
Sensory Involvement in Amyotrophic Lateral Sclerosis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. ALS as a Purely Motor Disease
3. Non-Motor and Non-Sensory Alterations in ALS
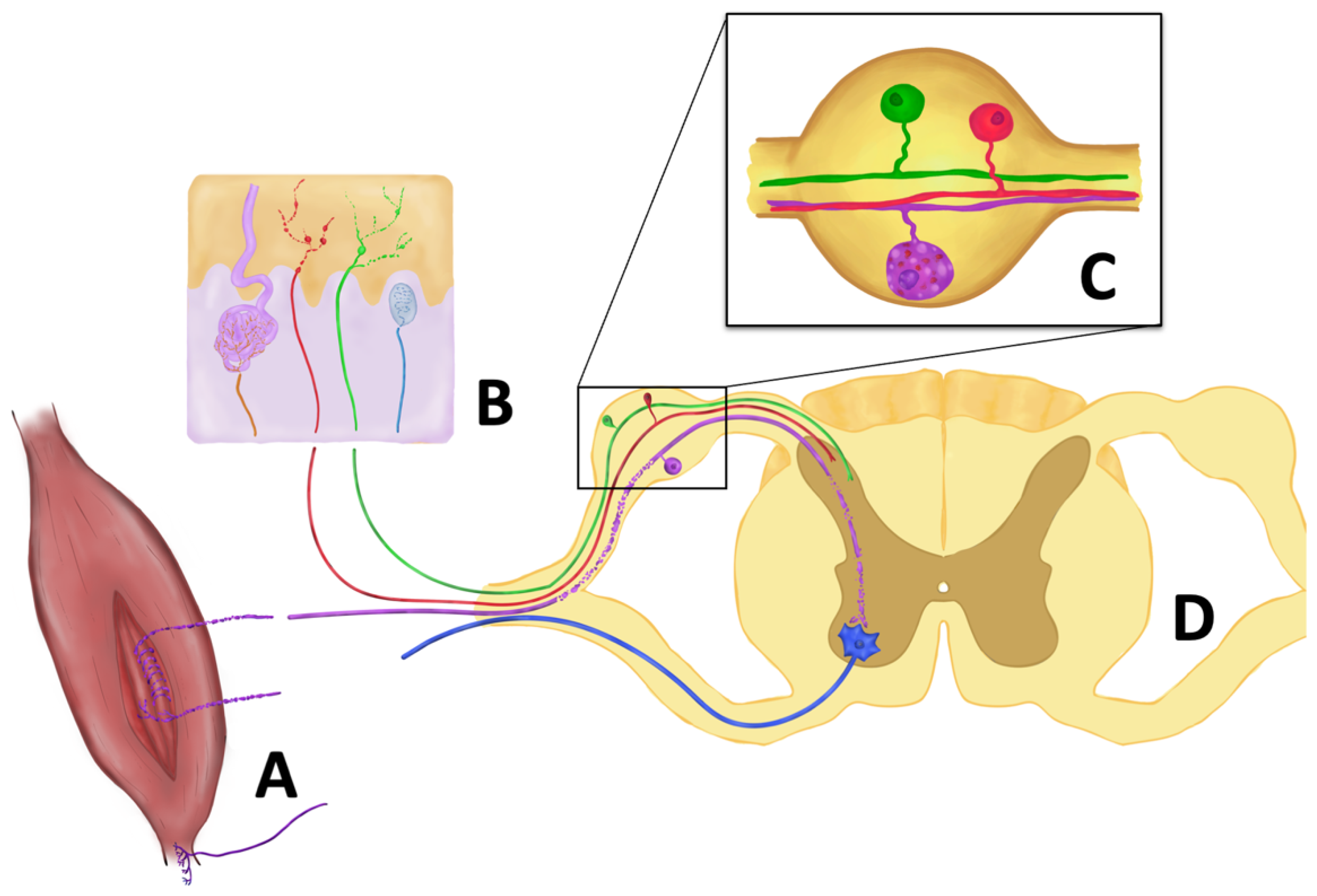
4. Involvement of the Somatosensory System in ALS
5. Somatosensory Cortex
6. Spinal Ascending Sensory Pathways
7. Dorsal Root Ganglia and Dorsal Roots
8. Peripheral Nerve Trunk
9. Proprioceptive Afferences
10. Small Sensory Nerve Fibers
11. Involvement of the Visual System in ALS
12. Involvement of the Olfactory System in ALS
13. Involvement of the Gustatory System in ALS
14. Involvement of the Auditory System in ALS
15. Involvement of the Autonomic Nervous System in ALS
16. Distal Axonopathy in ALS
17. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCombe, P.A.; Wray, N.R.; Henderson, R.D. Extra-motor abnormalities in amyotrophic lateral sclerosis: Another layer of heterogeneity. Expert Rev. Neurother. 2017, 17, 561–577. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.Q.; Wei, Q.; Wu, Z.Y. Sensory nerve disturbance in amyotrophic lateral sclerosis. Life Sci. 2018, 203, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Charcot, J.M.; Joffroy, A. Deux cas d’atrophie musculaire progressive: Avec lesions de la substance grise et des fiasceaux antérolatetéraux de la moell épinière. Arch. Physiol. Norm. Path. 1869, 2, 744–760. [Google Scholar]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L.; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J. Neurol. Sci. 1994, 124, 96–107. [Google Scholar] [PubMed]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef]
- Turner, M.R.; Swash, M. The expanding syndrome of amyotrophic lateral sclerosis: A clinical and molecular odyssey. J. Neurol. Neurosurg. Psychiatry 2015, 86, 667–673. [Google Scholar] [CrossRef]
- McCluskey, L.; Vandriel, S.; Elman, L.; Van Deerlin, V.M.; Powers, J.; Boller, A.; Wood, E.M.; Woo, J.; McMillan, C.T.; Rascovsky, K.; et al. ALS-Plus syndrome: Non-pyramidal features in a large ALS cohort. J. Neurol. Sci. 2014, 345, 118–124. [Google Scholar] [CrossRef]
- Fullmer, H.M.; Siedler, H.D.; Krooth, R.S.; Kurland, L.T. A cutaneous disorder of connective tissue in amyotrophic lateral sclerosis. A histochemical study. Neurology 1960, 10, 717–724. [Google Scholar] [CrossRef]
- Watanabe, S.; Yamada, K.; Ono, S.; Ishibashi, Y. Skin changes in patients with amyotrophic lateral sclerosis: Light and electron microscopic observations. J. Am. Acad. Dermatol. 1987, 17, 1006–1012. [Google Scholar] [CrossRef]
- Ono, S.; Toyokura, Y.; Mannen, T.; Ishibashi, Y. “Delayed return phenomenon” in amyotrophic lateral sclerosis. Acta Neurol. Scand. 1988, 77, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Toyokura, Y. Letter: Amyotrophic lateral sclerosis and bedsores. Lancet 1976, 1, 862. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Toyokura, Y.; Mannen, T.; Ishibashi, Y. Amyotrophic lateral sclerosis: Histologic, histochemical, and ultrastructural abnormalities of skin. Neurology 1986, 36, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Ono, S. The skin in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 191–199. [Google Scholar] [CrossRef]
- Paré, B.; Gros-Louis, F. Potential skin involvement in ALS: Revisiting Charcot’s observation—A review of skin abnormalities in ALS. Rev. Neurosci. 2017, 28, 551–572. [Google Scholar] [CrossRef] [PubMed]
- Beeldman, E.; Raaphorst, J.; Klein Twennaar, M.; de Visser, M.; Schmand, B.A.; de Haan, R.J. The cognitive profile of ALS: A systematic review and meta-analysis update. J. Neurol. Neurosurg. Psychiatry 2016, 87, 611–619. [Google Scholar] [CrossRef]
- Phukan, J.; Elamin, M.; Bede, P.; Jordan, N.; Gallagher, L.; Byrne, S.; Lynch, C.; Pender, N.; Hardiman, O. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: A population-based study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 102–108. [Google Scholar] [CrossRef]
- Montuschi, A.; Iazzolino, B.; Calvo, A.; Moglia, C.; Lopiano, L.; Restagno, G.; Brunetti, M.; Ossola, I.; Presti, A.L.; Cammarosano, S.; et al. Cognitive correlates in amyotrophic lateral sclerosis: A population-based study in Italy. J. Neurol. Neurosurg. Psychiatry 2015, 86, 168–173. [Google Scholar] [CrossRef]
- Xu, Z.; Alruwaili, A.R.S.; Henderson, R.D.; McCombe, P.A. Screening for cognitive and behavioural impairment in amyotrophic lateral sclerosis: Frequency of abnormality and effect on survival. J. Neurol. Sci. 2017, 376, 16–23. [Google Scholar] [CrossRef]
- Lomen-Hoerth, C. Characterization of amyotrophic lateral sclerosis and frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 2004, 17, 337–341. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134 Pt 9, 2456–2477. [Google Scholar] [CrossRef] [PubMed]
- Elamin, M.; Phukan, J.; Bede, P.; Jordan, N.; Byrne, S.; Pender, N.; Hardiman, O. Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology 2011, 76, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Westeneng, H.-J.; Debray, T.P.A.; Visser, A.E.; van Eijk, R.P.A.; Rooney, J.P.K.; Calvo, A.; Martin, S.; McDermott, C.J.; Thompson, A.G.; Pinto, S.; et al. Prognosis for patients with amyotrophic lateral sclerosis: Development and validation of a personalised prediction model. Lancet Neurol. 2018, 17, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, S.; Leigh, P.N.; Harvey, A.; Vythelingum, G.N.; Grisé, D.; Goldstein, L.H. Verbal fluency and executive dysfunction in amyotrophic lateral sclerosis (ALS). Neuropsychologia 2000, 38, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Olm, C.; McCluskey, L.; Elman, L.; Boller, A.; Moran, E.; Rascovsky, K.; Bisbing, T.; McMillan, C.; Grossman, M. Impaired cognitive flexibility in amyotrophic lateral sclerosis. Cogn. Behav. Neurol. 2015, 28, 17–26. [Google Scholar] [CrossRef]
- Stukovnik, V.; Zidar, J.; Podnar, S.; Repovs, G. Amyotrophic lateral sclerosis patients show executive impairments on standard neuropsychological measures and an ecologically valid motor-free test of executive functions. J. Clin. Exp. Neuropsychol. 2010, 32, 1095–1109. [Google Scholar] [CrossRef]
- Ash, S.; Olm, C.; McMillan, C.; Boller, A.; Irwin, D.; McCluskey, L.; Elman, L.; Grossman, M. Deficits in sentence expression in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 31–39. [Google Scholar] [CrossRef]
