
Cross-Talk between Transcriptome Analysis and Dynamic Changes of Carbohydrates Identifies Stage-Specific Genes during the Flower Bud Differentiation Process of Chinese Cherry (Prunus pseudocerasus L.)

 , ,
, ,
Abstract
1. Introduction
2. Results
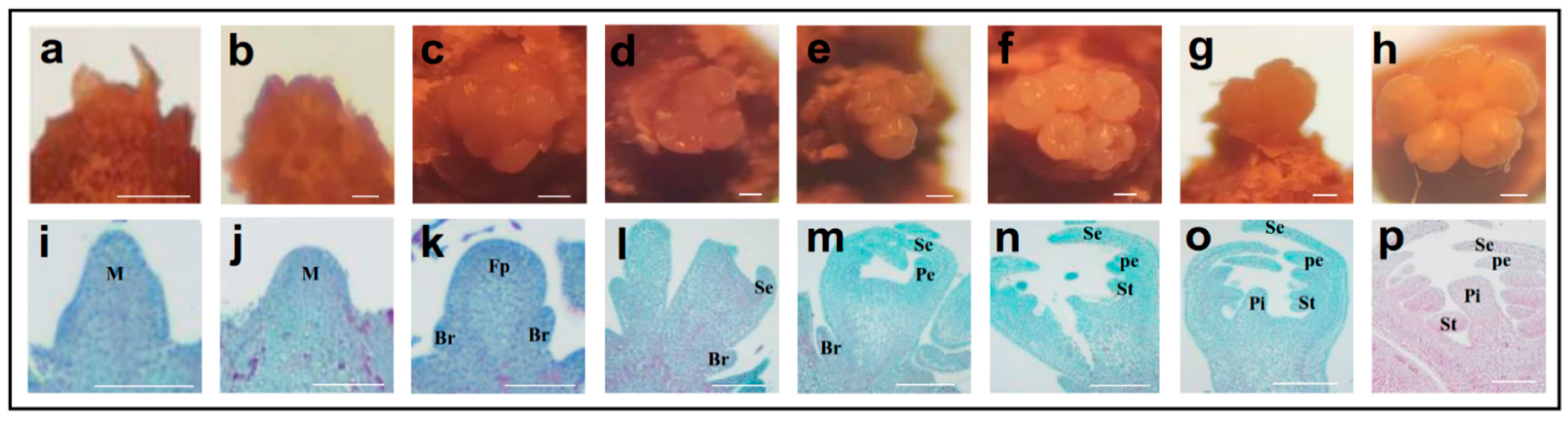
2.1. Morphological Changes during Flower Bud Differentiation
2.2. Global Analysis of RNA-Seq Data
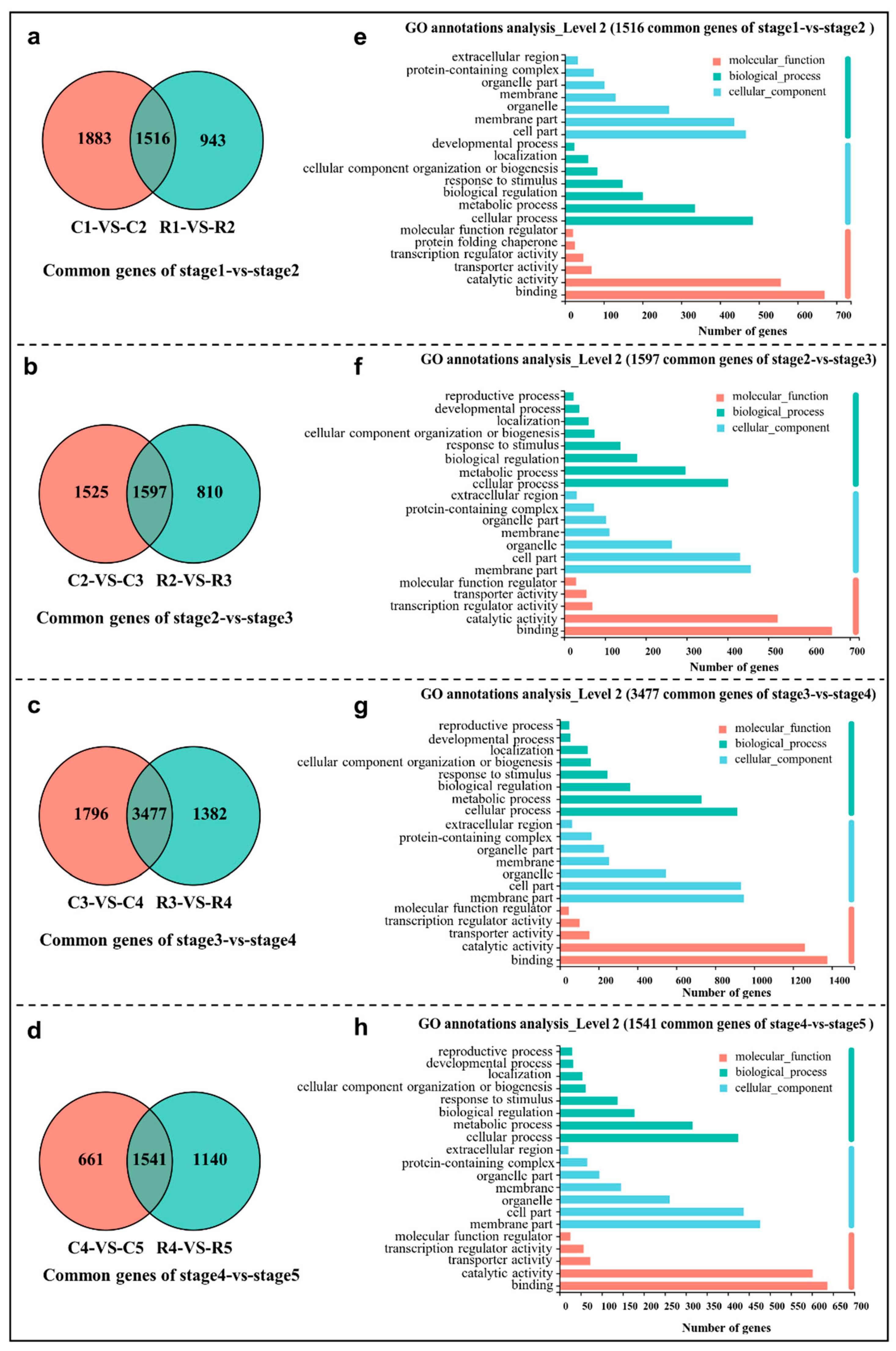
2.3. Comparative Transcriptomic Analysis of Adjacent Differentiation Stages
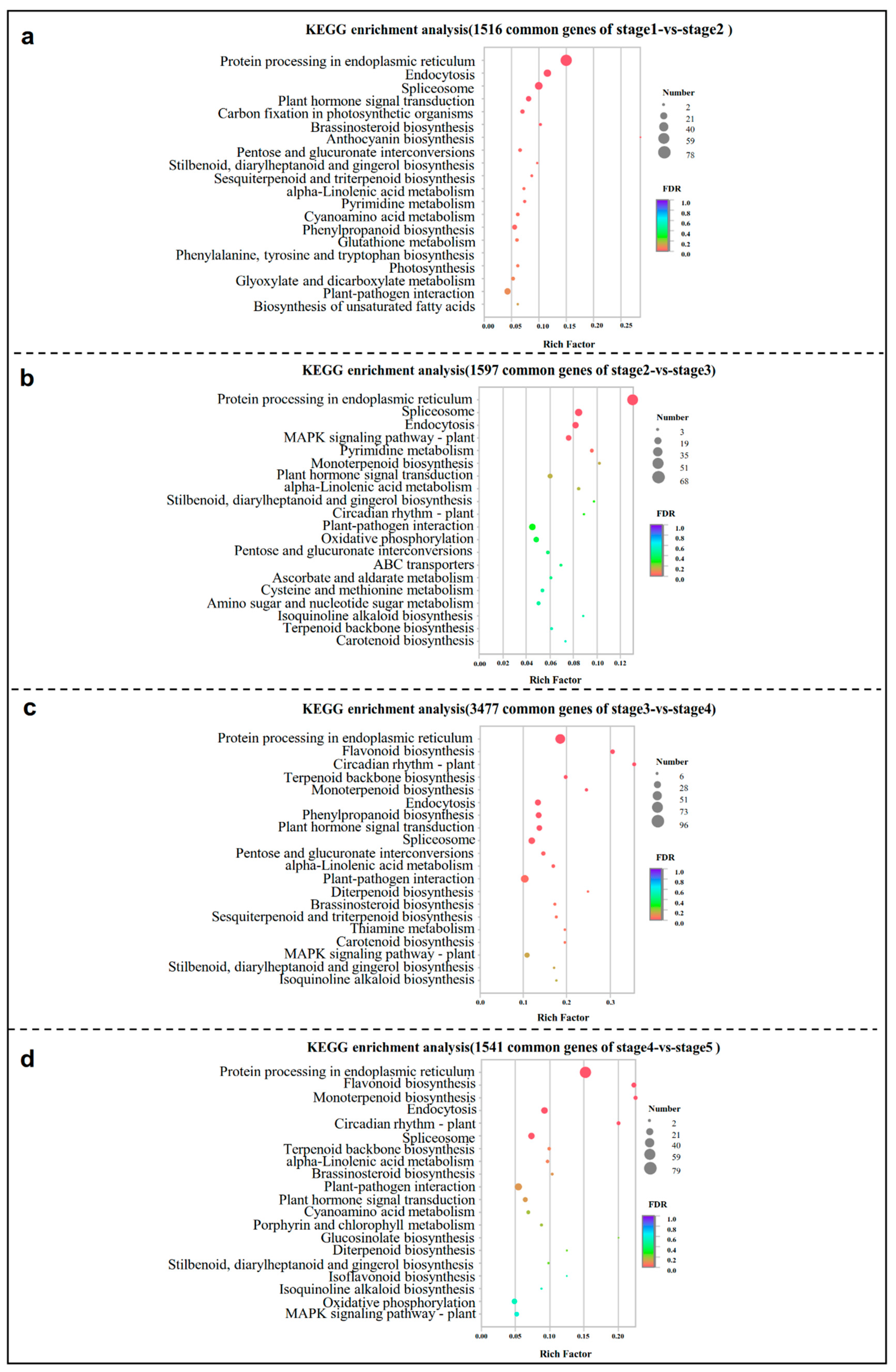
2.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis of Common DEGs at Four Comparison Groups
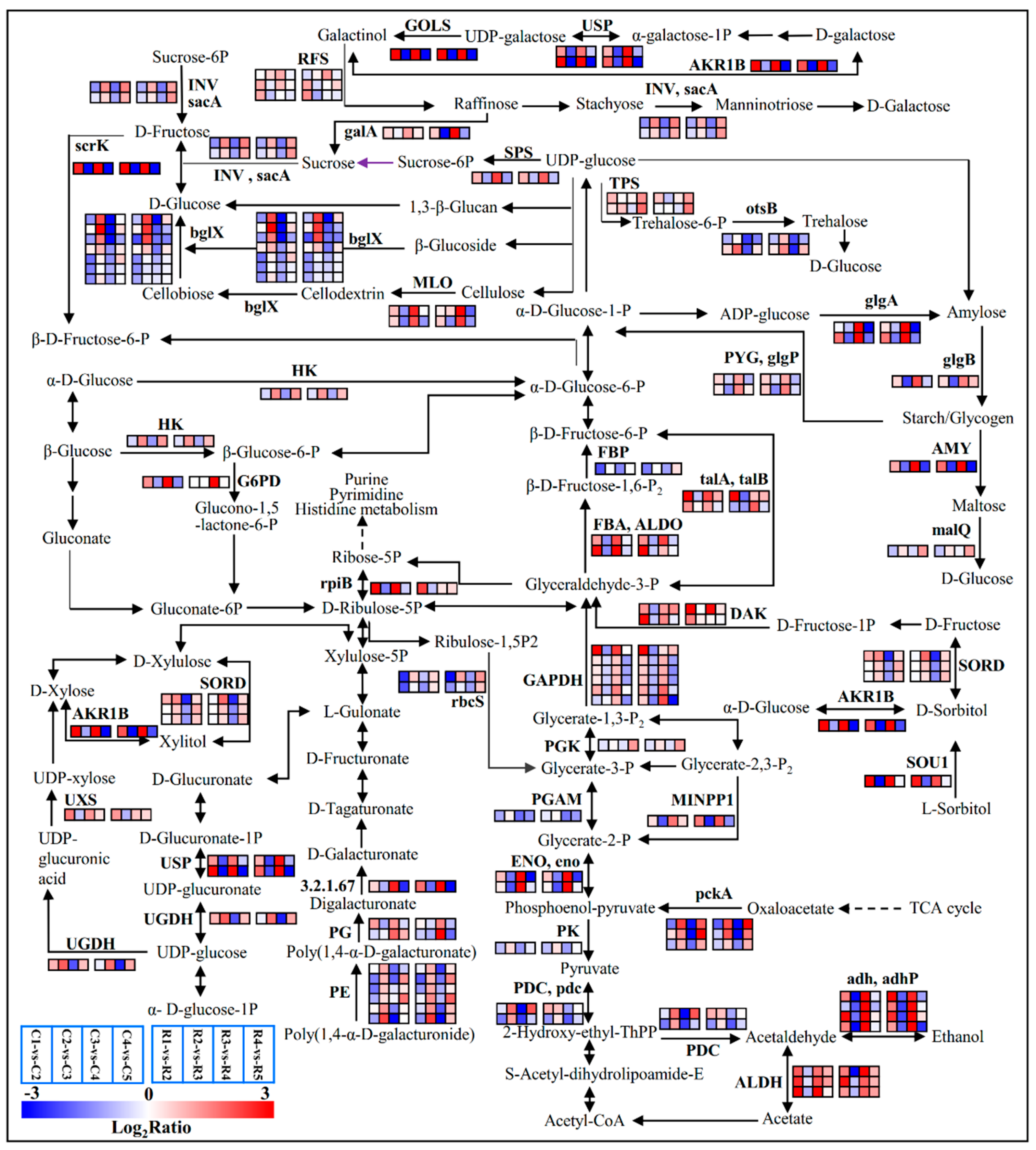
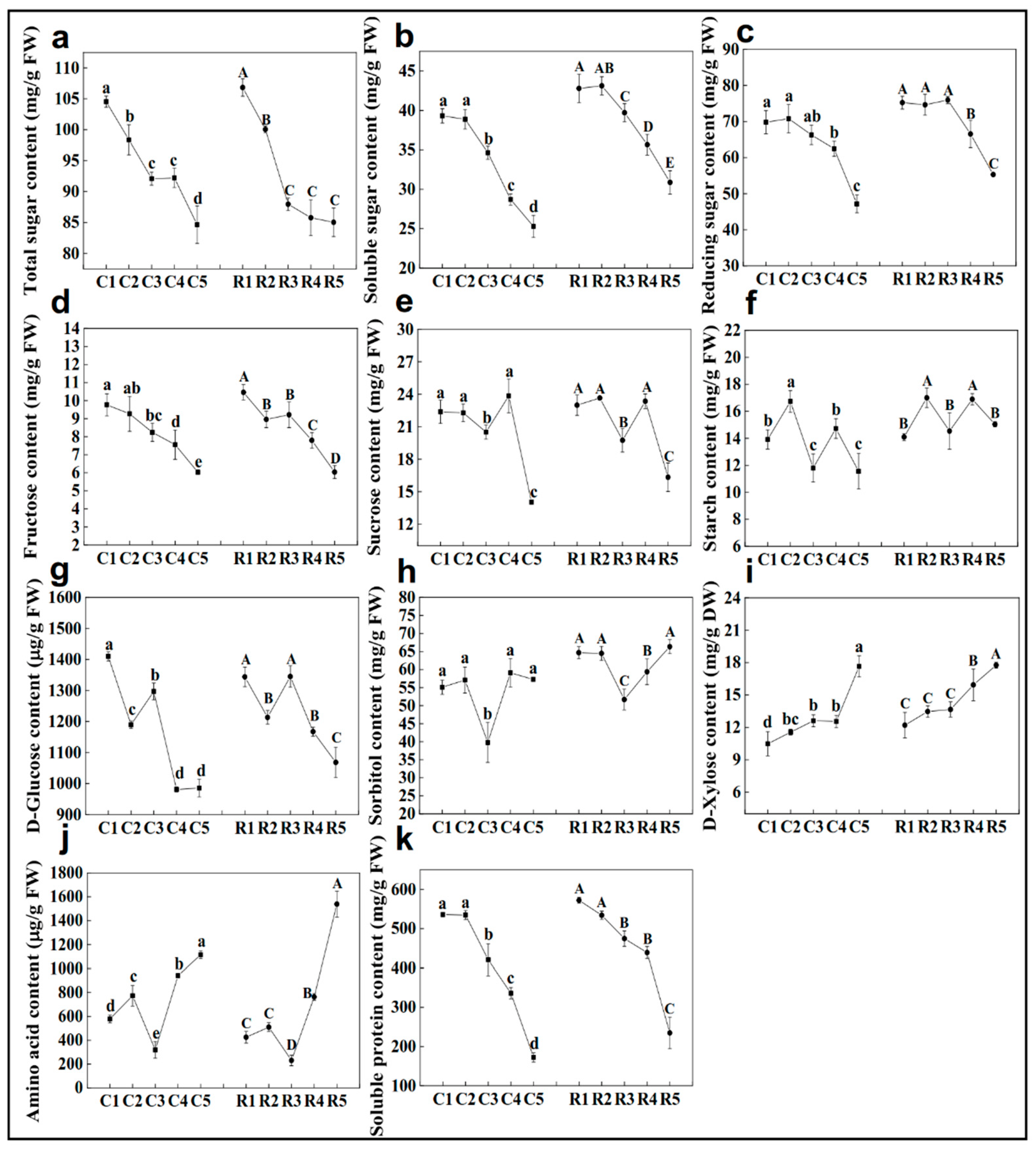
2.5. Expressions of Carbohydrate-Related Genes and Carbohydrate Contents in the Buds across Differentiation Stages
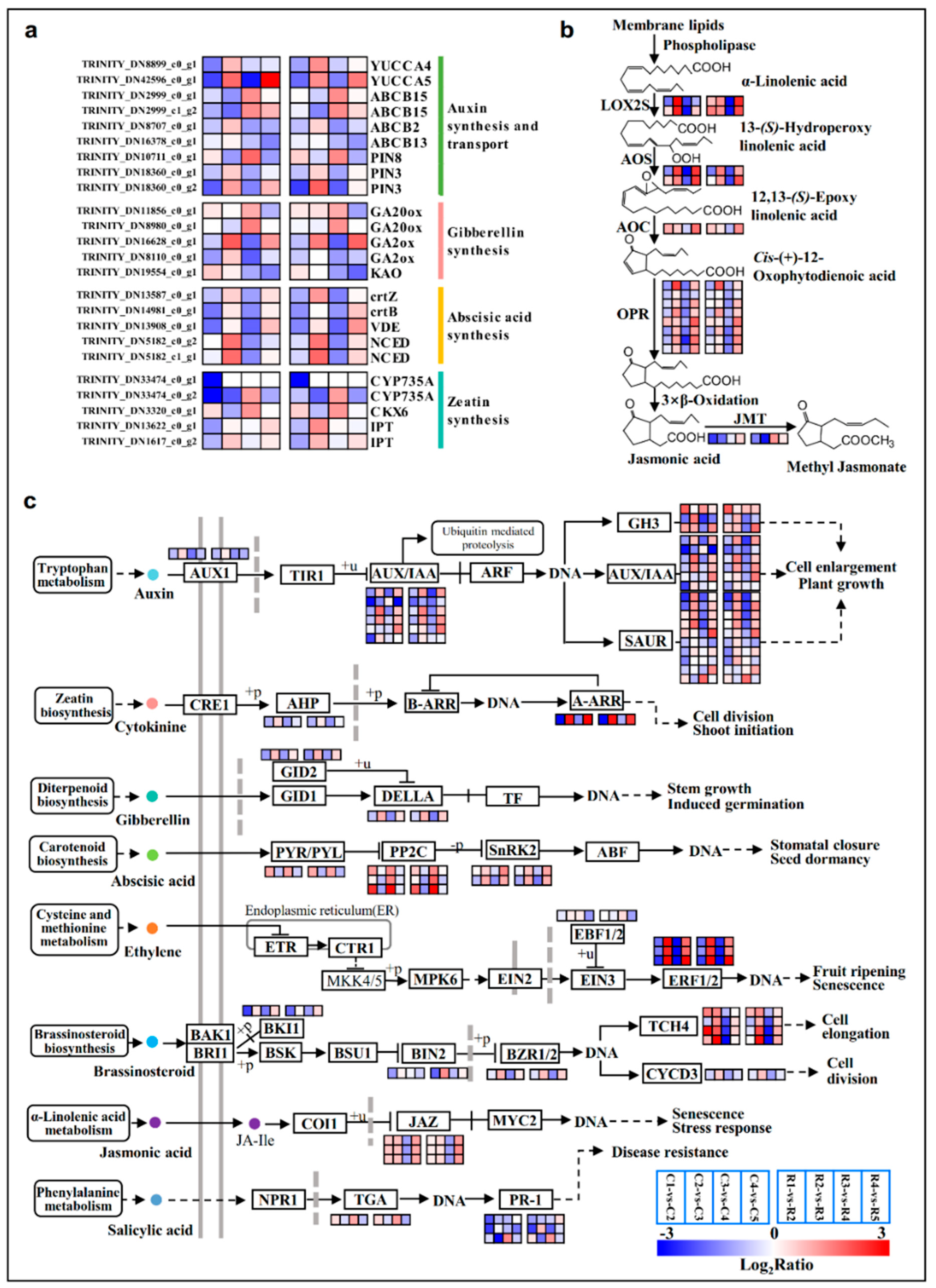
2.6. Stage-Dependent transcriptional Activation and Repression Were Associated with Several Conserved Pathways
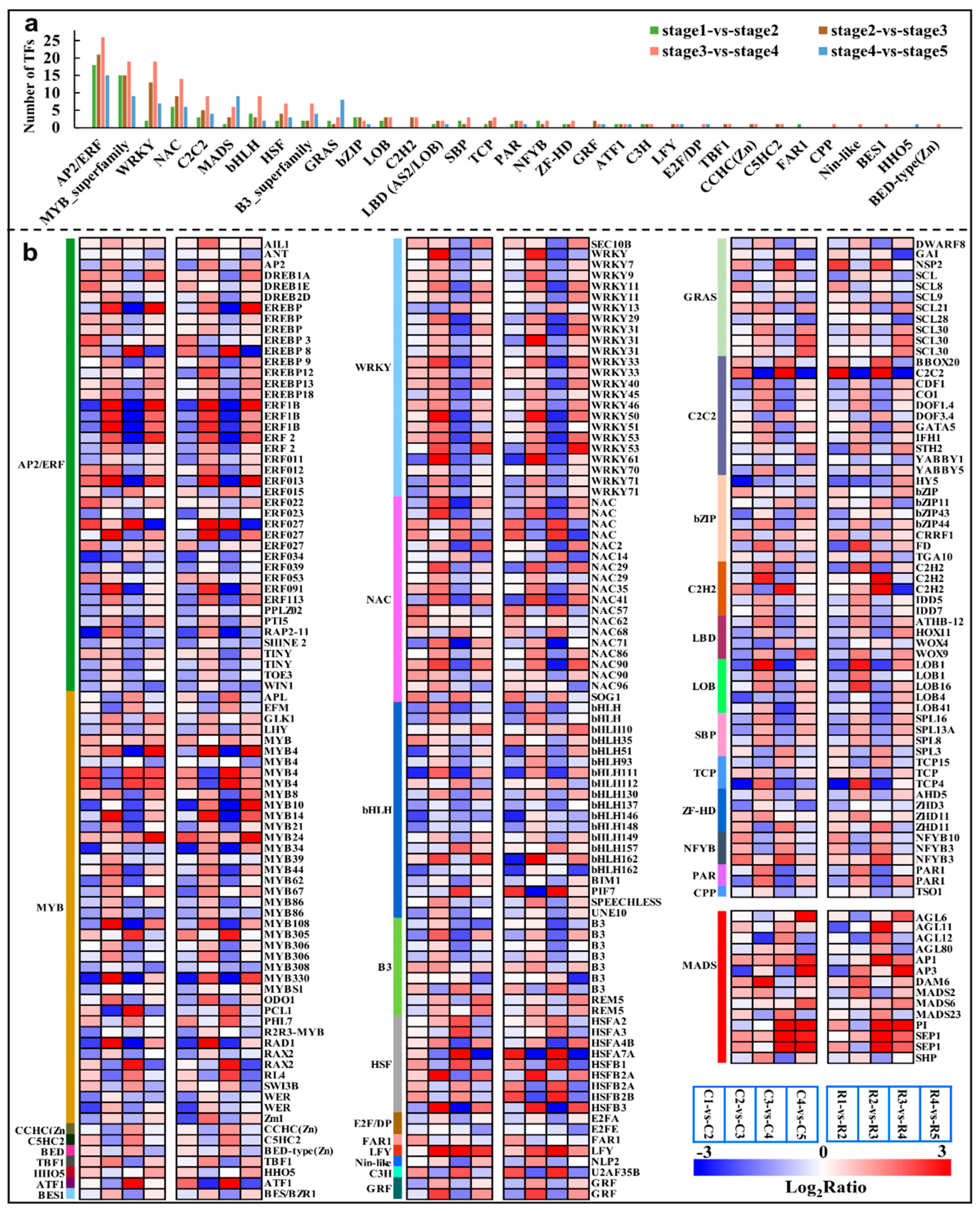
2.7. Major Transcription Factors Differentially Expressed during the Differentiation
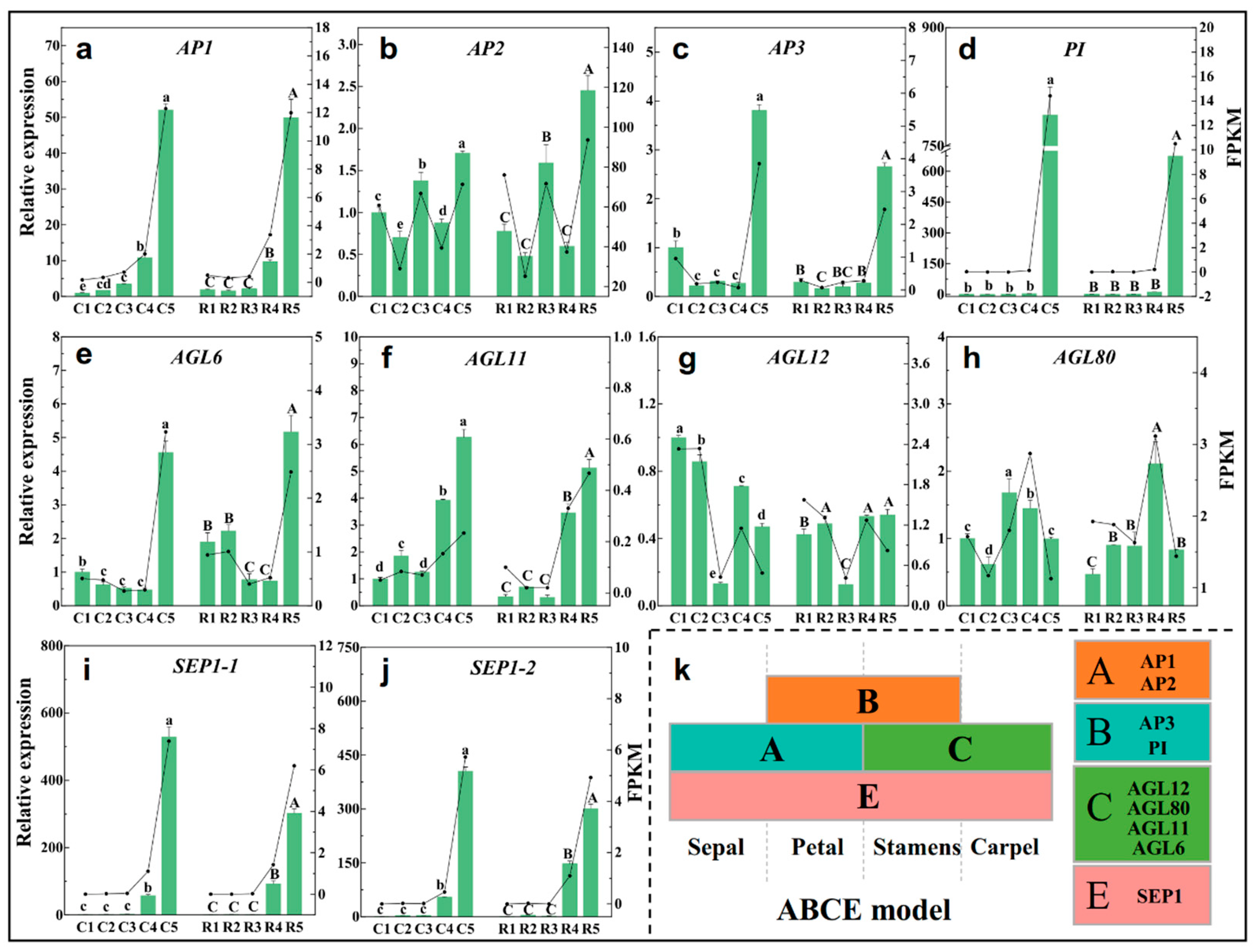
2.8. Expression Analysis of ABCE Model- and Flowering-Related Genes
2.9. RT-qPCR Validation of Differentially Expressed Genes (DEGs)
3. Discussion
3.1. Roles of Carbohydrate- and Hormone-Related Genes in the Flower Bud Differentiation of Chinese Cherries
3.2. Involvements of WRKYs and SBPs in Regulating the Vegetative-to-Reproductive Transition of Flower Bud Differentiation
3.3. The Implication of Floral Meristem Identity Genes and Flowering-Time Genes in Flower Bud Differentiation
4. Materials and Methods
4.1. Plant Materials and Sample Collection
4.2. Morphological Analysis of the Flower Bud Transition
4.3. Determination of Carbohydrates, Amino Acids, and Soluble Protein
4.4. RNA Extraction, Library Construction, and Sequencing
4.5. De Novo Assembly and Annotation
4.6. Mapping, Differential Expression Analysis, and Functional Enrichment
4.7. Validation of RNA-Seq Analysis by RT-qPCR
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, Y.; Liu, T.; Wang, X.Y.; Li, B.B.; Liang, C.L.; Cai, Y.L. Characterization of the complete chloroplast genome of the Chinese cherry Prunus pseudocerasus (Rosaceae). Conserv. Geneti. Resour. 2018, 10, 85–88. [Google Scholar] [CrossRef]
- Huang, X.J.; Wang, X.R.; Chen, T.; Tang, H.R. Research progress of germplasm diversity in Chinese cherry (Cerasus pseudocerasus). J. Fruit Sci. 2013, 30, 470–479. [Google Scholar] [CrossRef]
