.; van Dam, A.-M. Astrocyte-derived tissue Transglutaminase affects fibronectin deposition, but not aggregation, during cuprizone-induced demyelination. Sci. Rep. 2017, 7, srep40995. [Google Scholar] [CrossRef]
- Thomas, H.; Beck, K.; Adamczyk, M.; Aeschlimann, P.; Langley, M.; Oita, R.C.; Thiebach, L.; Hils, M.; Aeschlimann, D. Transglutaminase 6: A protein associated with central nervous system development and motor function. Amino Acids 2011, 44, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Rouvroye, M.D.; Zis, P.; Van Dam, A.-M.; Rozemuller, A.J.M.; Bouma, G.; Hadjivassiliou, M. The Neuropathology of Gluten-Related Neurological Disorders: A Systematic Review. Nutrients 2020, 12, 822. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Croall, I.D.; Zis, P.; Sarrigiannis, P.G.; Sanders, D.S.; Aeschlimann, P.; Grünewald, R.A.; Armitage, P.A.; Connolly, D.; Aeschlimann, D.; et al. Neurologic Deficits in Patients With Newly Diagnosed Celiac Disease Are Frequent and Linked With Autoimmunity to Transglutaminase 6. Clin. Gastroenterol. Hepatol. 2019, 17, 2678–2686.e2. [Google Scholar] [CrossRef] [PubMed]
- Coronel-Rodríguez, C.; Rodríguez-Martínez, A. Neurological Manifestations of the Celiac Disease in Children. OBM Neurobiol. 2020, 4, 1–15. [Google Scholar] [CrossRef]
- Zhu, J.; Li, Z.; Ji, Z.; Wu, Y.; He, Y.; Liu, K.; Chang, Y.; Peng, Y.; Lin, Z.; Wang, S.; et al. Glycocalyx is critical for blood-brain barrier integrity by suppressing caveolin1-dependent endothelial transcytosis following ischemic stroke. Brain Pathol. 2021, 32, e13006. [Google Scholar] [CrossRef]
- Barone, M.V.; Auricchio, R.; Nanayakkara, M.; Greco, L.; Troncone, R.; Auricchio, S. Pivotal Role of Inflammation in Celiac Disease. Int. J. Mol. Sci. 2022, 23, 7177. [Google Scholar] [CrossRef]
- Porpora, M.; Conte, M.; Lania, G.; Bellomo, C.; Rapacciuolo, L.; Chirdo, F.G.; Auricchio, R.; Troncone, R.; Auricchio, S.; Barone, M.V.; et al. Inflammation Is Present, Persistent and More Sensitive to Proinflammatory Triggers in Celiac Disease Enterocytes. Int. J. Mol. Sci. 2022, 23, 1973. [Google Scholar] [CrossRef]
- Severance, E.G.; Gressitt, K.L.; Alaedini, A.; Rohleder, C.; Enning, F.; Bumb, J.M.; Müller, J.K.; Schwarz, E.; Yolken, R.H.; Leweke, F.M. IgG dynamics of dietary antigens point to cerebrospinal fluid barrier or flow dysfunction in first-episode schizophrenia. Brain Behav. Immun. 2015, 44, 148–158. [Google Scholar] [CrossRef]
- Volta, U.; de Giorgio, R.; Petrolini, N.; Stanghellini, V.; Barbara, G.; Granito, A.; De Ponti, F.; Corinaldesi, R.; Bianchi, F.B. Clinical Findings and Anti-Neuronal Antibodies in Coeliac Disease with Neurological Disorders. Scand. J. Gastroenterol. 2002, 37, 1276–1281. [Google Scholar] [CrossRef]
- Cervio, E.; Volta, U.; Verri, M.; Boschi, F.; Pastoris, O.; Granito, A.; Barbara, G.; Parisi, C.; Felicani, C.; Tonini, M.; et al. Sera of Patients With Celiac Disease and Neurologic Disorders Evoke a Mitochondrial-Dependent Apoptosis In Vitro. Gastroenterology 2007, 133, 195–206. [Google Scholar] [CrossRef]
- Volta, U.; De Giorgio, R.; Granito, A.; Stanghellini, V.; Barbara, G.; Avoni, P.; Liguori, R.; Petrolini, N.; Fiorini, E.; Montagna, P.; et al. Anti-ganglioside antibodies in coeliac disease with neurological disorders. Dig. Liver Dis. 2006, 38, 183–187. [Google Scholar] [CrossRef]
- Granito, A.; Tovoli, F.; Raiteri, A.; Volta, U. Anti-ganglioside antibodies and celiac disease. Allergy Asthma Clin. Immunol. 2021, 17, 53. [Google Scholar] [CrossRef]
- Godschalk, P.C.R.; Heikema, A.P.; Gilbert, M.; Komagamine, T.; Ang, C.W.; Glerum, J.; Brochu, D.; Li, J.; Yuki, N.; Jacobs, B.C.; et al. The crucial role of Campylobacter jejuni genes in anti-ganglioside an-tibody induction in Guillain-Barre syndrome. J. Clin. Investig. 2004, 114, 1659–1665. [Google Scholar] [CrossRef]