- Ash, S.; Menaged, A.; Olm, C.; McMillan, C.; Boller, A.; Irwin, D.; McCluskey, L.; Elman, L.; Grossman, M. Narrative discourse deficits in amyotrophic lateral sclerosis. Neurology 2014, 83, 520–528. [Google Scholar] [CrossRef]
- Bambini, V.; Arcara, G.; Martinelli, I.; Bernini, S.; Alvisi, E.; Moro, A.; Cappa, S.F.; Ceroni, M. Communication and pragmatic breakdowns in amyotrophic lateral sclerosis patients. Brain Lang. 2016, 153–154, 1–12. [Google Scholar] [CrossRef]
- Woolley, S.C.; Rush, B.K. Considerations for clinical neuropsychological evaluation in amyotrophic lateral sclerosis. Arch. Clin. Neuropsychol. 2017, 32, 906–916. [Google Scholar] [CrossRef]
- Lillo, P.; Mioshi, E.; Zoing, M.C.; Kiernan, M.C.; Hodges, J.R. How common are behavioural changes in amyotrophic lateral sclerosis? Amyotroph. Lateral Scler. 2011, 12, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.; Pinto-Grau, M.; Lonergan, K.; Bede, P.; O’Sullivan, M.; Heverin, M.; Vajda, A.; McLaughlin, R.L.; Pender, N.; Hardiman, O. A Cross-sectional population-based investigation into behavioral change in amyotrophic lateral sclerosis: Subphenotypes, staging, cognitive predictors, and survival. Ann. Clin. Transl. Neurol. 2017, 4, 305–317. [Google Scholar] [CrossRef]
- Borrego-Écija, S.; Turon-Sans, J.; Ximelis, T.; Aldecoa, I.; Molina-Porcel, L.; Povedano, M.; Rubio, M.A.; Gámez, J.; Cano, A.; Paré-Curell, M.; et al. Cognitive decline in amyotrophic lateral sclerosis: Neuropathological substrate and genetic determinants. Brain Pathol. 2021, 31, e12942. [Google Scholar] [CrossRef] [PubMed]
- Gorges, M.; Vercruysse, P.; Müller, H.-P.; Huppertz, H.-J.; Rosenbohm, A.; Nagel, G.; Weydt, P.; Petersén, Å.; Ludolph, A.C.; Kassubek, J.; et al. Hypothalamic atrophy is related to body mass index and age at onset in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Christidi, F.; Karavasilis, E.; Riederer, F.; Zalonis, I.; Ferentinos, P.; Velonakis, G.; Xirou, S.; Rentzos, M.; Argiropoulos, G.; Zouvelou, V.; et al. Gray matter and white matter changes in non-demented amyotrophic lateral sclerosis patients with or without cognitive impairment: A combined voxel-based morphometry and tract-based spatial statistics whole-brain analysis. Brain Imaging Behav. 2018, 12, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Senda, J.; Atsuta, N.; Watanabe, H.; Bagarinao, E.; Imai, K.; Yokoi, D.; Riku, Y.; Masuda, M.; Nakamura, R.; Watanabe, H.; et al. Structural MRI correlates of amyotrophic lateral sclerosis progression. J. Neurol. Neurosurg. Psychiatry 2017, 88, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Menke, R.A.L.; Talbot, K.; Kiernan, M.C.; Turner, M.R. Regional thalamic MRI as a marker of widespread cortical pathology and progressive frontotemporal involvement in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1250–1258. [Google Scholar] [CrossRef]
- Bede, P.; Omer, T.; Finegan, E.; Chipika, R.H.; Iyer, P.M.; Doherty, M.A.; Vajda, A.; Pender, N.; McLaughlin, R.L.; Hutchinson, S.; et al. Connectivity-based characterisation of subcortical grey matter pathology in frontotemporal dementia and ALS: A multimodal neuroimaging study. Brain Imaging Behav. 2018, 12, 1696–1707. [Google Scholar] [CrossRef]
- Pradat, P.-F.; Bruneteau, G.; Munerati, E.; Salachas, F.; Le Forestier, N.; Lacomblez, L.; Lenglet, T.; Meininger, V. Extrapyramidal stiffness in patients with amyotrophic lateral sclerosis. Mov. Disord. 2009, 24, 2143–2148. [Google Scholar] [CrossRef]
- Waring, S.C.; Esteban-Santillan, C.; Reed, D.M.; Craig, U.K.; Labarthe, D.R.; Petersen, R.C.; Kurland, L.T. Incidence of amyotrophic lateral sclerosis and of the parkinsonism-dementia complex of Guam, 1950–1989. Neuroepidemiology 2004, 23, 192–200. [Google Scholar] [CrossRef]
- Floris, G.; Borghero, G.; Cannas, A.; Di Stefano, F.; Costantino, E.; Murru, M.R.; Brunetti, M.; Restagno, G.; Traynor, B.J.; Marrosu, M.G.; et al. Frontotemporal dementia with psychosis, parkinsonism, visuo-spatial dysfunction, upper motor neuron involvement associated to expansion of C9ORF72: A peculiar phenotype? J. Neurol. 2012, 259, 1749–1751. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wilke, C.; Pomper, J.K.; Biskup, S.; Puskás, C.; Berg, D.; Synofzik, M. Atypical parkinsonism in C9orf72 expansions: A case report and systematic review of 45 cases from the literature. J. Neurol. 2016, 263, 558–574. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.G.; Duno, M.; Batbayli, M.; Puschmann, A.; Braendgaard, H.; Mardosiene, S.; Svenstrup, K.; Pinborg, L.H.; Vestergaard, K.; Hjermind, L.E.; et al. Corticobasal and ataxia syndromes widen the spectrum of C9ORF72 hexanucleotide expansion disease. Clin. Genet. 2013, 83, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Borghero, G.; Floris, G.; Cannas, A.; Marrosu, M.G.; Murru, M.R.; Costantino, E.; Parish, L.D.; Pugliatti, M.; Ticca, A.; Traynor, B.J.; et al. A patient carrying a homozygous p.A382T TARDBP missense mutation shows a syndrome including ALS, extrapyramidal symptoms, and FTD. Neurobiol. Aging 2011, 32, 2327.e1–2327.e5. [Google Scholar] [CrossRef] [PubMed]
- Lattante, S.; Millecamps, S.; Stevanin, G.; Rivaud-Péchoux, S.; Moigneu, C.; Camuzat, A.; Da Barroca, S.; Mundwiller, E.; Couarch, P.; Salachas, F.; et al. Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology 2014, 83, 990–995. [Google Scholar] [CrossRef]
- Sproviero, W.; Shatunov, A.; Stahl, D.; Shoai, M.; van Rheenen, W.; Jones, A.R.; Al-Sarraj, S.; Andersen, P.M.; Bonini, N.M.; Conforti, F.L.; et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol. Aging 2017, 51, 178.e1–178.e9. [Google Scholar] [CrossRef]
- Watanabe, R.; Higashi, S.; Nonaka, T.; Kawakami, I.; Oshima, K.; Niizato, K.; Akiyama, H.; Yoshida, M.; Hasegawa, M.; Arai, T. Intracellular dynamics of Ataxin-2 in the human brains with normal and frontotemporal lobar degeneration with TDP-43 inclusions. Acta Neuropathol. Commun. 2020, 8, 176. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Hart, M.P.; Gitler, A.D. ALS-associated ataxin 2 polyQ expansions enhance stress-induced caspase 3 activation and increase TDP-43 pathological modifications. J. Neurosci. 2012, 32, 9133–9142. [Google Scholar] [CrossRef]
- Coco, D.L.; Puligheddu, M.; Mattaliano, P.; Congiu, P.; Borghero, G.; Fantini, M.L.; La Bella, V.; Ferri, R. REM sleep behavior disorder and periodic leg movements during sleep in ALS. Acta Neurol. Scand. 2017, 135, 219–224. [Google Scholar] [CrossRef]