- Ahmad, S.; Li, Y.S.; Yang, Y.J.; Zhou, Y.Z.; Zhao, K.; Zhang, Q.X. Isolation, functional characterization and evolutionary study of LFY1 gene in Prunus mume. Plant Cell Tissue Organ Cult. 2019, 136, 523–536. [Google Scholar] [CrossRef]
- Koutinas, N.; Pepelyankov, G.; Lichev, V. Flower induction and flower bud development in apple and sweet cherry. Biotechnol. Biotec. Eq. 2010, 24, 1549–1558. [Google Scholar] [CrossRef]
- Villar, L.; Lienqueo, I.; Llanes, A.; Rojas, P.; Perez, J.; Correa, F.; Sagredo, B.; Masciarelli, O.; Luna, V.; Almada, R. Comparative transcriptomic analysis reveals novel roles of transcription factors and hormones during the flowering induction and floral bud differentiation in sweet cherry trees (Prunus avium L. cv. Bing). PLoS ONE 2020, 15, e0230110. [Google Scholar] [CrossRef]
- Guo, Y.Y.; An, L.Z.; Yu, H.Y.; Yang, M.M. Endogenous hormones and biochemical changes during flower development and florescence in the buds and leaves of Lycium ruthenicum Murr. Forests 2022, 13, 763. [Google Scholar] [CrossRef]
- Zhang, W.E.; Li, J.J.; Zhang, W.L.; Njie, A.; Pan, X.J. The changes in C/N, carbohydrate, and amino acid content in leaves during female flower bud differentiation of Juglans sigillata. Acta Physiol. Plant. 2022, 44, 19. [Google Scholar] [CrossRef]
- Naor, V.; Kigel, J.; Ben-Tal, Y.; Ziv, M. Variation in endogenous gibberellins, abscisic acid, and carbohydrate content during the growth cycle of colored Zantedeschia spp., a tuberous geophyte. J. Plant Growth Regul. 2008, 27, 211–220. [Google Scholar] [CrossRef]
- Wan, C.Y.; Mi, L.; Chen, B.Y.; Li, J.F.; Huo, H.Z.; Xu, J.T.; Chen, X.P. Effects of nitrogen during nursery stage on flower bud differentiation and early harvest after transplanting in strawberry. Braz. J. Bot. 2018, 41, 1–10. [Google Scholar] [CrossRef]
- Xing, L.B.; Zhang, D.; Li, Y.M.; Shen, Y.W.; Zhao, C.P.; Ma, J.J.; An, N.; Han, M.Y. Transcription profiles reveal sugar and hormone signaling pathways mediating flower induction in apple (Malus domestica Borkh.). Plant Cell Physiol. 2015, 56, 2052–2068. [Google Scholar] [CrossRef]
- Smeekens, S.; Ma, J.; Hanson, J.; Rolland, F. Sugar signals and molecular networks controlling plant growth. Curr. Opin. Plant Biol. 2010, 13, 274–279. [Google Scholar] [CrossRef]
- Wang, J.X.; Luo, T.; Zhang, H.; Shao, J.Z.; Peng, J.Y.; Sun, J.S. Variation of endogenous hormones during flower and leaf buds development in ‘Tianhong 2′ apple. Hortscience 2020, 55, 1794–1798. [Google Scholar] [CrossRef]