- Perera, V.N.; Nachamkin, I.; Ung, H.; Patterson, J.H.; McConville, M.J.; Coloe, P.J.; Fry, B.N. Molecular mimicry inCampylobacter jejuni: Role of the lipo-oligosaccharide core oligosaccharide in inducing anti-ganglioside antibodies. FEMS Immunol. Med. Microbiol. 2007, 50, 27–36. [Google Scholar] [CrossRef]
- Cooke, W.T.; Smith, W.T. Neurological disorders associated with adult celiac disease. Brain 1966, 89, 683–722. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Grünewald, R.; Chattopadhyay, A.; Davies-Jones, G.; Gibson, A.; Jarratt, J.; Kandler, R.; Lobo, A.; Powell, T.; Smith, C. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet 1998, 352, 1582–1585. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Sanders, D.S.; Woodroofe, N.; Williamson, C.; Grünewald, R.A. Gluten ataxia. Cerebellum 2008, 7, 494–498. [Google Scholar] [CrossRef]
- Bushara, K.O.; Goebel, S.U.; Shill, H.; Goldfarb, L.G.; Hallett, M. Gluten sensitivity in sporadic and hereditary cerebellar ataxia. Ann. Neurol. 2001, 49, 540–543. [Google Scholar] [CrossRef]
- Pellecchia, M.T.; Scala, R.; Filla, A.; De Michele, G.; Ciacci, C.; Barone, P. Idiopathic cerebellar ataxia associated with celiac disease: Lack of distinctive neurological features. J. Neurol. Neurosurg. Psychiatry 1999, 66, 32–35. [Google Scholar] [CrossRef]
- Burk, K.; Bösch, S.; Müller, C.A.; Melms, A.; Zühlke, C.; Stern, M.; Besenthal, I.; Skalej, M.; Ruck, P.; Ferber, S.; et al. Sporadic cerebellar ataxia associated with gluten sensitivity. Brain 2001, 124, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Williamson, C.A.; Woodroofe, N. The immunology of gluten sensitivity: Beyond the gut. Trends Immunol. 2004, 25, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M. Immune-mediated acquired ataxias. Handb. Clin. Neurol. 2011, 103, 189–199. [Google Scholar] [CrossRef]
- Brucke, T.; Kollegger, H.; Schmidbauer, M.; Muller, C.; Podreka, I.; Deecke, L. Adult coeliac disease and brainstem encephalitis. J. Neurol. Neurosurg. Psychiatry 1988, 51, 456–457. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deconinck, N.; Scaillon, M.; Segers, V.; Groswasser, J.J.; Dan, B. Opsoclonus-Myoclonus Associated With Celiac Disease. Pediatr. Neurol. 2006, 34, 312–314. [Google Scholar] [CrossRef]
- Pereira, A.C.; Edwards, M.J.; Buttery, P.C.; Hawkes, C.H.; Quinn, N.P.; Giovannoni, G.; Hadjivassiliou, M.; Bhatia, K.P. Choreic syndrome and coeliac disease: A hitherto unrecognised association. Mov. Disord. 2003, 19, 478–482. [Google Scholar] [CrossRef]
- Wilkinson, I.D.; Hadjivassiliou, M.; Dickson, J.; Wallis, L.; Grünewald, R.A.; Coley, S.C.; Widjaja, E.; Griffiths, P.D. Cerebellar abnormalities on proton MR spectroscopy in gluten ataxia. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1011–1013. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Grünewald, R.; Sharrack, B.; Sanders, D.; Lobo, A.; Williamson, C.; Woodroofe, N.; Wood, N.; Davies-Jones, A. Gluten ataxia in perspective: Epidemiology, genetic susceptibility and clinical characteristics. Brain 2003, 126, 685–691. [Google Scholar] [CrossRef]
- Bushara, K.O.; Nance, M.; Gomez, C.M. Antigliadin antibodies in Huntington’s disease. Neurology 2004, 62, 132–133. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Grünewald, R.A.; Kandler, R.H.; Chattopadhyay, A.K.; Jarratt, J.A.; Sanders, D.S.; Sharrack, B.; Wharton, S.B.; Davies-Jones, G.A. Neuropathy associated with gluten sensitivity. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1262–1266. [Google Scholar] [CrossRef]
- Sapone, A.; Bai, J.C.; Ciacci, C.; Dolinsek, J.; Green, P.H.R.; Hadjivassiliou, M.; Kaukinen, K.; Rostami, K.; Sanders, D.S.; Schumann, M.; et al. Spectrum of gluten-related disorders: Consensus on new nomenclature and classification. BMC Med. 2012, 10, 13. [Google Scholar] [CrossRef]