- Ebben, M.R.; Shahbazi, M.; Lange, D.J.; Krieger, A.C. REM behavior disorder associated with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Sanjak, M.; Hirsch, M.A.; Bravver, E.K.; Bockenek, W.L.; Norton, H.J.; Brooks, B.R. Vestibular deficits leading to disequilibrium and falls in ambulatory amyotrophic lateral sclerosis. Arch. Phys. Med. Rehabil. 2014, 95, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, M.; Turkman, A.; Swash, M. Sensory modulation of fasciculation discharge frequency. Muscle Nerve 2019, 59, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Chipika, R.H.; Mulkerrin, G.; Murad, A.; Lope, J.; Hardiman, O.; Bede, P. Alterations in somatosensory, visual and auditory pathways in amyotrophic lateral sclerosis: An under-recognised facet of ALS. J. Integr. Neurosci. 2022, 21, 88. [Google Scholar] [CrossRef] [PubMed]
- Mezzapesa, D.M.; D’Errico, E.; Tortelli, R.; Distaso, E.; Cortese, R.; Tursi, M.; Federico, F.; Zoccolella, S.; Logroscino, G.; Dicuonzo, F.; et al. Cortical thinning and clinical heterogeneity in amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e80748. [Google Scholar] [CrossRef]
- Mioshi, E.; Lillo, P.; Yew, B.; Hsieh, S.; Savage, S.; Hodges, J.R.; Kiernan, M.C.; Hornberger, M. Cortical atrophy in ALS is critically associated with neuropsychiatric and cognitive changes. Neurology 2013, 80, 1117–1123. [Google Scholar] [CrossRef]
- Schuster, C.; Kasper, E.; Dyrba, M.; Machts, J.; Bittner, D.; Kaufmann, J.; Mitchell, A.J.; Benecke, R.; Teipel, S.; Vielhaber, S.; et al. Cortical thinning and its relation to cognition in amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 240–246. [Google Scholar] [CrossRef]
- Spinelli, E.G.; Agosta, F.; Ferraro, P.M.; Riva, N.; Lunetta, C.; Falzone, Y.M.; Comi, G.; Falini, A.; Filippi, M. Brain MR imaging in patients with lower motor neuron-predominant disease. Radiology 2016, 280, 545–556. [Google Scholar] [CrossRef]
- Christidi, F.; Karavasilis, E.; Velonakis, G.; Rentzos, M.; Zambelis, T.; Zouvelou, V.; Xirou, S.; Ferentinos, P.; Efstathopoulos, E.; Kelekis, N.; et al. Motor and extra-motor gray matter integrity may underlie neurophysiologic parameters of motor function in amyotrophic lateral sclerosis: A combined voxel-based morphometry and transcranial stimulation study. Brain Imaging Behav. 2018, 12, 1730–1741. [Google Scholar] [CrossRef]
- Rajagopalan, V.; Pioro, E.P. Corticospinal tract and related grey matter morphometric shape analysis in ALS phenotypes: A fractal dimension study. Brain Sci. 2021, 11, 371. [Google Scholar] [CrossRef]
- Ricciardi, D.; Todisco, V.; Tedeschi, G.; Trojsi, F.; Cirillo, G. Altered sensory-motor plasticity in amyotrophic lateral sclerosis and complex regional pain type I syndrome: A shared mechanism? Neurol. Sci. 2020, 41, 1919–1921. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Ma, L. Study of the features of proton MR spectroscopy ((1)H-MRS) on amyotrophic lateral sclerosis. J. Magn. Reson. Imaging 2010, 31, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Pagani, M.; Agosta, F.; Calvo, A.; Cistaro, A.; Filippi, M. Neuroimaging in amyotrophic lateral sclerosis: Insights into structural and functional changes. Lancet Neurol. 2014, 13, 1228–1240. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.L.; et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef]
- Agosta, F.; Canu, E.; Inuggi, A.; Chiò, A.; Riva, N.; Silani, V.; Calvo, A.; Messina, S.; Falini, A.; Comi, G.; et al. Resting state functional connectivity alterations in primary lateral sclerosis. Neurobiol. Aging 2014, 35, 916–925. [Google Scholar] [CrossRef]
- Agosta, F.; Valsasina, P.; Absinta, M.; Riva, N.; Sala, S.; Prelle, A.; Copetti, M.; Comola, M.; Comi, G.; Filippi, M. Sensorimotor functional connectivity changes in amyotrophic lateral sclerosis. Cereb. Cortex 2011, 21, 2291–2298. [Google Scholar] [CrossRef]
- Sala, A.; Iaccarino, L.; Fania, P.; Vanoli, E.G.; Fallanca, F.; Pagnini, C.; Cerami, C.; Calvo, A.; Canosa, A.; Pagani, M.; et al. Testing the diagnostic accuracy of [18F]FDG-PET in discriminating spinal- and bulbar-onset amyotrophic lateral sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1117–1131. [Google Scholar] [CrossRef]
- Menon, P.; Geevasinga, N.; Yiannikas, C.; Howells, J.; Kiernan, M.C.; Vucic, S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: A prospective study. Lancet Neurol. 2015, 14, 478–484, Correction in Lancet Neurol. 2015, 14, 566. [Google Scholar] [CrossRef]
- Shibuya, K.; Park, S.B.; Geevasinga, N.; Menon, P.; Howells, J.; Simon, N.G.; Huynh, W.; Noto, Y.-I.; Götz, J.; Kril, J.J.; et al. Motor cortical function determines prognosis in sporadic ALS. Neurology 2016, 87, 513–520. [Google Scholar] [CrossRef]
- Shimizu, T.; Bokuda, K.; Kimura, H.; Kamiyama, T.; Nakayama, Y.; Kawata, A.; Isozaki, E.; Ugawa, Y. Sensory cortex hyperexcitability predicts short survival in amyotrophic lateral sclerosis. Neurology 2018, 90, e1578–e1587. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Golaszewski, S.; Thomschewski, A.; Sebastianelli, L.; Versace, V.; Brigo, F.; Orioli, A.; Saltuari, L.; Höller, Y.; Trinka, E. Disinhibition of sensory cortex in patients with amyotrophic lateral sclerosis. Neurosci. Lett. 2020, 722, 134860. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Nakayama, Y.; Funai, A.; Morishima, R.; Hayashi, K.; Bokuda, K.; Nakata, Y.; Isozaki, E. Progressive deterioration of sensory cortex excitability in advanced amyotrophic lateral sclerosis with invasive ventilation. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Sangari, S.; Giron, A.; Marrelec, G.; Pradat, P.F.; Marchand-Pauvert, V. Abnormal cortical brain integration of somatosensory afferents in ALS. Clin. Neurophysiol. 2018, 129, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, Y.; Mizutani, T.; Shimizu, T.; Kawata, A. Proportional neuronal loss between the primary motor and sensory cortex in amyotrophic lateral sclerosis. Neurosci. Lett. 2011, 503, 73–75. [Google Scholar] [CrossRef]
- Oyanagi, K.; Mochizuki, Y.; Nakayama, Y.; Hayashi, K.; Shimizu, T.; Nagao, M.; Hashimoto, T.; Yamazaki, M.; Matsubara, S.; Komori, T. Marked preservation of the visual and olfactory pathways in ALS patients in a totally locked-in state. Clin. Neuropathol. 2015, 34, 267–274. [Google Scholar] [CrossRef]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef]
