
LMNA Co-Regulated Gene Expression as a Suitable Readout after Precise Gene Correction

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
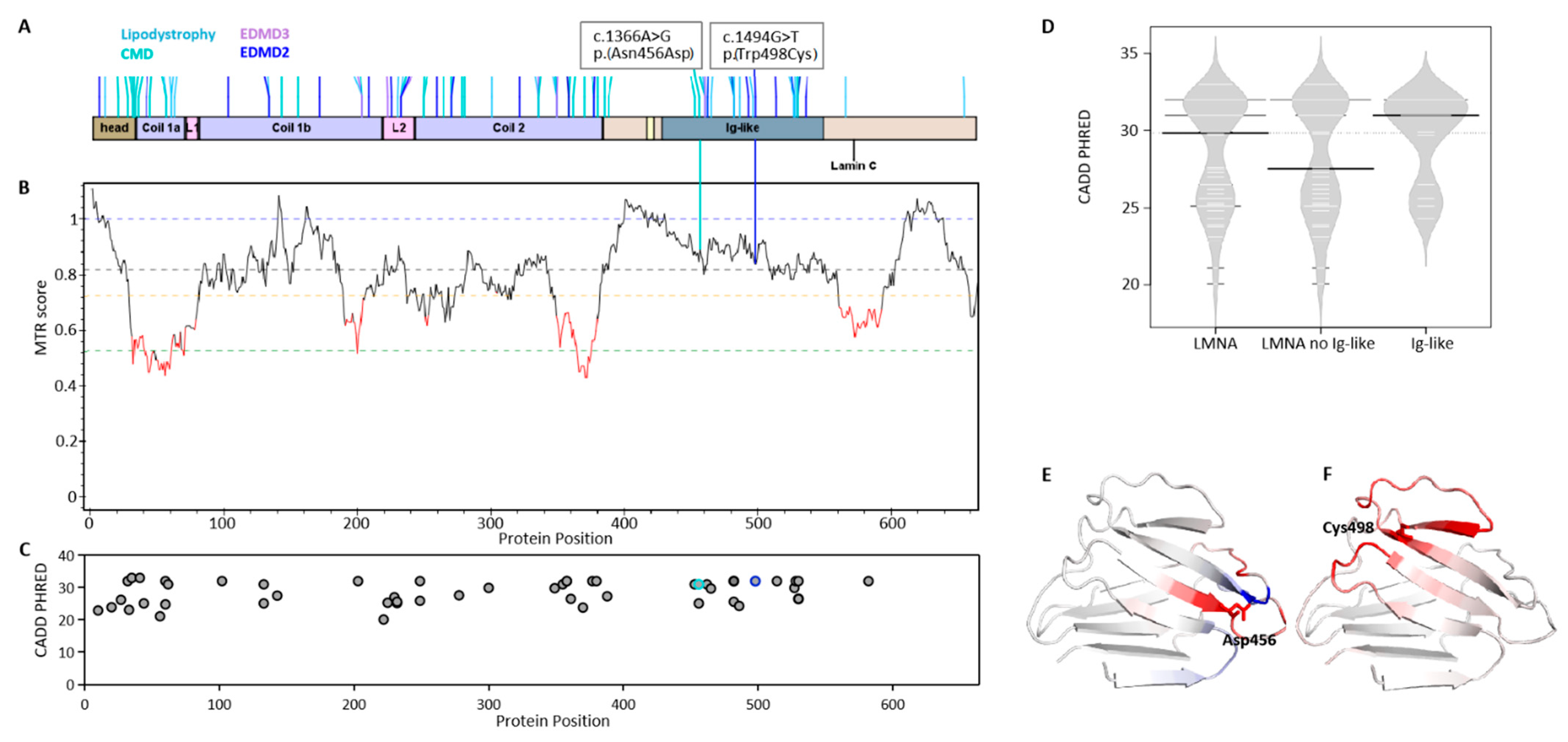
2.1. Mutation Impact on Lamin Proteins in LMNA-Related Muscular Dystrophy
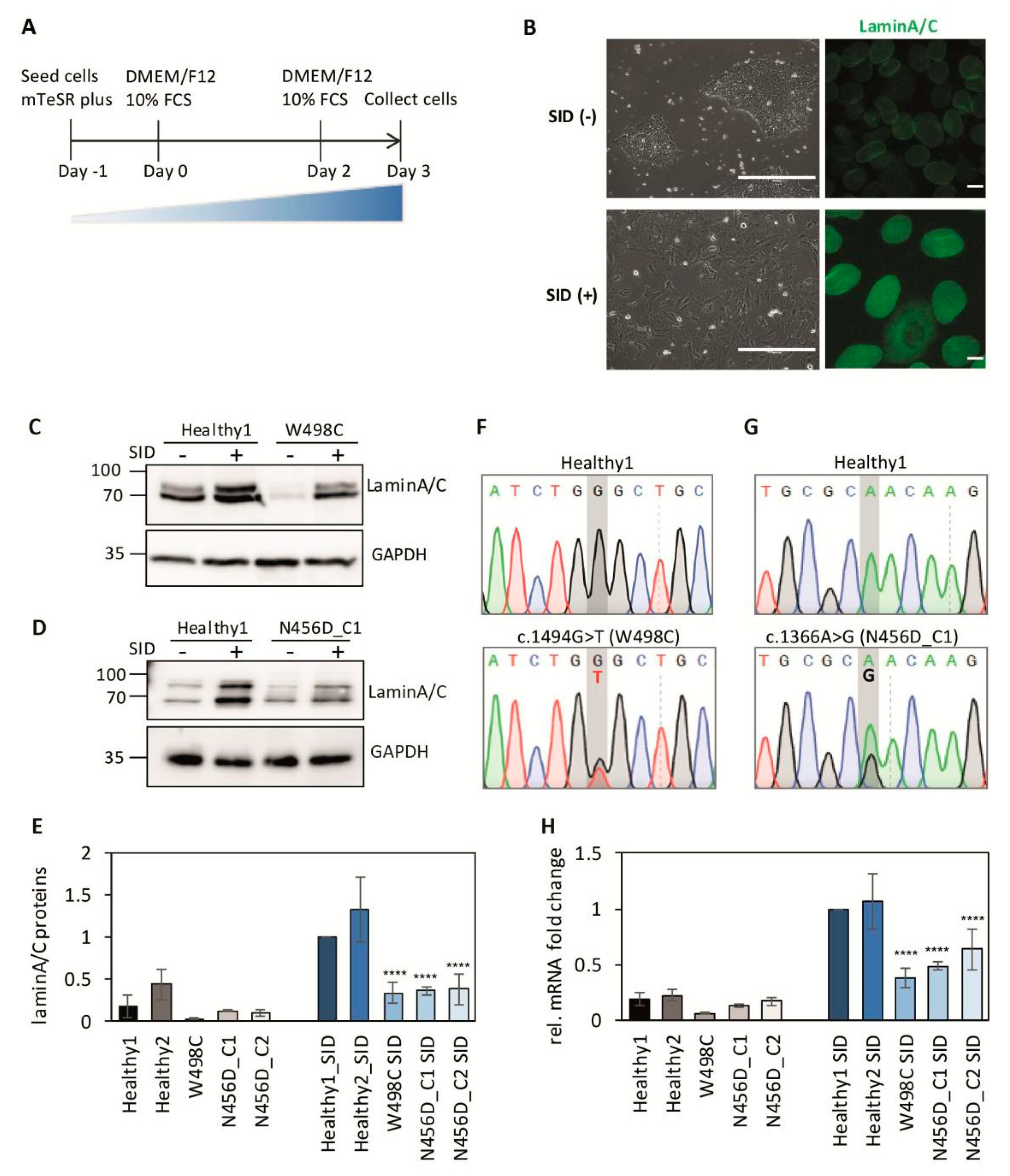
2.2. Impaired Expression of LMNA after Serum-Induced Differentiation in Patient-Derived iPSCs
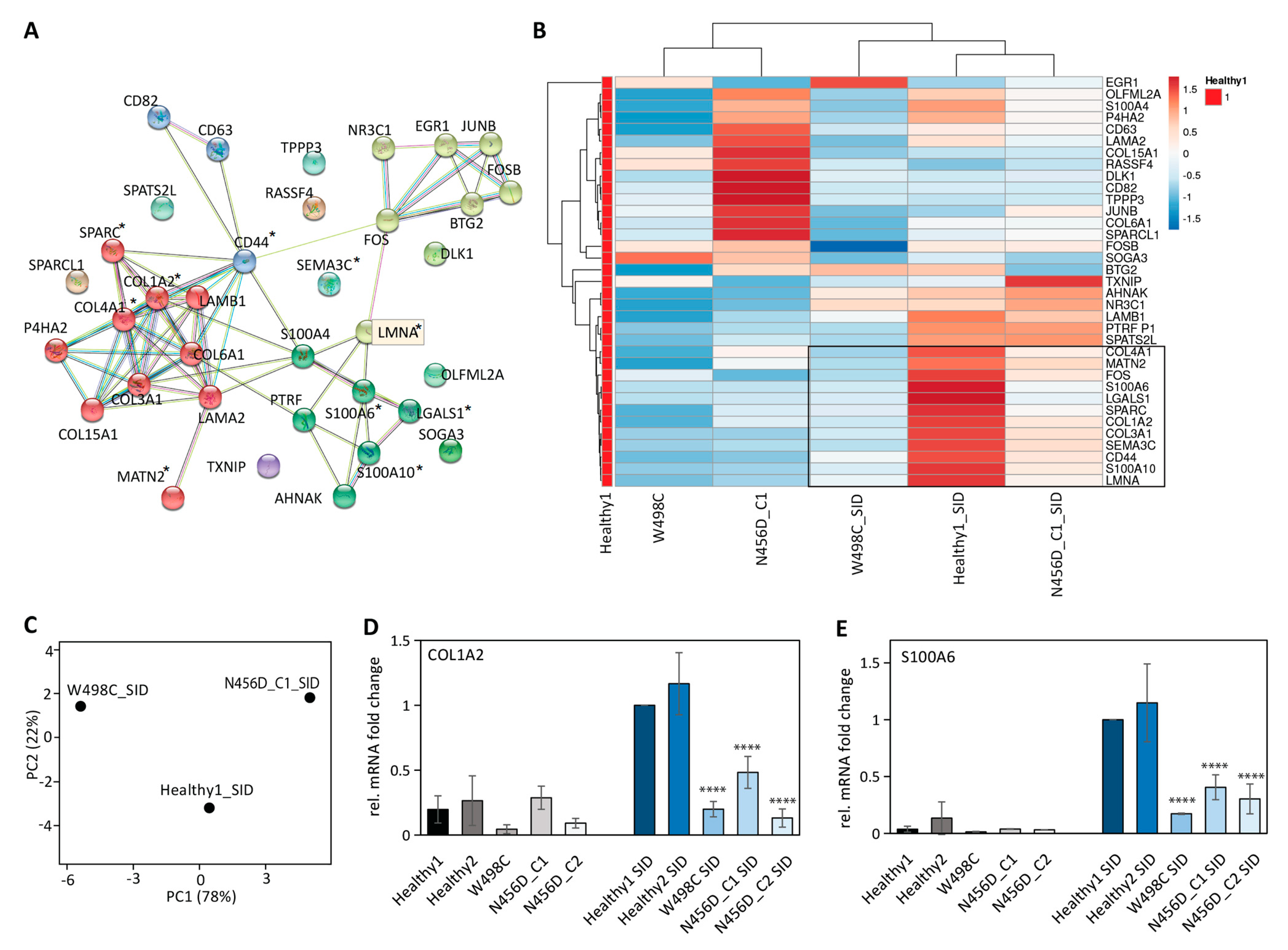
2.3. Impaired Expression of LMNA Co-Regulated Genes after SID in Patient-Derived iPSCs
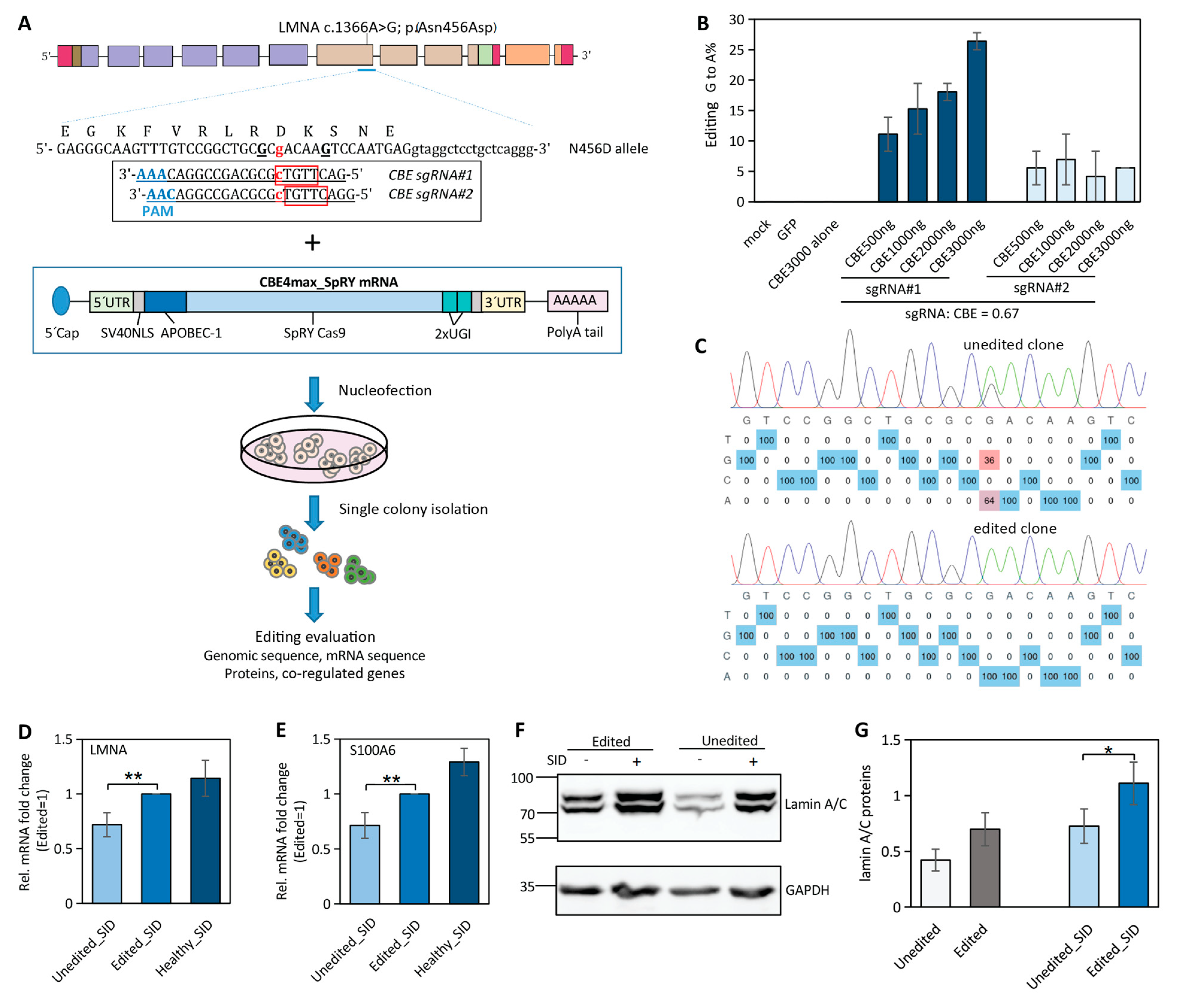
2.4. Near-PAMless Cytosine Base Editing of LMNA N456D Mutation in Patient-Derived iPSCs
2.5. Correction of LMNA Mutation in iPSCs Partially Restores LMNA and Its Co-Regulated Gene Expression
3. Discussion
4. Materials and Methods
4.1. iPSC Culture and Serum-Induced Differentiation (SID)
4.2. iPSC Nucleofection with mRNA CBE4max_SpRY and sgRNA
4.3. iPSC Transfection with Double Vectors
4.4. Genotype Sequencing and Analysis of Edited Cells
4.5. RT-PCR and qPCR
4.6. Western Blotting
4.7. Immunostaining
4.8. Statistical Analysis
4.9. Study Approval
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SID | serum-induced differentiation |
| iPSCs | induced pluripotent stem cells |
| CMD | congenital muscular dystrophy |
| EDMD2 | Emery–Dreifuss muscular dystrophy 2 |
| EDMD3 | Emery–Dreifuss muscular dystrophy 3 |
| MTR | missense tolerance ratio |
| CBE | cytosine base editor |
| PAM | protospacer-adjacent motif |