- Schrödl, D.; Kahlenberg, F.; Peter-Zimmer, K.; Hermann, W.; Kühn, H.-J.; Mothes, T. Intrathecal synthesis of autoantibodies against tissue transglutaminase. J. Autoimmun. 2004, 22, 335–340. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Grünewald, R.A.; Sanders, D.S.; Zis, P.; Croall, I.; Shanmugarajah, P.D.; Sarrigiannis, P.G.; Trott, N.; Wild, G.; Hoggard, N. The Significance of Low Titre Antigliadin Antibodies in the Diagnosis of Gluten Ataxia. Nutrients 2018, 10, 1444. [Google Scholar] [CrossRef]
- Kaukinen, K.; Collin, P.; Laurila, K.; Kaartinen, T.; Partanen, J.; Mäki, M. Resurrection of gliadin antibodies in coeliac disease. Deamidated gliadin peptide antibody test provides additional diagnostic benefit. Scand. J. Gastroenterol. 2007, 42, 1428–1433. [Google Scholar] [CrossRef]
- Marsh, M. The natural history of gluten sensitivity: Defining, refining and re-defining. QJM Int. J. Med. 1995, 88, 9–13. [Google Scholar] [CrossRef]
- Cataldo, F.; Marino, V.; Ventura, A.; Bottaro, G.; Corazza, G.R. The Italian Society of Paediatric Gastroenterology and Hepatology (SIGEP) “Club del Tenue” Working Groups on Coeliac Disease Prevalence and clinical features of selective immunoglobulin A deficiency in coeliac disease: An Italian multicentre study. Gut 1998, 42, 362–365. [Google Scholar] [CrossRef]
- Marzari, R.; Sblattero, D.; Florian, F.; Tongiorgi, E.; Not, T.; Tommasini, A.; Ventura, A.; Bradbury, A. Molecular Dissection of the Tissue Transglutaminase Autoantibody Response in Celiac Disease. J. Immunol. 2001, 166, 4170–4176. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Boscolo, S.; Davies–Jones, G.A.; Grünewald, R.A.; Not, T.; Sanders, D.S.; Simpson, J.E.; Tongiorgi, E.; Williamson, C.A.; Woodroofe, N.M. The humoral response in the pathogenesis of gluten ataxia. Neurology 2002, 58, 1221–1226. [Google Scholar] [CrossRef]
- VAN DE Wal, Y.; Kooy, Y.; VAN Veelen, P.; Vader, W.; Koning, F.; Peña, S. Coeliac disease: It takes three to tango! Gut 2000, 46, 734–737. [Google Scholar] [CrossRef][Green Version]
- Molberg, Ø.; Mcadam, S.N.; Körner, R.; Quarsten, H.; Kristiansen, C.; Madsen, L.; Fugger, L.; Scott, H.; Norén, O.; Roepstorff, P.; et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998, 4, 713–717. [Google Scholar] [CrossRef]
- Korponay-Szabo, I.R.; Halttunen, T.; Szalai, Z.; Laurila, K.; Király, R.; Kovács, J.B.; Fésüs, L.; Mäki, M. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut 2004, 53, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Sárdy, M.; Kárpáti, S.; Merkl, B.; Paulsson, M.; Smyth, N. Epidermal Transglutaminase (TGase 3) Is the Autoantigen of Dermatitis Herpetiformis. J. Exp. Med. 2002, 195, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Boscolo, S.; Sarich, A.; Lorenzon, A.; Passoni, M.; Rui, V.; Stebel, M.; Sblattero, D.; Marzari, R.; Hadjivassiliou, M.; Tongiorgi, E. Gluten Ataxia: Passive Transfer in a Mouse Model. Ann. N. Y. Acad. Sci. 2007, 1107, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Honnorat, J.; Saiz, A.; Giometto, B.; Vincent, A.; Brieva, L.; De Andres, C.; Maestre, J.; Fabien, N.; Vighetto, A.; Casamitjana, R.; et al. Cerebellar Ataxia With Anti–Glutamic Acid Decarboxylase Antibodies. Arch. Neurol. 2001, 58, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Krogsgaard-Larsen, P.; Frølund, B.; Kristiansen, U.; Ebart, B. Ligands for the GABAA Receptor Complex. In Glutamate and GABA Receptors of AMPA/Kainate Receptors and Transporters; Taylor & Francis: Milton Park, UK, 2002; Available online: https://drug.ku.dk/staff/researchersilf/?pure=en%2Fpublications%2Fligands-for-the-gabaa-receptor-complex(d03c7fb7-167a-4be6-8333-69afcb19e6d2)%2Fexport.html (accessed on 1 November 2022).