- Amini, S.; Rosli, K.; Abu-Bakar, M.F.; Alias, H.; Mat-Isa, M.N.; Juhari, M.A.A.; Haji-Adam, J.; Goh, H.H.; Wan, K.L. Transcriptome landscape of Rafflesia cantleyi floral buds reveals insights into the roles of transcription factors and phytohormones in flower development. PLoS ONE 2019, 14, e0226338. [Google Scholar] [CrossRef]
- Fornara, F.; de Montaigu, A.; Coupland, G. SnapShot: Control of flowering in Arabidopsis. Cell 2010, 141, 550. [Google Scholar] [CrossRef]
- Simon, R.; Igeno, M.I.; Coupland, G. Activation of floral meristem identity genes in Arabidopsis. Nature 1996, 384, 59–62. [Google Scholar] [CrossRef]
- Grandi, V.; Gregis, V.; Kater, M.M. Uncovering genetic and molecular interactions among floral meristem identity genes in Arabidopsis thaliana. Plant J. 2012, 69, 881–893. [Google Scholar] [CrossRef]
- Smaczniak, C.; Immink, R.G.H.; Muino, J.M.; Blanvillain, R.; Busscher, M.; Busscher-Lange, J.; Dinh, Q.D.; Liu, S.; Westphal, A.H.; Boeren, S.; et al. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc. Natl. Acad. Sci. USA 2012, 109, 1560–1565. [Google Scholar] [CrossRef]
- Tian, T.; Qiao, G.; Wen, Z.; Deng, B.; Qiu, Z.L.; Hong, Y.; Wen, X.P. Comparative transcriptome analysis reveals the molecular regulation underlying the adaptive mechanism of cherry (Cerasus pseudocerasus Lindl.) to shelter covering. BMC Plant Biol. 2020, 20, 27. [Google Scholar] [CrossRef]
- Wen, X.P.; Qiu, Z.L.; Hong, Y. Advances in physiological and molecular mechanisms underlying the fruit abscission of fruit trees. J. Mt. Agric. Biol. 2018, 37, 101. [Google Scholar] [CrossRef]
- Kanno, A. Molecular mechanism regulating floral architecture in monocotyledonous ornamental plants. Hort. J. 2016, 85, 8–22. [Google Scholar] [CrossRef]
- Bernier, G.; Perilleux, C. A physiological overview of the genetics of flowering time control. Plant Biotechnol. J. 2005, 3, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, F.M.; Goetz, K.P. Metabolites in cherry buds to detect winter dormancy. Metabolites 2022, 12, 247. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.H.; Li, S.H.; Nosarzewski, M.; Archbold, D.D. Sorbitol dehydrogenase gene expression and enzyme activity in apple: Tissue specificity during bud development and response to rootstock vigor and growth manipulation. J. Am. Soc. Hortic. Sci. 2010, 135, 379–387. [Google Scholar] [CrossRef]
- McQueen, J.C.; Minchin, P.E.H.; Silvester, W.B. Changes in non-structural carbohydrate concentration in 1-year-old shoots of ‘Braeburn’ apple (Malus domestica) over two consecutive years. N. Zeal. J. Crop Hort. Sci. 2004, 32, 319–323. [Google Scholar] [CrossRef]