References
- Donnaloja, F.; Carnevali, F.; Jacchetti, E.; Raimondi, M.T. Lamin A/C Mechanotransduction in Laminopathies. Cells 2020, 9, 1306. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Jo, I.; Kang, S.M.; Hong, S.; Kim, S.; Jeong, S.; Kim, Y.H.; Park, B.J.; Ha, N.C. Structural basis for lamin assembly at the molecular level. Nat. Commun. 2019, 10, 3757. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Fernández, O.; Osorio, F.G.; Quesada, V.; Rodríguez, F.; Basso, S.; Maeso, D.; Rolas, L.; Barkaway, A.; Nourshargh, S.; Folgueras, A.R.; et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat. Med. 2019, 25, 423–426. [Google Scholar] [CrossRef]
- Koblan, L.W.; Erdos, M.R.; Wilson, C.; Cabral, W.A.; Levy, J.M.; Xiong, Z.M.; Tavarez, U.L.; Davison, L.M.; Gete, Y.G.; Mao, X.; et al. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Earle, A.J.; Kirby, T.J.; Fedorchak, G.R.; Isermann, P.; Patel, J.; Iruvanti, S.; Moore, S.A.; Bonne, G.; Wallrath, L.L.; Lammerding, J. Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 2020, 19, 464–473. [Google Scholar] [CrossRef]
- Leong, E.L.; Khaing, N.T.; Cadot, B.; Hong, W.L.; Kozlov, S.; Werner, H.; Wong, E.S.M.; Stewart, C.L.; Burke, B.; Lee, Y.L. Nesprin-1 LINC complexes recruit microtubule cytoskeleton proteins and drive pathology in Lmna mutant striated muscle. Hum Mol Genet. 2022, ddac179. [Google Scholar] [CrossRef]