- Iglesias, C.; Sangari, S.; El Mendili, M.M.; Benali, H.; Marchand-Pauvert, V.; Pradat, P.F. Electrophysiological and spinal imaging evidences for sensory dysfunction in amyotrophic lateral sclerosis. BMJ Open 2015, 5, e007659. [Google Scholar] [CrossRef]
- Georgesco, M.; Salerno, A.; Carlander, B.; Leger, J.J.; Camu, W.; Billiard, M.; Cadilhac, J. Les potentiels évoqués somesthésiques dans la sclérose latérale amyotrophique et la sclérose latérale primaire [Somatosensory evoked potentials in amyotrophic lateral sclerosis and primary lateral sclerosis]. Rev. Neurol. 1994, 150, 292–298. [Google Scholar]
- Constantinovici, A. Abnormal somatosensory evoked potentials in amyotrophic lateral sclerosis. Rom. J. Neurol. Psychiatry 1993, 31, 273–278. [Google Scholar]
- Radtke, R.A.; Erwin, A.; Erwin, C.W. Abnormal sensory evoked potentials in amyotrophic lateral sclerosis. Neurology 1986, 36, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Dasheiff, R.M.; Drake, M.E.; Brendle, A.; Erwin, C.W. Abnormal somatosensory evoked potentials in amyotrophic lateral sclerosis. Electroencephalogr. Clin. Neurophysiol. 1985, 60, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Adad, J.; El Mendili, M.-M.; Morizot-Koutlidis, R.; Lehéricy, S.; Meininger, V.; Blancho, S.; Rossignol, S.; Benali, H.; Pradat, P.-F. Involvement of spinal sensory pathway in ALS and specificity of cord atrophy to lower motor neuron degeneration. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Mulder, D.W.; Bushek, W.; Spring, E.; Karnes, J.; Dyck, P.J. Motor neuron disease (ALS): Evaluation of detection thresholds of cutaneous sensation. Neurology 1983, 33, 1625–1627. [Google Scholar] [CrossRef]
- Dyck, P.J.; Stevens, J.C.; Mulder, D.W.; Espinosa, R.E. Frequency of nerve fiber degeneration of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. Morphometry of deep and superficial peroneal nerves. Neurology 1975, 25, 781–785. [Google Scholar] [CrossRef]
- Hammad, M.; Silva, A.; Glass, J.; Sladky, J.T.; Benatar, M. Clinical, electrophysiologic, and pathologic evidence for sensory abnormalities in ALS. Neurology 2007, 69, 2236–2242. [Google Scholar] [CrossRef]
- Ince, P.; Evans, J.; Knopp, M.; Forster, G.; Hamdalla, H.; Wharton, S.; Shaw, P. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology 2003, 60, 1252–1258. [Google Scholar] [CrossRef]
- Hirano, A.; Kurland, L.T.; Sayre, G.P. Familial amyotrophic lateral sclerosis. A subgroup characterized by posterior and spinocerebellar tract involvement and hyaline inclusions in the anterior horn cells. Arch. Neurol. 1967, 16, 232–243. [Google Scholar] [CrossRef]
- Lawyer, T., Jr.; Netsky, M.g. Amyotrophic lateral sclerosis. AMA Arch. Neurol. Psychiatry 1953, 69, 171–192. [Google Scholar] [CrossRef]
- Sasaki, S.; Tsutsumi, Y.; Yamane, K.; Sakuma, H.; Maruyama, S. Sporadic amyotrophic lateral sclerosis with extensive neurological involvement. Acta Neuropathol. 1992, 84, 211–215. [Google Scholar] [CrossRef]
- Kawamura, Y.; Dyck, P.J.; Shimono, M.; Okazaki, H.; Tateishi, J.; Doi, H. Morphometric comparison of the vulnerability of peripheral motor and sensory neurons in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1981, 40, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Camdessanché, J.-P.; Belzil, V.V.; Jousserand, G.; Rouleau, G.A.; Créac’h, C.; Convers, P.; Antoine, J.-C. Sensory and motor neuronopathy in a patient with the A382P TDP-43 mutation. Orphanet J. Rare Dis. 2011, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, F.; Abicht, A.; Kuepper, C.; Voelk, S.; Sonnenfeld, S.; Tonon, M.; Schaub, A.; Scholz, V.; Kleinle, S.; Erdmann, H.; et al. Sensory neuropathy due to RFC1 in a patient with ALS: More than a coincidence? J. Neurol. 2022, 269, 2774–2777. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Davis, A.A.; Tennant, P.; Wang, M.; Coleman, M.; Asress, S.; Adalbert, R.; Alexander, G.M.; Glass, J.D. The WldS gene modestly prolongs survival in the SOD1G93A fALS mouse. Neurobiol. Dis. 2005, 19, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-S.; Wu, D.-X.; Wu, H.-R.; Wu, S.-Y.; Yang, C.; Li, B.; Bu, H.; Zhang, Y.-S.; Li, C.-Y. Sensory involvement in the SOD1-G93A mouse model of amyotrophic lateral sclerosis. Exp. Mol. Med. 2009, 41, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Sábado, J.; Casanovas, A.; Tarabal, O.; Hereu, M.; Piedrafita, L.; Calderó, J.; Esquerda, J.E. Accumulation of misfolded SOD1 in dorsal root ganglion degenerating proprioceptive sensory neurons of transgenic mice with amyotrophic lateral sclerosis. Biomed. Res. Int. 2014, 2014, 852163. [Google Scholar] [CrossRef]
- Sassone, J.; Taiana, M.; Lombardi, R.; Porretta-Serapiglia, C.; Freschi, M.; Bonanno, S.; Marcuzzo, S.; Caravello, F.; Bendotti, C.; Lauria, G. ALS mouse model SOD1G93A displays early pathology of sensory small fibers associated to accumulation of a neurotoxic splice variant of peripherin. Hum. Mol. Genet. 2016, 25, 1588–1599. [Google Scholar] [CrossRef]
- Vaughan, S.K.; Sutherland, N.M.; Zhang, S.; Hatzipetros, T.; Vieira, F.; Valdez, G. The ALS-inducing factors, TDP43A315T and SOD1G93A, directly affect and sensitize sensory neurons to stress. Sci. Rep. 2018, 8, 16582. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Mech, A.M.; Aktar, T.; Zhang, Y.; Schiavo, G. Altered Sensory Neuron Development in CMT2D Mice Is Site-Specific and Linked to Increased GlyRS Levels. Front. Cell Neurosci. 2020, 14, 232. [Google Scholar] [CrossRef]
- Rubio, M.A.; Herrando-Grabulosa, M.; Gaja-Capdevila, N.; Vilches, J.J.; Navarro, X. Characterization of somatosensory neuron involvement in the SOD1G93A mouse model. Sci. Rep. 2022, 12, 7600. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Höller, Y.; Taylor, A.C.; Lochner, P.; Tezzon, F.; Golaszewski, S.; Brigo, F.; Trinka, E. Canine degenerative myelopathy: A model of human amyotrophic lateral sclerosis. Zoology 2016, 119, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Awano, T.; Johnson, G.S.; Wade, C.M.; Katz, M.L.; Johnson, G.C.; Taylor, J.F.; Perloski, M.; Biagi, T.; Baranowska, I.; Long, S.; et al. Genome-wide association analysis reveals a SOD1 mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 2794–2799. [Google Scholar] [CrossRef]