- Meng, D.; He, M.; Bai, Y.; Xu, H.; Dandekar, A.M.; Fei, Z.; Cheng, L. Decreased sorbitol synthesis leads to abnormal stamen development and reduced pollen tube growth via an MYB transcription factor, MdMYB39L, in apple (Malus domestica). New Phytol. 2018, 217, 641–656. [Google Scholar] [CrossRef]
- Ito, A.; Sugiura, T.; Sakamoto, D.; Moriguchi, T. Effects of dormancy progression and low-temperature response on changes in the sorbitol concentration in xylem sap of Japanese pear during winter season. Tree Physiol. 2013, 33, 398–408. [Google Scholar] [CrossRef]
- Chandler, J.W. The hormonal regulation of flower development. J. Plant Growth Regul. 2011, 30, 242–254. [Google Scholar] [CrossRef]
- Lo, S.F.; Yang, S.Y.; Chen, K.T.; Hsing, Y.L.; Zeevaart, J.A.D.; Chen, L.J.; Yu, S.M. A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. Plant Cell 2008, 20, 2603–2618. [Google Scholar] [CrossRef]
- Cheng, H.; Qin, L.; Lee, S.; Fu, X.; Richards, D.E.; Cao, D.; Luo, D.; Harberd, N.P.; Peng, J. Gibberellin regulates Arabidopsis floral development via suppression of DELLA protein function. Development 2004, 131, 1055–1064. [Google Scholar] [CrossRef]
- Lange, M.J.P.; Lange, T. Ovary-derived precursor gibberellin A(9) is essential for female flower development in cucumber. Development 2016, 143, 4425–4429. [Google Scholar] [CrossRef]
- An, H.S.; Jiang, S.; Zhang, J.Y.; Xu, F.J.; Zhang, X.Y. Comparative transcriptomic analysis of differentially expressed transcripts associated with flowering time of loquat (Eriobotya japonica Lindl.). Horticulturae 2021, 7, 171. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Li, A.; Liu, X.Q.; Sun, J.X.; Guo, W.J.; Zhang, J.W.; Lyu, Y.M. Changes in the morphology of the bud meristem and the levels of endogenous hormones after low temperature treatment of different Phalaenopsis cultivars. S. Afr. J. Bot. 2019, 125, 499–504. [Google Scholar] [CrossRef]
- Sun, P.; Li, J.R.; Du, G.G.; Han, W.J.; Fu, J.M.; Diao, S.F.; Suo, Y.J.; Zhang, Y.; Li, F.D. Endogenous phytohormone profiles in male and female floral buds of the persimmons (Diospyros kaki Thunb.) during development. Sci. Hortic. 2017, 218, 213–221. [Google Scholar] [CrossRef]
- Lunde, C.; Kimberlin, A.; Leiboff, S.; Koo, A.J.; Hake, S. Tasselseed5 overexpresses a wound-inducible enzyme, ZmCYP94B1, that affects jasmonate catabolism, sex determination, and plant architecture in maize. Commun. Biol. 2019, 266, 1501–1505. [Google Scholar] [CrossRef]