- Gilchrist, S.; Gilbert, N.; Perry, P.; Ostlund, C.; Worman, H.J.; Bickmore, W.A. Altered protein dynamics of disease-associated lamin A mutants. BMC Cell Biol. 2004, 5, 46. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, C.; Jafferali, M.H.; Gudise, S.; Markus, R.; Hallberg, E. An inner nuclear membrane protein induces rapid differentiation of human induced pluripotent stem cells. Stem Cell Res. 2017, 23, 33–38. [Google Scholar] [CrossRef]
- Xi, H.; Langerman, J.; Sabri, S.; Chien, P.; Young, C.S.; Younesi, S.; Hicks, M.; Gonzalez, K.; Fujiwara, W.; Marzi, J.; et al. A Human Skeletal Muscle Atlas Identifies the Trajectories of Stem and Progenitor Cells across Development and from Human Pluripotent Stem Cells. Cell Stem Cell. 2020, 27, 158–176.e10. [Google Scholar] [CrossRef] [PubMed]
- Spuler, S.; Geier, C.; Osterziel, K.J.; Gutberlet, M.; Genschel, J.; Lehmann, T.N.; Zinn-Justin, S.; Gilquin, B.; Schmidt, H. A new LMNA mutation causing limb girdle muscular dystrophy 1B. J. Neurol. 2005, 252, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, J.; Silk, M.; Wang, Q.; Berkovic, S.F.; Liu, L.; Ascher, D.B.; Balding, D.J.; Petrovski, S. Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome Res. 2017, 27, 1715–1729. [Google Scholar] [CrossRef]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—Improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 31. [Google Scholar] [CrossRef]