- Pugdahl, K.; Fuglsang-Frederiksen, A.; De Carvalho, M.; Johnsen, B.; Fawcett, P.R.W.; Labarre-Vila, A.; Liguori, R.; Nix, W.A.; Schofield, I.S. Generalised sensory system abnormalities in amyotrophic lateral sclerosis: A European multicentre study. J. Neurol. Neurosurg. Psychiatry 2007, 78, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.; Mills, K.; Donaghy, M. Progressive sensory nerve dysfunction in amyotrophic lateral sclerosis: A prospective clinical and neurophysiological study. J. Neurol. 1993, 240, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Mondelli, M.; Rossi, A.; Passero, S.; Guazzi, G.C. Involvement of peripheral sensory fibers in amyotrophic lateral sclerosis: Electrophysiological study of 64 cases. Muscle Nerve 1993, 16, 166–172. [Google Scholar] [CrossRef]
- Shefner, J.M.; Tyler, H.R.; Krarup, C. Abnormalities in the sensory action potential in patients with amyotrophic lateral sclerosis. Muscle Nerve 1991, 14, 1242–1246. [Google Scholar] [CrossRef]
- Theys, P.A.; Peeters, E.; Robberecht, W. Evolution of motor and sensory deficits in amyotrophic lateral sclerosis estimated by neurophysiological techniques. J. Neurol. 1999, 246, 438–442. [Google Scholar] [CrossRef]
- Pegat, A.; Bouhour, F.; Mouzat, K.; Vial, C.; Pegat, B.; Leblanc, P.; Broussolle, E.; Millecamps, S.; Lumbroso, S.; Bernard, E. Electrophysiological characterization of c9orf72-associated amyotrophic lateral sclerosis: A retrospective study. Eur. Neurol. 2019, 82, 106–112. [Google Scholar] [CrossRef]
- Kothari, M.J.; Rutkove, S.B.; Logigian, E.L.; Shefner, J.M. Coexistent entrapment neuropathies in patients with amyotrophic lateral sclerosis. Arch. Phys. Med. Rehabil. 1996, 77, 1186–1188. [Google Scholar] [CrossRef]
- Pugdahl, K.; Fuglsang-Frederiksen, A.; Johnsen, B.; de Carvalho, M.; Fawcett, P.R.; Labarre-Vila, A.; Liguori, R.; Nix, W.A.; Schofield, I.S. A prospective multicentre study on sural nerve action potentials in ALS. Clin. Neurophysiol. 2008, 119, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Isak, B.; Tankisi, H.; Johnsen, B.; Pugdahl, K.; Møller, A.T.; Finnerup, N.B.; Christensen, P.B.; Fuglsang-Frederiksen, A. Involvement of distal sensory nerves in amyotrophic lateral sclerosis. Muscle Nerve 2016, 54, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Sista, S.R.S.; Shelly, S.; Oskarsson, B.; Rubin, D.I.; Martinez-Thompson, J.M.; Parra-Cantu, C.; Staff, N.P.; Laughlin, R.S. Clinical and electrophysiological findings in C9ORF72 ALS. Muscle Nerve 2022, 66, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; Santos-Nogueira, E.; Osta, R.; Navarro, X. Electrophysiological analysis of a murine model of motoneuron disease. Clin. Neurophysiol. 2011, 122, 1660–1670. [Google Scholar] [CrossRef]
- Boërio, D.; Kalmar, B.; Greensmith, L.; Bostock, H. Excitability properties of mouse motor axons in the mutant SOD1(G93A) model of amyotrophic lateral sclerosis. Muscle Nerve 2010, 41, 774–784. [Google Scholar] [CrossRef]
- Kawata, A.; Kato, S.; Hayashi, H.; Hirai, S. Prominent sensory and autonomic disturbances in familial amyotrophic lateral sclerosis with a Gly93Ser mutation in the SOD1 gene. J. Neurol. Sci. 1997, 153, 82–85. [Google Scholar] [CrossRef]
- Bella, E.D.; Rigamonti, A.; Mantero, V.; Morbin, M.; Saccucci, S.; Gellera, C.; Mora, G.; Lauria, G. Heterozygous D90A-SOD1 mutation in a patient with facial onset sensory motor neuronopathy (FOSMN) syndrome: A bridge to amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1009–1011. [Google Scholar] [CrossRef]
- Ben Hamida, M.; Letaief, F.; Hentati, F.; Ben Hamida, C. Morphometric study of the sensory nerve in classical (or Charcot disease) and juvenile amyotrophic lateral sclerosis. J. Neurol. Sci. 1987, 78, 313–329. [Google Scholar] [CrossRef]
- Luigetti, M.; Conte, A.; Del Grande, A.; Bisogni, G.; Romano, A.; Sabatelli, M. Sural nerve pathology in ALS patients: A single-centre experience. Neurol. Sci. 2012, 33, 1095–1099. [Google Scholar] [CrossRef]
- Isaacs, J.D.; Dean, A.F.; Shaw, C.E.; Al-Chalabi, A.; Mills, K.R.; Leigh, P.N. Amyotrophic lateral sclerosis with sensory neuropathy: Part of a multisystem disorder? J. Neurol. Neurosurg. Psychiatry 2007, 78, 750–753. [Google Scholar] [CrossRef]
- Bradley, W.G.; Good, P.; Rasool, C.G.; Adelman, L.S. Morphometric and biochemical studies of peripheral nerves in amyotrophic lateral sclerosis. Ann. Neurol. 1983, 14, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Di Trapani, G.; David, P.; La Cara, A.; Servidei, S.; Tonali, P. Morphological studies of sural nerve biopsies in the pseudopolyneuropathic form of amyotrophic lateral sclerosis. Clin. Neuropathol. 1986, 5, 134–138. [Google Scholar] [PubMed]
- Di Trapani, G.; David, P.; La Cara, A.; Tonali, P.; Laurienzo, P. Light and ultrastructural studies in sural biopsies of the pseudopolyneuropathic form of ALS. Adv. Exp. Med. Biol. 1987, 209, 111–119. [Google Scholar] [PubMed]
- Dayan, A.D.; Graveson, G.S.; Illis, L.S.; Robinson, P.K. Schwann cell damage in motoneuron disease. Neurology 1969, 19, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Arnold, N.; Harriman, D.G. The incidence of abnormality in control human peripheral nerves studied by single axon dissection. J. Neurol. Neurosurg. Psychiatry 1970, 33, 55–61. [Google Scholar] [CrossRef][Green Version]
- Heads, T.; Pollock, M.; Robertson, A.; Sutherland, W.H.; Allpress, S. Sensory nerve pathology in amyotrophic lateral sclerosis. Acta Neuropathol. 1991, 82, 316–320. [Google Scholar] [CrossRef]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Sagare, A.P.; Zhao, Z.; Ma, Q.; Zuniga, E.; Wang, Y.; Zhong, Z.; Sullivan, J.S.; Griffin, J.H.; et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1035–E1042. [Google Scholar] [CrossRef]
- Garbuzova-Davis, S.; Hernandez-Ontiveros, D.G.; Rodrigues, M.C.; Haller, E.; Frisina-Deyo, A.; Mirtyl, S.; Sallot, S.; Saporta, S.; Borlongan, C.V.; Sanberg, P.R. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012, 1469, 114–128. [Google Scholar] [CrossRef]
- Alanne, M.H.; Pummi, K.; Heape, A.M.; Grènman, R.; Peltonen, J.; Peltonen, S. Tight junction proteins in human Schwann cell autotypic junctions. J. Histochem. Cytochem. 2009, 57, 523–529. [Google Scholar] [CrossRef]