- Niwa, T.; Suzuki, T.; Takebayashi, Y.; Ishiguro, R.; Higashiyama, T.; Sakakibara, H.; Ishiguro, S. Jasmonic acid facilitates flower opening and floral organ development through the upregulated expression of SlMYB21 transcription factor in tomato. Biosci. Biotechnol. Biochem. 2018, 82, 292–303. [Google Scholar] [CrossRef]
- Ishiguro, S.; Kawai-Oda, A.; Ueda, J.; Nishida, I.; Okada, K. The DEFECTIVE IN ANTHER DEHISCIENCE gene encodes a novel phospholipase A1 catalyzing the initial step of jasmonic acid biosynthesis, which synchronizes pollen maturation, anther dehiscence, and flower opening in Arabidopsis. Plant Cell 2001, 13, 2191–2209. [Google Scholar] [CrossRef]
- Huang, R.W.; Liu, D.F.; Huang, M.; Ma, J.; Li, Z.N.; Li, M.Y.; Sui, S.Z. CpWRKY71, a WRKY transcription factor gene of wintersweet (Chimonanthus praecox), promotes flowering and leaf senescence in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 5325. [Google Scholar] [CrossRef]
- Yu, Y.C.; Liu, Z.H.; Wang, L.; Kim, S.G.; Seo, P.J.; Qiao, M.; Wang, N.; Li, S.; Cao, X.F.; Park, C.M.; et al. WRKY71 accelerates flowering via the direct activation of FLOWERING LOCUS T and LEAFY in Arabidopsis thaliana. Plant J. 2016, 85, 96–106. [Google Scholar] [CrossRef]
- Khan, M.A.; Kang, D.; Wu, Y.F.; Wang, Y.; Ai, P.H.; Wang, Z.C. Characterization of WRKY gene family in whole-genome and exploration of flowering improvement genes in Chrysanthemum lavandulifolium. Front. Plant Sci. 2022, 13, 861193. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.P.; Yu, D.Q. Arabidopsis WRKY transcription factors WRKY12 and WRKY13 oppositely regulate flowering under short-day conditions. Mol. Plant 2016, 9, 1492–1503. [Google Scholar] [CrossRef]
- Poethig, R.S. Vegetative Phase Change and Shoot Maturation in Plants. Curr. Top. Dev. Biol. 2013, 105, 125–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Mo, Z.H.; Lin, R.Z.; Zhu, C.C. Characterization and expression analysis of the SPL gene family during floral development and abiotic stress in pecan (Carya illinoinensis). Peerj 2021, 9, e12490. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Wu, M.F.; Yang, L.; Wu, G.; Poethig, R.S.; Wagner, D. The microRNA-regulated SBP-box transcription factor SPL3 is a direct upstream activator of LEAFY, FRUITFULL, and APETALA1. Dev. Cell 2009, 17, 268–278. [Google Scholar] [CrossRef]
- Gou, J.; Tang, C.; Chen, N.; Wang, H.; Debnath, S.; Sun, L.; Flanagan, A.; Tang, Y.; Jiang, Q.; Allen, R.D.; et al. SPL7 and SPL8 represent a novel flowering regulation mechanism in switchgrass. New Phytol. 2019, 222, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Schwarz, S.; Saedler, H.; Huijser, P. SPL8, a local regulator in a subset of gibberellin-mediated developmental processes in Arabidopsis. Plant Mol. Biol. 2007, 63, 429–439. [Google Scholar] [CrossRef]