- Etich, J.; Leßmeier, L.; Rehberg, M.; Sill, H.; Zaucke, F.; Netzer, C.; Semler, O. Osteogenesis imperfecta-pathophysiology and therapeutic options. Mol. Cell Pediatr. 2020, 7, 9. [Google Scholar] [CrossRef]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 proteins in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef]
- Mandinova, A.; Atar, D.; Schafer, B.W.; Spiess, M.; Aebi, U.; Heizmann, C.W. Distinct subcellular localization of calcium binding S100 proteins in human smooth muscle cells and their relocation in response to rises in intracellular calcium. J. Cell Sci. 1998, 111 Pt 14, 2043–2054. [Google Scholar] [CrossRef]
- Shah, D.; Virtanen, L.; Prajapati, C.; Kiamehr, M.; Gullmets, J.; West, G.; Kreutzer, J.; Pekkanen-Mattila, M.; Heliö, T.; Kallio, P.; et al. Modeling of LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Cells 2019, 8, 594. [Google Scholar] [CrossRef]
- Morales Rodriguez, B.; Domínguez-Rodríguez, A.; Benitah, J.P.; Lefebvre, F.; Marais, T.; Mougenot, N.; Beauverger, P.; Bonne, G.; Briand, V.; Gómez, A.M.; et al. Activation of sarcolipin expression and altered calcium cycling in LMNA cardiomyopathy. Biochem. Biophys Rep. 2020, 22, 100767. [Google Scholar] [CrossRef]
- Bal, N.C.; Gupta, S.C.; Pant, M.; Sopariwala, D.H.; Gonzalez-Escobedo, G.; Turner, J.; Gunn, J.S.; Pierson, C.R.; Harper, S.Q.; Rafael-Fortney, J.A.; et al. Is Upregulation of Sarcolipin Beneficial or Detrimental to Muscle Function? Front. Physiol. 2021, 12, 633058. [Google Scholar] [CrossRef] [PubMed]