- Riva, N.; Gentile, F.; Cerri, F.; Gallia, F.; Podini, P.; Dina, G.; Falzone, Y.M.; Fazio, R.; Lunetta, C.; Calvo, A.; et al. Phosphorylated TDP-43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis. Brain 2022, 145, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Simmatis, L.; Atallah, G.; Scott, S.H.; Taylor, S. The feasibility of using robotic technology to quantify sensory, motor, and cognitive impairments associated with ALS. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Krieg, I.; Dalin, D.; Heimbach, B.; Wiesmeier, I.K.; Maurer, C. Abnormal trunk control determines postural abnormalities in Amyotrophic Lateral Sclerosis. NeuroRehabilitation 2019, 44, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Nardone, A.; Galante, M.; Lucas, B.; Schieppati, M. Stance control is not affected by paresis and reflex hyperexcitability: The case of spastic patients. J. Neurol. Neurosurg. Psychiatry 2001, 70, 635–643. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ruoppolo, G.; Onesti, E.; Gori, M.C.; Schettino, I.; Frasca, V.; Biasiotta, A.; Giordano, C.; Ceccanti, M.; Cambieri, C.; Greco, A.; et al. Laryngeal sensitivity in patients with amyotrophic lateral sclerosis. Front. Neurol. 2016, 7, 212. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.K.; Kemp, Z.; Hatzipetros, T.; Vieira, F.; Valdez, G. Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis-causing mutations. J. Comp. Neurol. 2015, 523, 2477–2494. [Google Scholar] [CrossRef]
- Seki, S.; Yamamoto, T.; Quinn, K.; Spigelman, I.; Pantazis, A.; Olcese, R.; Wiedau-Pazos, M.; Chandler, S.H.; Venugopal, S. Circuit-specific early impairment of proprioceptive sensory neurons in the SOD1G93A mouse model for ALS. J. Neurosci. 2019, 39, 8798–8815. [Google Scholar] [CrossRef]
- Held, A.; Major, P.; Sahin, A.; Reenan, R.A.; Lipscombe, D.; Wharton, K.A. Circuit dysfunction in SOD1-ALS model first detected in sensory feedback prior to motor neuron degeneration is alleviated by BMP signaling. J. Neurosci. 2019, 39, 2347–2364. [Google Scholar] [CrossRef]
- Wootz, H.; FitzSimons-Kantamneni, E.; Larhammar, M.; Rotterman, T.M.; Enjin, A.; Patra, K.; André, E.; Van Zundert, B.; Kullander, K.; Alvarez, F.J. Alterations in the motor neuron-renshaw cell circuit in the Sod1(G93A) mouse model. J. Comp. Neurol. 2013, 521, 1449–1469. [Google Scholar] [CrossRef]
- Siembab, V.C.; Gomez-Perez, L.; Rotterman, T.M.; Shneider, N.A.; Alvarez, F.J. Role of primary afferents in the developmental regulation of motor axon synapse numbers on Renshaw cells. J. Comp. Neurol. 2016, 524, 1892–1919. [Google Scholar] [CrossRef]
- Nagao, M.; Misawa, H.; Kato, S.; Hirai, S. Loss of cholinergic synapses on the spinal motor neurons of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 1998, 57, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Casas, C.; Herrando-Grabulosa, M.; Manzano, R.; Mancuso, R.; Osta, R.; Navarro, X. Early presymptomatic cholinergic dysfunction in a murine model of amyotrophic lateral sclerosis. Brain Behav. 2013, 3, 145–158, Correction in Brain Behav. 2013, 3, 328. [Google Scholar] [CrossRef] [PubMed]
- Murayama, S.; Ookawa, Y.; Mori, H.; Nakano, I.; Ihara, Y.; Kuzuhara, S.; Tomonaga, M. Immunocytochemical and ultrastructural study of Lewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathol. 1989, 78, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Kozlowski, M.A.; Hinton, D.R.; Miller, C.A. Degeneration of spinocerebellar neurons in amyotrophic lateral sclerosis. Ann. Neurol. 1990, 27, 215–225. [Google Scholar] [CrossRef]
- Kokubo, Y.; Kuzuhara, S.; Narita, Y.; Kikugawa, K.; Nakano, R.; Inuzuka, T.; Tsuji, S.; Watanabe, M.; Miyazaki, T.; Murayama, S.; et al. Accumulation of neurofilaments and SOD1-immunoreactive products in a patient with familial amyotrophic lateral sclerosis with I113T SOD1 mutation. Arch. Neurol. 1999, 56, 1506–1508. [Google Scholar] [CrossRef]
- Fujita, Y.; Ikeda, M.; Yanagisawa, T.; Senoo, Y.; Okamoto, K. Different clinical and neuropathologic phenotypes of familial ALS with A315E TARDBP mutation. Neurology 2011, 77, 1427–1431. [Google Scholar] [CrossRef]
- Ilieva, H.S.; Yamanaka, K.; Malkmus, S.; Kakinohana, O.; Yaksh, T.; Marsala, M.; Cleveland, D.W. Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc. Natl. Acad. Sci. USA 2008, 105, 12599–12604. [Google Scholar] [CrossRef]
- Lalancette-Hebert, M.; Sharma, A.; Lyashchenko, A.K.; Shneider, N.A. Gamma motor neurons survive and exacerbate alpha motor neuron degeneration in ALS. Proc. Natl. Acad. Sci. USA 2016, 113, E8316–E8325. [Google Scholar] [CrossRef]
- Mentis, G.Z.; Blivis, D.; Liu, W.; Drobac, E.; Crowder, M.E.; Kong, L.; Alvarez, F.J.; Sumner, C.J.; O’Donovan, M.J. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 2011, 69, 453–467. [Google Scholar] [CrossRef]
- Pagnini, F.; Lunetta, C.; Banfi, P.; Rossi, G.; Fossati, F.; Marconi, A.; Castelnuovo, G.; Corbo, M.; Molinari, E. Pain in amyotrophic lateral sclerosis: A psychological perspective. Neurol. Sci. 2012, 33, 1193–1196. [Google Scholar] [CrossRef]
- Pizzimenti, A.; Aragona, M.; Onesti, E.; Inghilleri, M. Depression, pain and quality of life in patients with amyotrophic lateral sclerosis: A cross-sectional study. Funct. Neurol. 2013, 28, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, F.; Skudlarek, A.; Berndt, J.; Kornhuber, M.E. Characteristics of pain in amyotrophic lateral sclerosis. Brain Behav. 2015, 5, e00296. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Canosa, A.; Gallo, S.; Moglia, C.; Ilardi, A.; Cammarosano, S.; Papurello, D.; Calvo, A. Pain in amyotrophic lateral sclerosis: A population-based controlled study. Eur. J. Neurol. 2012, 19, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.S.; Zhang, J.; Zheng, J.Y.; Zhang, S.; Kang, D.X.; Fan, D.S. Fully intact contact heat evoked potentials in patients with amyotrophic lateral sclerosis. Muscle Nerve 2009, 39, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Simone, I.L.; Tortelli, R.; Samarelli, V.; D’Errico, E.; Sardaro, M.; Difruscolo, O.; Calabrese, R.; Francesco, V.D.V.; Livrea, P.; de Tommaso, M. Laser evoked potentials in amyotrophic lateral sclerosis. J. Neurol. Sci. 2010, 288, 106–111. [Google Scholar] [CrossRef]
- Deepika, J.; Manvir, B.; Sumit, S.; Vinay, G.; Trilochan, S.; Garima, S.; Padma, M.V.; Madhuri, B. Quantitative thermal sensory testing in patients with amyotrophic lateral sclerosis using reaction time exclusive method of levels (MLE). Electromyogr. Clin. Neurophysiol. 2006, 46, 145–148. [Google Scholar]