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Akobeng, A.K.; Singh, P.; Kumar, M.; Al Khodor, S. Role of the gut microbiota in the pathogenesis of coeliac disease and potential therapeutic implications. Eur. J. Nutr. 2020, 59, 3369–3390. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Singh, P.; Kumar, M.; Al Khodor, S. Vitamin D Deficiency in the Gulf Cooperation Council: Exploring the Triad of Genetic Predisposition, the Gut Microbiome and the Immune System. Front. Immunol. 2019, 10, 1042. [Google Scholar] [CrossRef]
- Pecora, F.; Persico, F.; Gismondi, P.; Fornaroli, F.; Iuliano, S.; de’Angelis, G.L.; Esposito, S. Gut Microbiota in Celiac Disease: Is There Any Role for Probiotics? Front. Immunol. 2020, 11, 957. [Google Scholar] [CrossRef]
- Giuffrè, M.; Campigotto, M.; Campisciano, G.; Comar, M.; Crocè, L.S. A story of liver and gut microbes: How does the intestinal flora affect liver disease? A review of the literature. Am. J. Physiol. Liver Physiol. 2020, 318, G889–G906. [Google Scholar] [CrossRef]
- Giuffrè, M.; Moretti, R.; Campisciano, G.; Da Silveira, A.B.M.; Monda, V.M.; Comar, M.; Di Bella, S.; Antonello, R.M.; Luzzati, R.; Crocè, L.S. You Talking to Me? Says the Enteric Nervous System (ENS) to the Microbe. How Intestinal Microbes Interact with the ENS. J. Clin. Med. 2020, 9, 3705. [Google Scholar] [CrossRef]
- Laterza, L.; Rizzatti, G.; Gaetani, E.; Chiusolo, P.; Gasbarrini, A. The gut microbiota and immune system relationship in human graft-versus-host disease. Mediterr. J. Hematol. Infect. Dis. 2016, 8, 2016025. [Google Scholar] [CrossRef]
- Caminero, A.; Herrán, A.R.; Nistal, E.; Pérez-Andrés, J.; Vaquero, L.; Vivas, S.; De Morales, J.M.G.R.; Albillos, S.M.; Casqueiro, J. Diversity of the cultivable human gut microbiome involved in gluten metabolism: Isolation of microorganisms with potential interest for coeliac disease. FEMS Microbiol. Ecol. 2014, 88, 309–319. [Google Scholar] [CrossRef]
- Olivares, M.; Laparra, M.; Sanz, Y. Influence of Bifidobacterium longum CECT 7347 and Gliadin Peptides on Intestinal Epithelial Cell Proteome. J. Agric. Food Chem. 2011, 59, 7666–7671. [Google Scholar] [CrossRef]
- Caminero, A.; Galipeau, H.J.; McCarville, J.L.; Johnston, C.W.; Bernier, S.P.; Russell, A.K.; Jury, J.; Herran, A.R.; Casqueiro, J.; Tye-Din, J.A.; et al. Duodenal Bacteria From Patients With Celiac Disease and Healthy Subjects Distinctly Affect Gluten Breakdown and Immunogenicity. Gastroenterology 2016, 151, 670–683. [Google Scholar] [CrossRef]
- Nadal, I.; Donant, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 2007, 56, 1669–1674. [Google Scholar] [CrossRef]
- De Palma, G.; Nadal, I.; Medina, M.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol. 2010, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalances in faecal and duodenal Bifidobacterium species composition in active and non-active coeliac disease. BMC Microbiol. 2008, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J. Clin. Pathol. 2008, 62, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Ou, G.; Hedberg, M.; Hörstedt, P.; Baranov, V.; Forsberg, G.; Drobni, M.; Sandström, O.; Wai, S.N.; Johansson, I.; Hammarström, M.-L.; et al. Proximal Small Intestinal Microbiota and Identification of Rod-Shaped Bacteria Associated With Childhood Celiac Disease. Am. J. Gastroenterol. 2009, 104, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Di Cagno, R.; De Angelis, M.; De Pasquale, I.; Ndagijimana, M.; Vernocchi, P.; Ricciuti, P.; Gagliardi, F.; Laghi, L.; Crecchio, C.; Guerzoni, M.E.; et al. Duodenal and faecal microbiota of celiac children: Molecular, phenotype and metabolome characterization. BMC Microbiol. 2011, 11, 219. [Google Scholar] [CrossRef]
- Heyman, M.; Abed, J.; Lebreton, C.; Cerf-Bensussan, N. Intestinal permeability in coeliac disease: Insight into mechanisms and relevance to pathogenesis. Gut 2011, 61, 1355–1364. [Google Scholar] [CrossRef]
