
Immunomodulation of HDAC Inhibitor Entinostat Potentiates the Anticancer Effects of Radiation and PD-1 Blockade in the Murine Lewis Lung Carcinoma Model

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Entinostat Enhances Radiation-Induced Tumor Growth Delay in a Lewis Lung Cancer Model
2.2. Entinostat Reshapes Immune Response in the Tumors That Receive Radiotherapy
2.3. Transcriptomic Analysis Reveals That Entinostat Alters Radiation-Mediated Immune Response in the Tumor Microenvironment
2.4. Entinostat Boosts the Combined Effect of Radiation and Anti-PD-1 Immunotherapy
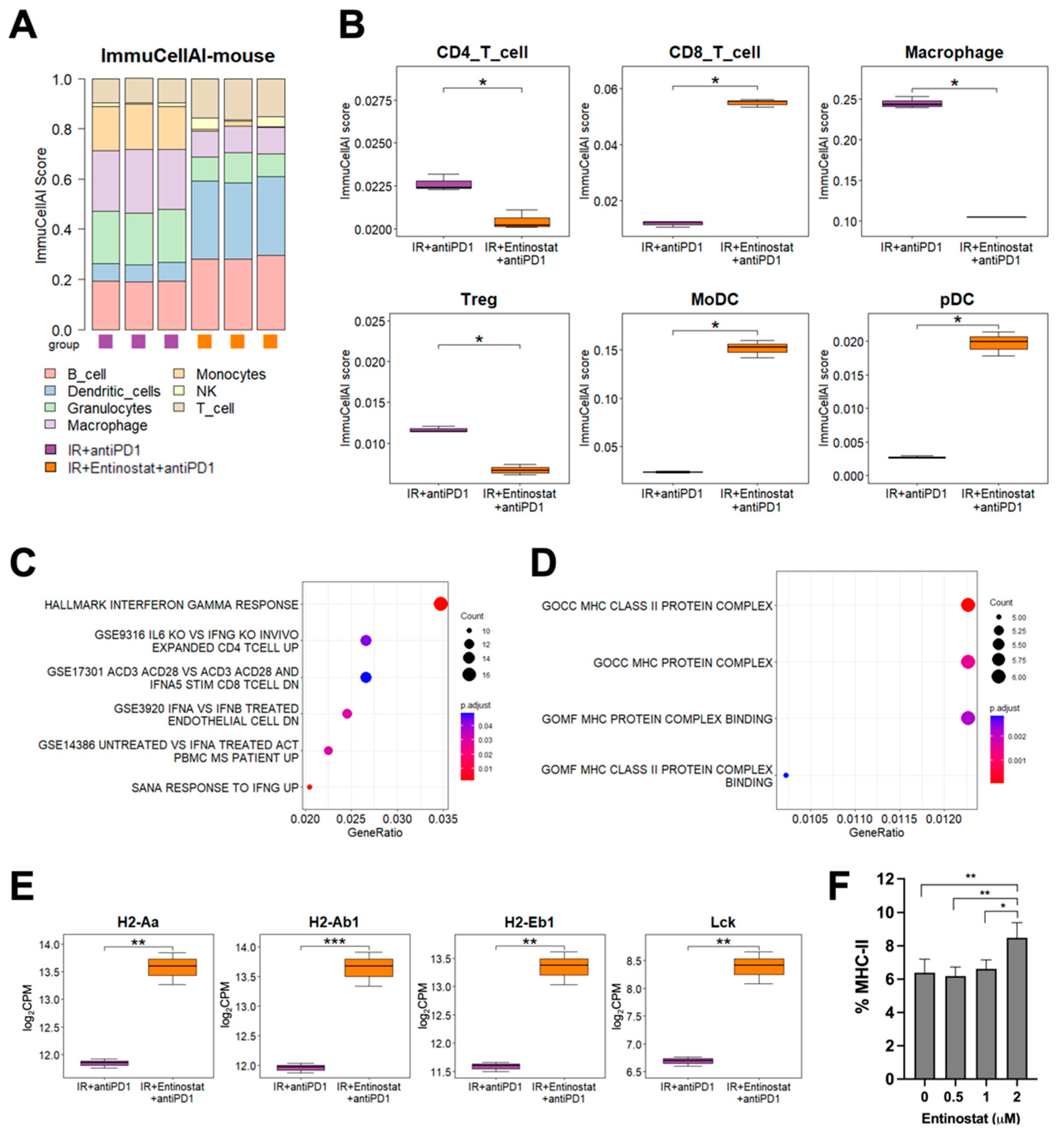
2.5. Potentiation of IR plus Anti-PD1 Combination with Entinostat May Be Caused by Upregulating Type II Interferon Signaling and MHC-II Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animal Models
4.3. Irradiation
4.4. Flow Cytometry
4.5. RNA Sequence Analysis
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.; Liu, X.; Chen, D.; Yu, J. Radiotherapy combined with immunotherapy: The dawn of cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Faivre-Finn, C.; Gray, J.E.; Vicente, D.; Planchard, D.; Paz-Ares, L.; Vansteenkiste, J.F.; Garassino, M.C.; Hui, R.; Quantin, X.; et al. Five-Year Survival Outcomes From the PACIFIC Trial: Durvalumab After Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Diao, H.; Dong, N.; Su, X.; Wang, B.; Mo, Q.; Yu, H.; Wang, X.; Chen, C. Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cells via mitochondrial injury and p53 up-acetylation. Cell Biol. Toxicol. 2016, 32, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-A.; Wen, W.-L.; Chang, J.-W.; Wei, T.-T.; Tan, Y.-H.C.; Salunke, S.; Chen, C.-T.; Chen, C.-S.; Wang, Y.-C. A Novel Histone Deacetylase Inhibitor Exhibits Antitumor Activity via Apoptosis Induction, F-Actin Disruption and Gene Acetylation in Lung Cancer. PLoS ONE 2010, 5, e12417. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; et al. Carboplatin and Paclitaxel in Combination With Either Vorinostat or Placebo for First-Line Therapy of Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar] [CrossRef]
- Reguart, N.; Rosell, R.; Cardenal, F.; Cardona, A.F.; Isla, D.; Palmero, R.; Moran, T.; Rolfo, C.; Pallarès, M.C.; Insa, A.; et al. Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer 2014, 84, 161–167. [Google Scholar] [CrossRef]
- Munshi, A.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Richon, V.M.; Meyn, R.E. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of γ-H2AX foci. Mol. Cancer Ther. 2006, 5, 1967–1974. [Google Scholar] [CrossRef]
- Rivera, S.; Leteur, C.; Mégnin, F.; Law, F.; Martins, I.; Kloos, I.; Depil, S.; Modjtahedi, N.; Perfettini, J.L.; Hennequin, C.; et al. Time dependent modulation of tumor radiosensitivity by a pan HDAC inhibitor: Abexinostat. Oncotarget 2017, 8, 56210–56227. [Google Scholar] [CrossRef]