- Lopes, L.; Galhardoni, R.; Silva, V.; Jorge, F.; Yeng, L.; Callegaro, D.; Chadi, G.; Teixeira, M.; de Andrade, D.C. Beyond weakness: Characterization of pain, sensory profile and conditioned pain modulation in patients with motor neuron disease: A controlled study. Eur. J. Pain 2018, 22, 72–83. [Google Scholar] [CrossRef]
- Truini, A.; Biasiotta, A.; Onesti, E.; Di Stefano, G.; Ceccanti, M.; La Cesa, S.; Pepe, A.; Giordano, C.; Cruccu, G.; Inghilleri, M. Small-fibre neuropathy related to bulbar and spinal-onset in patients with ALS. J. Neurol. 2015, 262, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Treede, R.D.; Meyer, R.A.; Campbell, J.N. Myelinated mechanically insensitive afferents from monkey hairy skin: Heat-response properties. J. Neurophysiol. 1998, 80, 1082–1093. [Google Scholar] [CrossRef]
- Weis, J.; Katona, I.; Muller-Newen, G.; Sommer, C.; Necula, G.; Hendrich, C.; Ludolph, A.C.; Sperfeld, A.D. Small-fiber neuropathy in patients with ALS. Neurology 2011, 76, 2024–2029. [Google Scholar] [CrossRef]
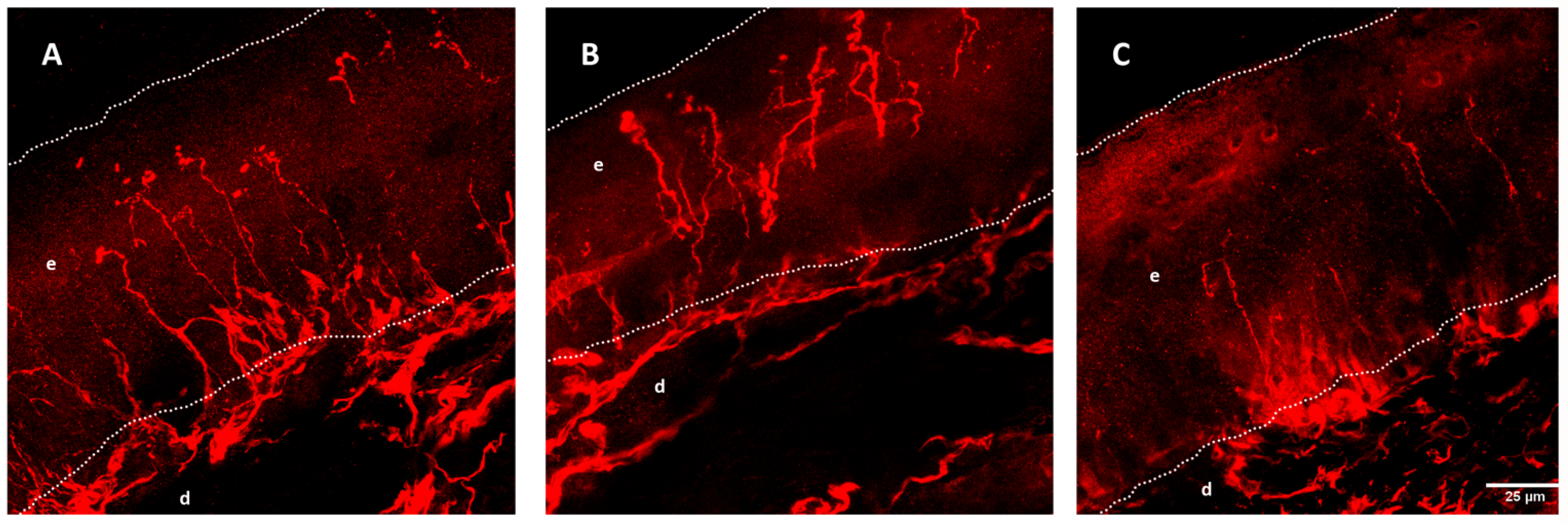
- Nolano, M.; Provitera, V.; Manganelli, F.; Iodice, R.; Caporaso, G.; Stancanelli, A.; Marinou, K.; Lanzillo, B.; Santoro, L.; Mora, G. Non-motor involvement in amyotrophic lateral sclerosis: New insight from nerve and vessel analysis in skin biopsy. Neuropathol. Appl. Neurobiol. 2017, 43, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Isak, B.; Pugdahl, K.; Karlsson, P.; Tankisi, H.; Finnerup, N.B.; Furtula, J.; Johnsen, B.; Sunde, N.; Jakobsen, J.; Fuglsang-Frederiksen, A. Quantitative sensory testing and structural assessment of sensory nerve fibres in amyotrophic lateral sclerosis. J. Neurol. Sci. 2017, 373, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Dalla Bella, E.; Lombardi, R.; Porretta-Serapiglia, C.; Ciano, C.; Gellera, C.; Pensato, V.; Cazzato, D.; Lauria, G. Amyotrophic lateral sclerosis causes small fiber pathology. Eur. J. Neurol. 2016, 23, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.A.; Herrando-Grabulosa, M.; Velasco, R.; Blasco, I.; Povedano, M.; Navarro, X. TDP-43 Cytoplasmic Translocation in the Skin Fibroblasts of ALS Patients. Cells 2022, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Liu, W.; Li, Y.; Sun, B.; Li, Y.; Yang, F.; Wang, H.; Li, M.; Cui, F.; Huang, X. Cutaneous somatic and autonomic nerve TDP-43 deposition in amyotrophic lateral sclerosis. J. Neurol. 2018, 265, 1753–1763. [Google Scholar] [CrossRef]
- Genç, B.; Lagrimas, A.K.B.; Kuru, P.; Hess, R.; Tu, M.W.; Menichella, D.M.; Miller, R.J.; Paller, A.S.; Ozdinler, P.H. Visualization of sensory neurons and their projections in an upper motor neuron reporter line. PLoS ONE 2015, 10, e0132815. [Google Scholar] [CrossRef]
- Rubio, M.A.; Herrando-Grabulosa, M.; Vilches, J.J.; Navarro, X. Involvement of sensory innervation in the skin of SOD1(G93A) ALS mice. J. Peripher. Nerv. Syst. 2016, 21, 88–95. [Google Scholar] [CrossRef]
- Ferrari, G.; Grisan, E.; Scarpa, F.; Efazio, R.; Ecomola, M.; Quattrini, A.; Ecomi, G.; Rama, P.; Eriva, N. Corneal confocal microscopy reveals trigeminal small sensory fiber neuropathy in amyotrophic lateral sclerosis. Front. Aging Neurosci. 2014, 6, 278. [Google Scholar] [CrossRef]
- Amin, M.R.; Harris, D.; Cassel, S.G.; Grimes, E.; Heiman-Patterson, T. Sensory testing in the assessment of laryngeal sensation in patients with amyotrophic lateral sclerosis. Ann. Otol. Rhinol. Laryngol. 2006, 115, 528–534. [Google Scholar] [CrossRef]
- Tabor-Gray, L.; Vasilopoulos, T.; Wheeler-Hegland, K.; Wymer, J.; Plowman, E.K. Reflexive airway sensorimotor responses in individuals with amyotrophic lateral sclerosis. Dysphagia 2021, 36, 574–582. [Google Scholar] [CrossRef]
- Gwathmey, K.G. Sensory neuronopathies. Muscle Nerve 2016, 53, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Averbuch-Heller, L.; Helmchen, C.; Horn, A.K.; Leigh, R.J.; Büttner-Ennerver, J.A. Slow vertical saccades in motor neuron disease: Correlation of structure and function. Ann. Neurol. 1998, 44, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Gizzi, M.; DiRocco, A.; Sivak, M.; Cohen, B. Ocular motor function in motor neuron disease. Neurology 1992, 42, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Puligheddu, M.; Congiu, P.; Aricò, D.; Rundo, F.; Borghero, G.; Marrosu, F.; Fantini, M.L.; Ferri, R. Isolated rapid eye movement sleep without atonia in amyotrophic lateral sclerosis. Sleep Med. 2016, 26, 16–22. [Google Scholar] [CrossRef]
- Münte, T.F.; Tröger, M.C.; Nusser, I.; Wieringa, B.M.; Johannes, S.; Matzke, M.; Dengler, R. Alteration of early components of the visual evoked potential in amyotrophic lateral sclerosis. J. Neurol. 1998, 245, 206–210. [Google Scholar] [CrossRef]
- Rojas, P.; Ramírez, A.I.; Fernández-Albarral, J.A.; López-Cuenca, I.; Salobrar-García, E.; Cadena, M.; Elvira-Hurtado, L.; Salazar, J.J.; De Hoz, R.; Ramírez, J.M. Amyotrophic lateral sclerosis: A neurodegenerative motor neuron disease with ocular involvement. Front. Neurosci. 2020, 14, 566858. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, H.; Fan, D.; Wang, W. Comparison of optical coherence tomography findings and visual field changes in patients with primary open-angle glaucoma and amyotrophic lateral sclerosis. J. Clin. Neurosci. 2018, 48, 233–237. [Google Scholar] [CrossRef]
- Volpe, N.J.; Simonett, J.; Fawzi, A.A.; Siddique, T. Ophthalmic manifestations of amyotrophic lateral sclerosis (an American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc. 2015, 113, T12. [Google Scholar]
- Moss, H.E.; Samelson, M.; Mohan, G.; Jiang, Q.L. High and low contrast visual acuity are not affected in amyotrophic lateral sclerosis. PLoS ONE 2016, 11, e0168714. [Google Scholar] [CrossRef]
- Rojas, P.; de Hoz, R.; Ramírez, A.; Ferreras, A.; Salobrar-Garcia, E.; Muñoz-Blanco, J.; Urcelay-Segura, J.; Salazar, J.; Ramírez, J. Changes in retinal OCT and their correlations with neurological disability in early ALS patients, a follow-up study. Brain Sci. 2019, 9, 337. [Google Scholar] [CrossRef]
- Moss, H.E.; McCluskey, L.; Elman, L.; Hoskins, K.; Talman, L.; Grossman, M.; Balcer, L.J.; Galetta, S.L.; Liu, G.T. Cross-sectional evaluation of clinical neuro-ophthalmic abnormalities in an amyotrophic lateral sclerosis population. J. Neurol. Sci. 2012, 314, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, A.A.; Simonett, J.M.; Purta, P.; Moss, H.; Lowry, J.L.; Deng, H.-X.; Siddique, N.; Sufit, R.; Bigio, E.H.; Volpe, N.J.; et al. Clinicopathologic report of ocular involvement in ALS patients with C9orf72 mutation. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, A.; Mazzalovo, E.; Locatelli, C.; Zibetti, A.; Zaffaroni, M.; Montanini, R. Multimodality evoked potentials in amyotrophic lateral sclerosis. Acta Neurol. Scand. 1989, 79, 353–356. [Google Scholar] [CrossRef]