- Lindfors, K.; Blomqvist, T.; Juuti-Uusitalo, K.; Stenman, S.; Venäläinen, J.; Mäki, M.; Kaukinen, K. Live probiotic Bifidobacterium lactis bacteria inhibit the toxic effects induced by wheat gliadin in epithelial cell culture. Clin. Exp. Immunol. 2008, 152, 552–558. [Google Scholar] [CrossRef]
- Medina, M.; De Palma, G.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Bifidobacterium strains suppress in vitro the pro-inflammatory milieu triggered by the large intestinal microbiota of coeliac patients. J. Inflamm. 2008, 5, 19. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Stolp, H.; Dziegielewska, K.M.; Ek, C.J.; Habgood, M.D.; Lane, M.; Potter, A.M.; Saunders, N.R. Breakdown of the blood–brain barrier to proteins in white matter of the developing brain following systemic inflammation. Cell Tissue Res. 2005, 320, 369–378. [Google Scholar] [CrossRef]
- Jiang, S.; Xia, R.; Jiang, Y.; Wang, L.; Gao, F. Vascular Endothelial Growth Factors Enhance the Permeability of the Mouse Blood-brain Barrier. PLoS ONE 2014, 9, e86407. [Google Scholar] [CrossRef]
- Gu, Y.; Dee, C.M.; Shen, J. Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front. Biosci. 2011, S3, 1216–1231. [Google Scholar] [CrossRef] [PubMed]
- Matisz, C.; Gruber, A. Neuroinflammatory remodeling of the anterior cingulate cortex as a key driver of mood disorders in gastrointestinal disease and disorders. Neurosci. Biobehav. Rev. 2022, 133, 104497. [Google Scholar] [CrossRef] [PubMed]
- Delpech, J.-C.; Madore, C.; Joffre, C.; Aubert, A.; Kang, J.X.; Nadjar, A.; Layé, S. Transgenic Increase in n-3/n-6 Fatty Acid Ratio Protects Against Cognitive Deficits Induced by an Immune Challenge through Decrease of Neuroinflammation. Neuropsychopharmacology 2014, 40, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Rawat, A.; Al-Jarrah, B.; Saraswathi, S.; Gad, H.; Elawad, M.; Hussain, K.; Hendaus, M.A.; Al-Masri, W.; Malik, R.A.; et al. Distinctive Microbial Signatures and Gut-Brain Crosstalk in Pediatric Patients with Coeliac Disease and Type 1 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 1511. [Google Scholar] [CrossRef]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; De Oliveira, A.C.P. Lipopolysaccharide-Induced Neuroinflammation as a Bridge to Understand Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.-S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Bozzato, A.M.; Martingano, P.; Mucelli, R.A.P.; Cavallaro, M.F.M.; Cesarotto, M.; Marcello, C.; Tiribelli, C.; Pascut, D.; Pizzolato, R.; Mucelli, F.P.; et al. MicroRNAs Related to TACE Treatment Response: A Review of the Literature from a Radiological Point of View. Diagnostics 2022, 12, 374. [Google Scholar] [CrossRef]
- Magni, S.; Comani, G.B.; Elli, L.; Vanessi, S.; Ballarini, E.; Nicolini, G.; Rusconi, M.; Castoldi, M.; Meneveri, R.; Muckenthaler, M.U.; et al. miRNAs Affect the Expression of Innate and Adaptive Immunity Proteins in Celiac Disease. Am. J. Gastroenterol. 2014, 109, 1662–1674. [Google Scholar] [CrossRef]
- Vaira, V.; Roncoroni, L.; Barisani, D.; Gaudioso, G.; Bosari, S.; Bulfamante, G.; Doneda, L.; Conte, D.; Tomba, C.; Bardella, M.T.; et al. microRNA profiles in coeliac patients distinguish different clinical phenotypes and are modulated by gliadin peptides in primary duodenal fibroblasts. Clin. Sci. 2013, 126, 417–423. [Google Scholar] [CrossRef]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs Are Differentially Expressed in Ulcerative Colitis and Alter Expression of Macrophage Inflammatory Peptide-2α. Gastroenterology 2008, 135, 1624–1635.e24. [Google Scholar] [CrossRef]
- Capuano, M.; Iaffaldano, L.; Tinto, N.; Montanaro, D.; Capobianco, V.; Izzo, V.; Tucci, F.; Troncone, G.; Greco, L.; Sacchetti, L. MicroRNA-449a Overexpression, Reduced NOTCH1 Signals and Scarce Goblet Cells Characterize the Small Intestine of Celiac Patients. PLoS ONE 2011, 6, e29094. [Google Scholar] [CrossRef]