- Dridi, H.; Wu, W.; Reiken, S.R.; Ofer, R.M.; Liu, Y.; Yuan, Q.; Sittenfeld, L.; Kushner, J.; Muchir, A.; Worman, H.J.; et al. Ryanodine receptor remodeling in cardiomyopathy and muscular dystrophy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2021, 29, 3919–3934. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, R.; Wu, J.; Xiong, Y.C.; Wei, J.; Zhang, S.; Yang, B.; Chen, J.; Yang, L. Comparison of cytosine base editors and development of the BEable-GPS database for targeting pathogenic SNVs. Genome Biol. 2019, 20, 218. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Wang, T.; Randolph, P.B.; Arbab, M.; Shen, M.W.; Huang, T.P.; Matuszek, Z.; Newby, G.A.; Rees, H.A.; Liu, D.R. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol. 2020, 38, 471–481. [Google Scholar] [CrossRef]
- Zhang, W.; Yin, J.; Zhang-Ding, Z.; Xin, C.; Liu, M.; Wang, Y.; Ai, C.; Hu, J. In-depth assessment of the PAM compatibility and editing activities of Cas9 variants. Nucleic Acids Res. 2021, 49, 8785–8795. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.; Sánchez-Bolaños, C.; Moreno-Sánchez, I.; Brena, D.; Vejnar, C.E.; Kukhtar, D.; Ruiz-López, M.; Cots-Ponjoan, M.; Rubio, A.; Melero, N.R.; et al. Genome editing in animals with minimal PAM CRISPR-Cas9 enzymes. Nat. Commun. 2022, 13, 2601. [Google Scholar] [CrossRef]
- De Masi, C.; Spitalieri, P.; Murdocca, M.; Novelli, G.; Sangiuolo, F. Application of CRISPR/Cas9 to human-induced pluripotent stem cells: From gene editing to drug discovery. Hum. Genom. 2020, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Magli, A.; Perlingeiro, R.R.C. Myogenic progenitor specification from pluripotent stem cells. Semin. Cell Dev. Biol. 2017, 72, 87–98. [Google Scholar] [CrossRef]