- Choi, C.; Lee, G.; Son, A.; Yoo, G.; Yu, J.; Park, H. Downregulation of Mcl-1 by Panobinostat Potentiates Proton Beam Therapy in Hepatocellular Carcinoma Cells. Cells 2021, 10, 554. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.I.; Choi, C.; Shin, S.-W.; Son, A.; Lee, G.-H.; Kim, S.-Y.; Park, H.C. Valproic Acid Sensitizes Hepatocellular Carcinoma Cells to Proton Therapy by Suppressing NRF2 Activation. Sci. Rep. 2017, 7, 14986. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.; Fan, K.; Paschen, A.; Hadrup, S.R.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 2290. [Google Scholar] [CrossRef] [PubMed]
- Truong, A.S.; Zhou, M.; Krishnan, B.; Utsumi, T.; Manocha, U.; Stewart, K.G.; Beck, W.; Rose, T.L.; Milowsky, M.I.; He, X.; et al. Entinostat induces antitumor immune responses through immune editing of tumor neoantigens. J. Clin. Investig. 2021, 131, e138560. [Google Scholar] [CrossRef]
- Briere, D.; Sudhakar, N.; Woods, D.M.; Hallin, J.; Engstrom, L.D.; Aranda, R.; Chiang, H.; Sodré, A.L.; Olson, P.; Weber, J.S.; et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol. Immunother. 2018, 67, 381–392. [Google Scholar] [CrossRef]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.A.; Sotomayor, E.M.; Weber, J. HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef]
- Li, X.; Su, X.; Liu, R.; Pan, Y.; Fang, J.; Cao, L.; Feng, C.; Shang, Q.; Chen, Y.; Shao, C.; et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene 2021, 40, 1836–1850. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Kirkwood, J.; Dent, P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 2017, 8, 83155–83170. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-F.; Ning, F.; Liu, Z.-C.; Wu, L.; Li, Z.-Q.; Qi, Y.-F.; Zhang, G.; Wang, H.-S.; Cai, S.-H.; Du, J. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol. Immunother. 2017, 66, 355–366. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef]
- Hashimoto, A.; Fukumoto, T.; Zhang, R.; Gabrilovich, D. Selective targeting of different populations of myeloid-derived suppressor cells by histone deacetylase inhibitors. Cancer Immunol. Immunother. 2020, 69, 1929–1936. [Google Scholar] [CrossRef]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef]
- Borcoman, E.; Kamal, M.; Marret, G.; Dupain, C.; Castel-Ajgal, Z.; Le Tourneau, C. HDAC Inhibition to Prime Immune Checkpoint Inhibitors. Cancers 2021, 14, 66. [Google Scholar] [CrossRef]
- Kim, Y.; Choi, C.; Park, J.H.; Ahn, W.-G.; Shin, S.-W.; Kim, S.-Y.; Noh, J.M. Immunomodulatory effect of splenectomy in lung cancer mouse xenograft models receiving radiation therapy. Radiat. Oncol. J. 2022, 40, 53–65. [Google Scholar] [CrossRef]
- Altorki, N.K.; McGraw, T.E.; Borczuk, A.C.; Saxena, A.; Port, J.L.; Stiles, B.M.; Lee, B.E.; Sanfilippo, N.J.; Scheff, R.J.; Pua, B.B.; et al. Neoadjuvant durvalumab with or without stereotactic body radiotherapy in patients with early-stage non-small-cell lung cancer: A single-centre, randomised phase 2 trial. Lancet Oncol. 2021, 22, 824–835. [Google Scholar] [CrossRef]