- Unte, U.S.; Sorensen, A.-M.; Pesaresi, P.; Gandikota, M.; Leister, D.; Saedler, H.; Huijser, P. SPL8, an SBP-box gene that affects pollen sac development in Arabidopsis. Plant Cell 2003, 15, 1009–1019. [Google Scholar] [CrossRef]
- Hepworth, S.R.; Klenz, J.E.; Haughn, G.W. UFO in the Arabidopsis inflorescence apex is required for floral-meristem identity and bract suppression. Planta 2006, 223, 769–778. [Google Scholar] [CrossRef]
- Ratcliffe, O.J.; Bradley, D.J.; Coen, E.S. Separation of shoot and floral identity in Arabidopsis. Development 1999, 126, 1109–1120. [Google Scholar] [CrossRef]
- Savidge, B.; Rounsley, S.D.; Yanofsky, M.F. Temporal relationship between the transcription of two Arabidopsis MADS box genes and the floral organ identity genes. Plant Cell 1995, 7, 721–733. [Google Scholar] [CrossRef]
- Samach, A.; Klenz, J.E.; Kohalmi, S.E.; Risseeuw, E.; Haughn, G.W.; Crosby, W.L. The unusual floral organs gene of Arabidopsis thaliana is an F-box protein required for normal patterning and growth in the floral meristem. Plant J. 1999, 20, 433–445. [Google Scholar] [CrossRef]
- Chae, E.; Tan, Q.K.G.; Hill, T.A.; Irish, V.F. An Arabidopsis F-box protein acts as a transcriptional co-factor to regulate floral development. Development 2008, 135, 1235–1245. [Google Scholar] [CrossRef]
- Moyroud, E.; Tichtinsky, G.; Parcy, F. The LEAFY floral regulators in angiosperms: Conserved proteins with diverse roles. J. Plant Biol. 2009, 52, 177–185. [Google Scholar] [CrossRef]
- Winter, C.M.; Austin, R.S.; Blanvillain-Baufume, S.; Reback, M.A.; Monniaux, M.; Wu, M.F.; Sang, Y.; Yamaguchi, A.; Yamaguchi, N.; Parker, J.E.; et al. LEAFY target genes reveal floral regulatory logic, cis motifs, and a link to biotic stimulus response. Dev. Cell 2011, 20, 430–443. [Google Scholar] [CrossRef]
- Sundstrom, J.F.; Nakayama, N.; Glimelius, K.; Irish, V.F. Direct regulation of the floral homeotic APETALA1 gene by APETALA3 and PISTILLATA in Arabidopsis. Plant J. 2006, 46, 593–600. [Google Scholar] [CrossRef]
- Serrano-Mislata, A.; Goslin, K.; Zheng, B.; Rae, L.; Wellmer, F.; Graciet, E.; Madueno, F. Regulatory interplay between LEAFY, APETALA1/CAULIFLOWER AND TERMINAL FLOWER1: New insights into an old relationship. Plant Signal. Behav. 2017, 12, e1370164. [Google Scholar] [CrossRef]