- Chal, J.; Oginuma, M.; Al Tanoury, Z.; Gobert, B.; Sumara, O.; Hick, A.; Bousson, F.; Zidouni, Y.; Mursch, C.; Moncuquet, P.; et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 2015, 33, 962–969. [Google Scholar] [CrossRef]
- Shelton, M.; Metz, J.; Liu, J.; Carpenedo, R.L.; Demers, S.P.; Stanford, W.L.; Skerjanc, I.S. Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell. Rep. 2014, 3, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Steele-Stallard, H.B.; Pinton, L.; Sarcar, S.; Ozdemir, T.; Maffioletti, S.M.; Zammit, P.S.; Tedesco, F.S. Modeling Skeletal Muscle Laminopathies Using Human Induced Pluripotent Stem Cells Carrying Pathogenic LMNA Mutations. Front Physiol. 2018, 9, 1332. [Google Scholar] [CrossRef]
- Metzler, E.; Telugu, N.; Diecke, S.; Spuler, S.; Escobar, H. Generation of three age and gender matched pairs of human induced pluripotent stem cells derived from myoblasts (MDCi011-A, MDCi012-A, MDCi013-A) and from peripheral blood mononuclear cells (MDCi011-B, MDCi012-B, MDCi013-B) from the same donor. Stem Cell Res. 2020, 48, 101987. [Google Scholar] [CrossRef] [PubMed]
- Metzler, E.; Telugu, N.; Diecke, S.; Spuler, S.; Escobar, H. Generation of two human induced pluripotent stem cell lines derived from myoblasts (MDCi014-A) and from peripheral blood mononuclear cells (MDCi014-B) from the same donor. Stem Cell Res. 2020, 48, 101998. [Google Scholar] [CrossRef] [PubMed]