- Amr, K.S.; Bayoumi, F.S.; Eissa, E.; Abu-Zekry, M. Circulating microRNAs as potential non-invasive biomarkers in pediatric patients with celiac disease. Eur. Ann. Allergy Clin. Immunol. 2019, 51, 159–164. [Google Scholar] [CrossRef]
- Ramírez-Sánchez, A.D.; Tan, I.L.; Gonera-De Jong, B.C.; Visschedijk, M.C.; Jonkers, I.; Withoff, S. Molecular Biomarkers for Celiac Disease: Past, Present and Future. Int. J. Mol. Sci. 2020, 21, 8528. [Google Scholar] [CrossRef]
- Mills, J.R.; Murray, J.A. Contemporary celiac disease diagnosis: Is a biopsy avoidable? Curr. Opin. Gastroenterol. 2016, 32, 80–85. [Google Scholar] [CrossRef]
- McKeon, A.; Lennon, V.A.; Pittock, S.J.; Kryzer, T.J.; Murray, J. The neurologic significance of celiac disease biomarkers. Neurology 2014, 83, 1789–1796. [Google Scholar] [CrossRef]
- Samaroo, D.; Dickerson, F.; Kasarda, D.D.; Green, P.H.; Briani, C.; Yolken, R.H.; Alaedini, A. Novel immune response to gluten in individuals with schizophrenia. Schizophr. Res. 2010, 118, 248–255. [Google Scholar] [CrossRef]
- Leffler, D.A.; Green, P.H.R.; Fasano, A. Extraintestinal manifestations of coeliac disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 561–571. [Google Scholar] [CrossRef]
- Campagna, G.; Pesce, M.; Tatangelo, R.; Rizzuto, A.; La Fratta, I.; Grilli, A. The progression of coeliac disease: Its neurological and psychiatric implications. Nutr. Res. Rev. 2016, 30, 25–35. [Google Scholar] [CrossRef]
- Vives-Pi, M.; Takasawa, S.; Pujol-Autonell, I.; Planas, R.; Cabre, E.; Ojanguren, I.; Montraveta, M.; Santos, A.L.; Ruiz-Ortiz, E. Biomarkers for Diagnosis and Monitoring of Celiac Disease. J. Clin. Gastroenterol. 2013, 47, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Parisi, P.; Principessa, L.; Ferretti, A.; D’Onofrio, D.; Del Giudice, E.; Pacchiarotti, C.; Villa, M.P. “EEG abnormalities” may represent a confounding factor in celiac disease: A 4-year follow-up family report. Epilepsy Behav. Rep. 2014, 2, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Işıkay, S.; Kocamaz, H.; Sezer, S.; Özkars, M.Y.; Işıkay, N.; Filik, B.; Şan, M.; Kanmaz, A. The Frequency of Epileptiform Discharges in Celiac Disease. Pediatr. Neurol. 2015, 53, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Işikay, S.; Hizli, Ş.; Çoşkun, S.; Yilmaz, K. Increased tissue transglutaminase levels are associated with increased epileptiform activity in electroencephalography among patients with celiac disease. Arq. Gastroenterol. 2015, 52, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Di Lazzaro, V.; Pilato, F.; Batocchi, A.P.; Restuccia, D.; Cammarota, G.; Profice, P. Tired legs-a gut diagnosis. Lancet 2010, 376, 1798. [Google Scholar] [CrossRef]
- Pawlak-Osińska, K.; Kaźmierczak, H.; Kuczyńska, R.; Szaflarska-Popławska, A. Looking for the auditory and vestibular pathology in celiac disease. Otolaryngol. Pol. 2007, 61, 178–183. [Google Scholar] [CrossRef]
- Aksoy, E.; Teber, S.; Kansu, A.; Deda, G.; Kartal, A. Neurological findings spectrum in celiac disease. Turk. J. Pediatr. 2016, 58, 233–240. [Google Scholar] [CrossRef]
- Pellecchia, M.T.; Scala, R.; Perretti, A.; De Michele, G.; Santoro, L.; Filla, A.; Ciacci, C.; Barone, P. Cerebellar ataxia associated with subclinical celiac disease responding to gluten-free diet. Neurology 1999, 53, 1606. [Google Scholar] [CrossRef]
- Lanza, G.; Fisicaro, F.; Dubbioso, R.; Ranieri, F.; Chistyakov, A.V.; Cantone, M.; Pennisi, M.; Grasso, A.A.; Bella, R.; Di Lazzaro, V. A comprehensive review of transcranial magnetic stimulation in secondary dementia. Front. Aging Neurosci. 2022, 14, 995000. [Google Scholar] [CrossRef]