- Saini, P.; Yadav, R.K. C-terminal domain of APETALA1 is essential for its functional divergence from CAULIFLOWER in Arabidopsis. J. Plant Biochem. Biot. 2020, 29, 824–831. [Google Scholar] [CrossRef]
- Smyth, D.R.; Bowman, J.L.; Meyerowitz, E.M. Early flower development in Arabidopsis. Plant Cell 1990, 2, 755–767. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Z.; Yong, X.; Ahmad, S.; Yang, W.; Cheng, T.; Wang, J.; Zhang, Q. SEP-class genes in Prunus mume and their likely role in floral organ development. Bmc Plant Biol. 2017, 17, 10. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, N.; Guo, C.C.; Xu, G.X.; Kong, H.Z.; Shan, H.Y. Evolutionary divergence of the APETALA1 and CAULIFLOWER proteins. J. Syst. Evol. 2012, 50, 502–511. [Google Scholar] [CrossRef]
- Ohmori, S.; Kimizu, M.; Sugita, M.; Miyao, A.; Hirochika, H.; Uchida, E.; Nagato, Y.; Yoshida, H. MOSAIC FLORAL ORGANS1, an AGL6-Like MADS box gene, regulates floral organ identity and meristem fate in rice. Plant Cell 2009, 21, 3008–3025. [Google Scholar] [CrossRef]
- Rijpkema, A.S.; Zethof, J.; Gerats, T.; Vandenbussche, M. The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant J. 2009, 60, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Morel, P.; Chambrier, P.; Boltz, V.; Chamot, S.; Rozier, F.; Bento, S.R.; Trehin, C.; Monniaux, M.; Zethof, J.; Vandenbussche, M. Divergent functional diversification patterns in the SEP/AGL6/AP1 MADS-Box transcription factor superclade. Plant Cell 2019, 31, 3033–3056. [Google Scholar] [CrossRef] [PubMed]
- Kania, T.; Russenberger, D.; Peng, S.; Apel, K.; Melzer, S. FPF1 promotes flowering in Arabidopsis. Plant Cell 1997, 9, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Melzer, S.; Kampmann, G.; Chandler, J.; Apel, K. FPF1 modulates the competence to flowering in Arabidopsis. Plant J. 1999, 18, 395–405. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Qiu, Z.L.; He, M.Q.; Wen, Z.; Yang, K.; Hong, Y.; Wen, X.P. Selection and validation of reference genes in sweet cherry flower bud at different development stages. Seed 2020, 39, 37–43. (In Chinese) [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Date (Month/Day) | Cultivation Patterns | Physiological Differentiation | Differentiation Initiation | Floral | Sepal | Petal | Stamen | Pistil |
|---|---|---|---|---|---|---|---|---|
| Primordia Differentiation | ||||||||
| 10 May (stage1) | C | 100.0 | - | - | - | - | - | - |
| R | 100.0 | - | - | - | - | - | - | |
| 20 May | C | 90.0 | 10.0 | - | - | - | - | - |
| R | 93.3 | 6.7 | - | - | - | - | - | |