- Kim, J.H.; Shin, J.H.; Kim, I.H. Susceptibility and radiosensitization of human glioblastoma cells to trichostatin A, a histone deacetylase inhibitor. Int. J. Radiat. Oncol. 2004, 59, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jung, M.; Dritschilo, A.; Jung, M. Enhancement of Radiation Sensitivity of Human Squamous Carcinoma Cells by Histone Deacetylase Inhibitors. Radiat. Res. 2004, 161, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Cuneo, K.C.; Fu, A.; Tu, T.; Atadja, P.W.; Hallahan, D.E. Histone Deacetylase (HDAC) Inhibitor LBH589 Increases Duration of γ-H2AX Foci and Confines HDAC4 to the Cytoplasm in Irradiated Non–Small Cell Lung Cancer. Cancer Res. 2006, 66, 11298–11304. [Google Scholar] [CrossRef] [PubMed]
- Shirbhate, E.; Patel, P.; Patel, V.K.; Veerasamy, R.; Sharma, P.C.; Rajak, H. The combination of histone deacetylase inhibitors and radiotherapy: A promising novel approach for cancer treatment. Futur. Oncol. 2020, 16, 2457–2469. [Google Scholar] [CrossRef]
- Groselj, B.; Sharma, N.L.; Hamdy, F.C.; Kerr, M.; Kiltie, A.E. Histone deacetylase inhibitors as radiosensitisers: Effects on DNA damage signalling and repair. Br. J. Cancer 2013, 108, 748–754. [Google Scholar] [CrossRef]
- Camphausen, K.; Burgan, W.; Cerra, M.; Oswald, K.A.; Trepel, J.B.; Lee, M.-J.; Tofilon, P.J. Enhanced Radiation-Induced Cell Killing and Prolongation of γH2AX Foci Expression by the Histone Deacetylase Inhibitor MS-275. Cancer Res. 2004, 64, 316–321. [Google Scholar] [CrossRef]
- Cassandri, M.; Pomella, S.; Rossetti, A.; Petragnano, F.; Milazzo, L.; Vulcano, F.; Camero, S.; Codenotti, S.; Cicchetti, F.; Maggio, R.; et al. MS-275 (Entinostat) Promotes Radio-Sensitivity in PAX3-FOXO1 Rhabdomyosarcoma Cells. Int. J. Mol. Sci. 2021, 22, 10671. [Google Scholar] [CrossRef] [PubMed]
- Sidiropoulos, D.N.; Rafie, C.I.; Jang, J.K.; Castanon, S.; Baugh, A.G.; Gonzalez, E.; Christmas, B.J.; Narumi, V.H.; Davis-Marcisak, E.F.; Sharma, G.; et al. Entinostat Decreases Immune Suppression to Promote Antitumor Responses in a HER2+ Breast Tumor Microenvironment. Cancer Immunol. Res. 2022, 10, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I Histone Deacetylase Inhibitor Entinostat Suppresses Regulatory T Cells and Enhances Immunotherapies in Renal and Prostate Cancer Models. PLoS ONE 2012, 7, e30815. [Google Scholar] [CrossRef] [PubMed]
- Akimova, T.; Ge, G.; Golovina, T.; Mikheeva, T.; Wang, L.; Riley, J.L.; Hancock, W.W. Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin. Immunol. 2010, 136, 348–363. [Google Scholar] [CrossRef]