- Lanza, G.; Bella, R.; Giuffrida, S.; Cantone, M.; Pennisi, G.; Spampinato, C.; Giordano, D.; Malaguarnera, G.; Raggi, A.; Pennisi, M. Preserved Transcallosal Inhibition to Transcranial Magnetic Stimulation in Nondemented Elderly Patients with Leukoaraiosis. BioMed Res. Int. 2013, 2013, 351680. [Google Scholar] [CrossRef]
- Bella, R.; Ferri, R.; Cantone, M.; Pennisi, M.; Lanza, G.; Malaguarnera, G.; Spampinato, C.; Giordano, D.; Raggi, A.; Pennisi, G. Motor cortex excitability in vascular depression. Int. J. Psychophysiol. 2011, 82, 248–253. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Oliviero, A.; Mazzone, P.; Insola, A.; Pilato, F.; Saturno, E.; Accurso, A.; Tonali, P.; Rothwell, J. Comparison of descending volleys evoked by monophasic and biphasic magnetic stimulation of the motor cortex in conscious humans. Exp. Brain Res. 2001, 141, 121–127. [Google Scholar] [CrossRef]
- Bella, R.; Ferri, R.; Lanza, G.; Cantone, M.; Pennisi, M.; Puglisi, V.; Vinciguerra, L.; Spampinato, C.; Mazza, T.; Malaguarnera, G.; et al. TMS follow-up study in patients with vascular cognitive impairment-no dementia. Neurosci. Lett. 2013, 534, 155–159. [Google Scholar] [CrossRef]
- Pennisi, G.; Lanza, G.; Giuffrida, S.; Vinciguerra, L.; Puglisi, V.; Cantone, M.; Pennisi, M.; D’Agate, C.C.; Naso, P.; Aprile, G.; et al. Excitability of the Motor Cortex in De Novo Patients with Celiac Disease. PLoS ONE 2014, 9, e102790. [Google Scholar] [CrossRef]
- Bella, R.; Lanza, G.; Cantone, M.; Giuffrida, S.; Puglisi, V.; Vinciguerra, L.; Pennisi, M.; Ricceri, R.; D’Agate, C.C.; Malaguarnera, G.; et al. Effect of a Gluten-Free Diet on Cortical Excitability in Adults with Celiac Disease. PLoS ONE 2015, 10, e0129218. [Google Scholar] [CrossRef]
- Pennisi, M.; Lanza, G.; Cantone, M.; Ricceri, R.; Ferri, R.; D’Agate, C.C.; Pennisi, G.; Di Lazzaro, V.; Bella, R. Cortical involvement in celiac disease before and after long-term gluten-free diet: A Transcranial Magnetic Stimulation study. PLoS ONE 2017, 12, e0177560. [Google Scholar] [CrossRef]
- Fisicaro, F.; Lanza, G.; D’Agate, C.; Ferri, R.; Cantone, M.; Falzone, L.; Pennisi, G.; Bella, R.; Pennisi, M. Intracortical and Intercortical Motor Disinhibition to Transcranial Magnetic Stimulation in Newly Diagnosed Celiac Disease Patients. Nutrients 2021, 13, 1530. [Google Scholar] [CrossRef]
- Lanza, G.; Fisicaro, F.; D’Agate, C.C.; Ferri, R.; Cantone, M.; Falzone, L.; Pennisi, G.; Bella, R.; Hadjivassiliou, M.; Pennisi, M. Preserved central cholinergic functioning to transcranial magnetic stimulation in de novo patients with celiac disease. PLoS ONE 2021, 16, e0261373. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Croall, I.; Grünewald, R.; Trott, N.; Sanders, D.; Hoggard, N. Neurological Evaluation of Patients with Newly Diagnosed Coeliac Disease Presenting to Gastroenterologists: A 7-Year Follow-Up Study. Nutrients 2021, 13, 1846. [Google Scholar] [CrossRef]
- Sharma, V.K.; Wong, K.S.; Alexandrov, A.V. Transcranial Doppler. In Intracranial Atherosclerosis: Pathophysiology, Diagnosis and Treatment; Kim, J.S., Caplan, L.R., Wong, K.S., Eds.; Karger: Basel, Switzerland, 2016; Volume 40, pp. 124–140. [Google Scholar] [CrossRef]
- Hakimi, R.; Alexandrov, A.V.; Garami, Z. Neuro-ultrasonography. Neurol. Clin. 2019, 38, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, V.; Bramanti, A.; Lanza, G.; Cantone, M.; Vinciguerra, L.; Pennisi, M.; Bonanno, L.; Pennisi, G.; Bella, R. Impaired Cerebral Haemodynamics in Vascular Depression: Insights From Transcranial Doppler Ultrasonography. Front. Psychiatry 2018, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, F.; Lanza, G.; D’Agate, C.C.; Pennisi, M.; Cantone, M.; Pennisi, G.; Hadjivassiliou, M.; Bella, R. Cerebral hemodynamic changes to transcranial Doppler sonography in celiac disease: A pilot study. Front. Hum. Neurosci. 2022, 16, 931727. [Google Scholar] [CrossRef] [PubMed]