| 30 May | C | 46.7 | 53.3 | - | - | - | - | - |
| R | 50.0 | 50.0 | - | - | - | - | - | |
| 10 June (stage2) | C | 16.7 | 83.3 | - | - | - | - | - |
| R | 23.3 | 76.7 | - | - | - | - | - | |
| 20 June | C | - | 50.0 | 46.7 | 3.3 | - | - | - |
| R | - | 53.3 | 46.7 | - | - | - | - | |
| 1 July (stage3) | C | - | 6.7 | 46.7 | 36.6 | 10.0 | - | - |
| R | - | 20.0 | 43.4 | 33.3 | 3.3 | - | - | |
| 15 July | C | - | - | 16.7 | 53.3 | 30.0 | - | - |
| R | - | - | 23.3 | 56.7 | 20.0 | - | - | |
| 30 July (stage4) | C | - | - | - | 10.0 | 56.7 | 30.0 | 3.3 |
| R | - | - | - | 20.0 | 53.3 | 26.7 | - | |
| 15 August | C | - | - | - | - | 30 | 46.7 | 23.3 |
| R | - | - | - | - | 36.7 | 43.3 | 20.0 | |
| 30 August (stage5) | C | - | - | - | - | 3.3 | 66.7 | 30.0 |
| R | - | - | - | - | 10.0 | 63.3 | 26.7 | |
| 15 September | C | - | - | - | - | - | 16.7 | 83.3 |
| R | - | - | - | - | - | 20.0 | 80.0 | |
| 30 September | C | - | - | - | - | - | - | 100.0 |
| R | - | - | - | - | - | - | 100.0 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, C.; Cao, X.; Tian, T.; Hou, Q.; Wen, Z.; Qiao, G.; Wen, X. Cross-Talk between Transcriptome Analysis and Dynamic Changes of Carbohydrates Identifies Stage-Specific Genes during the Flower Bud Differentiation Process of Chinese Cherry (Prunus pseudocerasus L.). Int. J. Mol. Sci. 2022, 23, 15562. https://doi.org/10.3390/ijms232415562
Shang C, Cao X, Tian T, Hou Q, Wen Z, Qiao G, Wen X. Cross-Talk between Transcriptome Analysis and Dynamic Changes of Carbohydrates Identifies Stage-Specific Genes during the Flower Bud Differentiation Process of Chinese Cherry (Prunus pseudocerasus L.). International Journal of Molecular Sciences. 2022; 23(24):15562. https://doi.org/10.3390/ijms232415562
Chicago/Turabian StyleShang, Chunqiong, Xuejiao Cao, Tian Tian, Qiandong Hou, Zhuang Wen, Guang Qiao, and Xiaopeng Wen. 2022. "Cross-Talk between Transcriptome Analysis and Dynamic Changes of Carbohydrates Identifies Stage-Specific Genes during the Flower Bud Differentiation Process of Chinese Cherry (Prunus pseudocerasus L.)" International Journal of Molecular Sciences 23, no. 24: 15562. https://doi.org/10.3390/ijms232415562
APA StyleShang, C., Cao, X., Tian, T., Hou, Q., Wen, Z., Qiao, G., & Wen, X. (2022). Cross-Talk between Transcriptome Analysis and Dynamic Changes of Carbohydrates Identifies Stage-Specific Genes during the Flower Bud Differentiation Process of Chinese Cherry (Prunus pseudocerasus L.). International Journal of Molecular Sciences, 23(24), 15562. https://doi.org/10.3390/ijms232415562