- Escobar, H.; Krause, A.; Keiper, S.; Kieshauer, J.; Müthel, S.; de Paredes, M.G.; Metzler, E.; Kühn, R.; Heyd, F.; Spuler, S. Base editing repairs an SGCA mutation in human primary muscle stem cells. JCI Insight 2021, 6, e145994. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Krause, A.; Escobar, H.; Müthel, S.; Metzler, E.; Spuler, S. LMNA Co-Regulated Gene Expression as a Suitable Readout after Precise Gene Correction. Int. J. Mol. Sci. 2022, 23, 15525. https://doi.org/10.3390/ijms232415525
Wang H, Krause A, Escobar H, Müthel S, Metzler E, Spuler S. LMNA Co-Regulated Gene Expression as a Suitable Readout after Precise Gene Correction. International Journal of Molecular Sciences. 2022; 23(24):15525. https://doi.org/10.3390/ijms232415525
Chicago/Turabian StyleWang, Haicui, Anne Krause, Helena Escobar, Stefanie Müthel, Eric Metzler, and Simone Spuler. 2022. "LMNA Co-Regulated Gene Expression as a Suitable Readout after Precise Gene Correction" International Journal of Molecular Sciences 23, no. 24: 15525. https://doi.org/10.3390/ijms232415525
APA StyleWang, H., Krause, A., Escobar, H., Müthel, S., Metzler, E., & Spuler, S. (2022). LMNA Co-Regulated Gene Expression as a Suitable Readout after Precise Gene Correction. International Journal of Molecular Sciences, 23(24), 15525. https://doi.org/10.3390/ijms232415525