- Vagli, C.; Fisicaro, F.; Vinciguerra, L.; Puglisi, V.; Rodolico, M.S.; Giordano, A.; Ferri, R.; Lanza, G.; Bella, R. Cerebral Hemodynamic Changes to Transcranial Doppler in Asymptomatic Patients with Fabry’s Disease. Brain Sci. 2020, 10, 546. [Google Scholar] [CrossRef]
- Lanza, G.; Bella, R.; Cantone, M.; Pennisi, G.; Ferri, R.; Pennisi, M. Cognitive Impairment and Celiac Disease: Is Transcranial Magnetic Stimulation a Trait d’Union between Gut and Brain? Int. J. Mol. Sci. 2018, 19, 2243. [Google Scholar] [CrossRef]


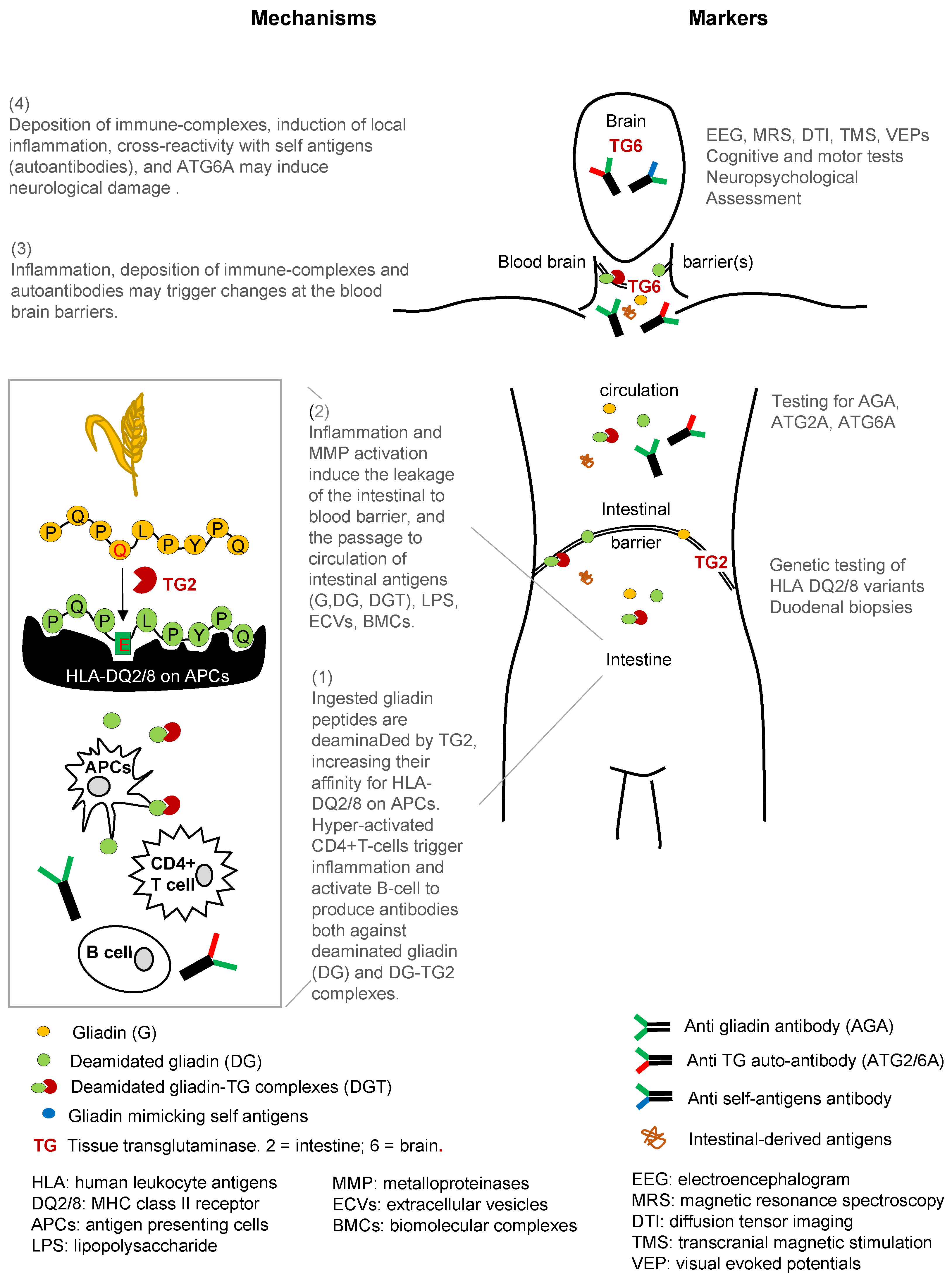{kind=link}
| Tissue, Organ, or System | Antigen Target | Response | Effects | Biomarker Tool |
|---|---|---|---|---|
| Intestine | Gluten Gliadin Peptides | TG → gliadin-TG complexes | APCs (high affinity to antigens in HLA-DQ2/DQ8 individuals), Th1 cells. | Genetic testing: HLA-DQ2/DQ8 duodenal biopsy (gold standard) |
| Intestinal Barrier | Gliadin Gliadin peptides Deamidated gliadin Peptides | APCs, Th2 cells → proinflammatory cytokines and chemokines ROS, LPS; downregulation of TJ & PPAR-γ EGFR pathway activation | APCs, Th1 cells, B cells Antibodies to gliadin Autoantibodies gliadin-TG complex → deposited in tissues | Genetic testing: HLA-DQ2/DQ8 duodenal biopsy intestinal barrier leakage tests |
| Blood/Systemic Circulation | Gliadin Gliadin peptides Deamidated gliadin Peptides | Proinflammatory cytokines ROS, LPS Biomolecular complexes (BMCs) Extracellular vesicles (ECVs) | Antibodies to gliadin Autoantibodies gliadin-TG complex → deposited in tissues | Genetic Testing: HLA-DQ2/DQ8 Anti-gliadin antibodies Anti-endomysium antibodies Abs: Anti-TGs, Anti-TG2, Anti-TG6 |
| Blood Brain Barrier | Gliadin Gliadin peptides Deamidated gliadin Peptides | Proinflammatory cytokines ROS, LPS Stripping of glycocalyx in endothelial cells → integrity loss | Microglia activation Antibodies to gliadin Autoantibodies gliadin-TG complex Anti-TG6 antibodies | Autoantibodies to gliadin-TG complex |
| Brain | TG6 expression in CNS structures | Low grade inflammation → increased sensitivity Calcification in occipital lobes | Microglia activation Antibodies to gliadin Autoantibodies gliadin-TG complex Anti-TG6 antibodies | Epileptiform changes, cortical excitability, white matter lesions Using various brain imaging tools: EEG, MRS, DTI, TMS, VEPs |
| Cerebellar | TG6 expression in cerebellum | Low grade inflammation → increased sensitivity | Anti-TG6 antibodies | Motor dysfunction |
| Non-Cerebellar | TG6 expression in brainstem, forceps major of corpus callosum, segment of superior longitudinal fasciculus, thalamic white matter | Low grade inflammation → increased sensitivity | Anti-TG6 antibodies | Cognitive tests Neuropsychological assessment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuffrè, M.; Gazzin, S.; Zoratti, C.; Llido, J.P.; Lanza, G.; Tiribelli, C.; Moretti, R. Celiac Disease and Neurological Manifestations: From Gluten to Neuroinflammation. Int. J. Mol. Sci. 2022, 23, 15564. https://doi.org/10.3390/ijms232415564
Giuffrè M, Gazzin S, Zoratti C, Llido JP, Lanza G, Tiribelli C, Moretti R. Celiac Disease and Neurological Manifestations: From Gluten to Neuroinflammation. International Journal of Molecular Sciences. 2022; 23(24):15564. https://doi.org/10.3390/ijms232415564
Chicago/Turabian StyleGiuffrè, Mauro, Silvia Gazzin, Caterina Zoratti, John Paul Llido, Giuseppe Lanza, Claudio Tiribelli, and Rita Moretti. 2022. "Celiac Disease and Neurological Manifestations: From Gluten to Neuroinflammation" International Journal of Molecular Sciences 23, no. 24: 15564. https://doi.org/10.3390/ijms232415564
APA StyleGiuffrè, M., Gazzin, S., Zoratti, C., Llido, J. P., Lanza, G., Tiribelli, C., & Moretti, R. (2022). Celiac Disease and Neurological Manifestations: From Gluten to Neuroinflammation. International Journal of Molecular Sciences, 23(24), 15564. https://doi.org/10.3390/ijms232415564