- Ma, S.; Liu, T.; Xu, L.; Wang, Y.; Zhou, J.; Huang, T.; Li, P.; Liu, H.; Zhang, Y.; Zhou, X.; et al. Histone deacetylases inhibitor MS-275 suppresses human esophageal squamous cell carcinoma cell growth and progression via the PI3K/Akt/mTOR pathway. J. Cell. Physiol. 2019, 234, 22400–22410. [Google Scholar] [CrossRef]
- Smith, H.J.; McCaw, T.R.; Londono, A.I.; Katre, A.A.; Meza-Perez, S.; Yang, E.S.; Forero, A.; Buchsbaum, N.J.; Randall, T.D.; Straughn, J.J.M.; et al. The antitumor effects of entinostat in ovarian cancer require adaptive immunity. Cancer 2018, 124, 4657–4666. [Google Scholar] [CrossRef]
- Turner, T.B.; Meza-Perez, S.; Londoño, A.; Katre, A.; Peabody, J.E.; Smith, H.J.; Forero, A.; Norian, L.A.; Michael Straughn, J., Jr.; Buchsbaum, D.J.; et al. Epigenetic modifiers upregulate MHC II and impede ovarian cancer tumor growth. Oncotarget 2017, 8, 44159–44170. [Google Scholar] [CrossRef]
- Johnson, A.M.; Bullock, B.; Neuwelt, A.J.; Poczobutt, J.M.; Kaspar, R.E.; Li, H.Y.; Kwak, J.W.; Hopp, K.; Weiser-Evans, M.C.M.; Heasley, L.E.; et al. Cancer Cell–Intrinsic Expression of MHC Class II Regulates the Immune Microenvironment and Response to Anti–PD-1 Therapy in Lung Adenocarcinoma. J. Immunol. 2020, 204, 2295–2307. [Google Scholar] [CrossRef]
- du Sert, N.P.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLOS Biol. 2020, 18, e3000411. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.-R.; Xia, M.; Luo, M.; Luo, T.; Yang, M.; Guo, A.-Y. ImmuCellAI-mouse: A tool for comprehensive prediction of mouse immune cell abundance and immune microenvironment depiction. Bioinformatics 2021, 38, 785–791. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Park, K.; Kim, Y.J.; Shin, S.-W.; Kim, Y.J.; Choi, C.; Noh, J.M. Immunomodulation of HDAC Inhibitor Entinostat Potentiates the Anticancer Effects of Radiation and PD-1 Blockade in the Murine Lewis Lung Carcinoma Model. Int. J. Mol. Sci. 2022, 23, 15539. https://doi.org/10.3390/ijms232415539
Kim Y, Park K, Kim YJ, Shin S-W, Kim YJ, Choi C, Noh JM. Immunomodulation of HDAC Inhibitor Entinostat Potentiates the Anticancer Effects of Radiation and PD-1 Blockade in the Murine Lewis Lung Carcinoma Model. International Journal of Molecular Sciences. 2022; 23(24):15539. https://doi.org/10.3390/ijms232415539
Chicago/Turabian StyleKim, Yeeun, Kyunghee Park, Yeon Jeong Kim, Sung-Won Shin, Yeon Joo Kim, Changhoon Choi, and Jae Myoung Noh. 2022. "Immunomodulation of HDAC Inhibitor Entinostat Potentiates the Anticancer Effects of Radiation and PD-1 Blockade in the Murine Lewis Lung Carcinoma Model" International Journal of Molecular Sciences 23, no. 24: 15539. https://doi.org/10.3390/ijms232415539
APA StyleKim, Y., Park, K., Kim, Y. J., Shin, S.-W., Kim, Y. J., Choi, C., & Noh, J. M. (2022). Immunomodulation of HDAC Inhibitor Entinostat Potentiates the Anticancer Effects of Radiation and PD-1 Blockade in the Murine Lewis Lung Carcinoma Model. International Journal of Molecular Sciences, 23(24), 15539. https://doi.org/10.3390/ijms232415539