- Palma, V.; Guadagnino, M.; Brescia Morra, V.; Nolfe, G. Multimodality evoked potentials in sporadic amyotrophic lateral sclerosis: A statistical approach. Electromyogr. Clin. Neurophysiol. 1993, 33, 167–171. [Google Scholar] [PubMed]
- Matheson, J.K.; Harrington, H.J.; Hallett, M. Abnormalities of multimodality evoked potentials in amyotrophic lateral sclerosis. Arch. Neurol. 1986, 43, 338–340. [Google Scholar] [CrossRef]
- Lulé, D.; Diekmann, V.; Müller, H.P.; Kassubek, J.; Ludolph, A.C.; Birbaumer, N. Neuroimaging of multimodal sensory stimulation in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Ringer, C.; Weihe, E.; Schütz, B. SOD1G93A mutant mice develop a neuroinflammation-independent dendropathy in excitatory neuronal subsets of the olfactory bulb and retina. J. Neuropathol. Exp. Neurol. 2017, 76, 769–778. [Google Scholar] [CrossRef]
- Kushner, P.D.; Stephenson, D.T.; Wright, S. Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J. Neuropathol. Exp. Neurol. 1991, 50, 263–277. [Google Scholar] [CrossRef]
- Albrecht, P.; Müller, A.-K.; Südmeyer, M.; Ferrea, S.; Ringelstein, M.; Cohn, E.; Aktas, O.; Dietlein, T.; Lappas, A.; Foerster, A.; et al. Optical coherence tomography in parkinsonian syndromes. PLoS ONE 2012, 7, e34891. [Google Scholar] [CrossRef]
- Siger, M.; Dziegielewski, K.; Jasek, L.; Bieniek, M.; Nicpan, A.; Nawrocki, J.; Selmaj, K. Optical coherence tomography in multiple sclerosis: Thickness of the retinal nerve fiber layer as a potential measure of axonal loss and brain atrophy. J. Neurol. 2008, 255, 1555–1560. [Google Scholar] [CrossRef]
- Mukherjee, N.; McBurney-Lin, S.; Kuo, A.; Bedlack, R.; Tseng, H. Retinal thinning in amyotrophic lateral sclerosis patients without ophthalmic disease. PLoS ONE 2017, 12, e0185242. [Google Scholar] [CrossRef] [PubMed]
- Rohani, M.; Meysamie, A.; Zamani, B.; Sowlat, M.M.; Akhoundi, F.H. Reduced retinal nerve fiber layer (RNFL) thickness in ALS patients: A window to disease progression. J. Neurol. 2018, 265, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Hübers, A.; Müller, H.P.; Dreyhaupt, J.; Böhm, K.; Lauda, F.; Tumani, H.; Kassubek, J.; Ludolph, A.C.; Pinkhardt, E.H. Retinal involvement in amyotrophic lateral sclerosis: A study with optical coherence tomography and diffusion tensor imaging. J. Neural Transm. 2016, 123, 281–287. [Google Scholar] [CrossRef]
- Simonett, J.M.; Huang, R.; Siddique, N.; Farsiu, S.; Siddique, T.; Volpe, N.J.; Fawzi, A.A. Macular sub-layer thinning and association with pulmonary function tests in Amyotrophic Lateral Sclerosis. Sci. Rep. 2016, 6, 29187. [Google Scholar] [CrossRef] [PubMed]
- Cerveró, A.; Casado, A.; Riancho, J. Retinal changes in amyotrophic lateral sclerosis: Looking at the disease through a new window. J. Neurol. 2021, 268, 2083–2089. [Google Scholar] [CrossRef]
- Roth, N.M.; Saidha, S.; Zimmermann, H.; Brandt, A.U.; Oberwahrenbrock, T.; Maragakis, N.J.; Tumani, H.; Ludolph, A.C.; Meyer, T.; Calabresi, P.; et al. Optical coherence tomography does not support optic nerve involvement in amyotrophic lateral sclerosis. Eur. J. Neurol. 2013, 20, 1170–1176. [Google Scholar] [CrossRef]
- Ward, M.E.; Taubes, A.; Chen, R.; Miller, B.L.; Sephton, C.F.; Gelfand, J.M.; Minami, S.; Boscardin, J.; Martens, L.H.; Seeley, W.W.; et al. Early retinal neurodegeneration and impaired Ran-mediated nuclear import of TDP-43 in progranulin-deficient FTLD. J. Exp. Med. 2014, 211, 1937–1945. [Google Scholar] [CrossRef]
- Sharma, K.; Amin, M.A.M.; Gupta, N.; Zinman, L.; Zhou, X.; Irving, H.; Yücel, Y. Retinal spheroids and axon pathology identified in amyotrophic lateral sclerosis. Investig. Ophthalmol. Vis. Sci. 2020, 61, 30. [Google Scholar] [CrossRef]
- Cho, K.I.; Yoon, D.; Yu, M.; Peachey, N.S.; Ferreira, P.A. Microglial activation in an amyotrophic lateral sclerosis-like model caused by Ranbp2 loss and nucleocytoplasmic transport impairment in retinal ganglion neurons. Cell Mol. Life Sci. 2019, 76, 3407–3432. [Google Scholar] [CrossRef]
- Ferreira, P.A. The coming-of-age of nucleocytoplasmic transport in motor neuron disease and neurodegeneration. Cell Mol. Life Sci. 2019, 76, 2247–2273, Correction in Cell Mol. Life Sci. 2019, 76, 2275. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Hashemi, S.H.; Anderson, S.K.; Huang, Y.; Moreira, M.C.; Lynch, D.R.; Glass, I.A.; Chance, P.F.; Bennett, C.L. Senataxin, the yeast Sen1p orthologue: Characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol. Dis. 2006, 23, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Abdelhak, A.; Hübers, A.; Böhm, K.; Ludolph, A.C.; Kassubek, J.; Pinkhardt, E.H. In vivo assessment of retinal vessel pathology in amyotrophic lateral sclerosis. J. Neurol. 2018, 265, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Boyce, J.M.; Shone, G.R. Effects of ageing on smell and taste. Postgrad. Med. J. 2006, 82, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Landis, B.N.; Konnerth, C.G.; Hummel, T. A study on the frequency of olfactory dysfunction. Laryngoscope 2004, 114, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Rouby, C.; Thomas-Danguin, T.; Vigouroux, M.; Ciuperca, G.; Jiang, T.; Alexanian, J.; Barges, M.; Gallice, I.; Degraix, J.-L.; Sicard, G. The lyon clinical olfactory test: Validation and measurement of hyposmia and anosmia in healthy and diseased populations. Int. J. Otolaryngol. 2011, 2011, 203805. [Google Scholar] [CrossRef]
- Growdon, M.E.; Schultz, A.P.; Dagley, A.S.; Amariglio, R.E.; Hedden, T.; Rentz, D.M.; Johnson, K.A.; Sperling, R.A.; Albers, M.W.; Marshall, G.A. Odor identification and Alzheimer disease biomarkers in clinically normal elderly. Neurology 2015, 84, 2153–2160. [Google Scholar] [CrossRef]
- Jennings, D.; Siderowf, A.; Stern, M.; Seibyl, J.; Eberly, S.; Oakes, D.; Marek, K. Imaging prodromal Parkinson disease: The Parkinson Associated Risk Syndrome Study. Neurology 2014, 83, 1739–1746. [Google Scholar] [CrossRef]
- Elian, M. Olfactory impairment in motor neuron disease: A pilot study. J. Neurol. Neurosurg. Psychiatry 1991, 54, 927–928. [Google Scholar] [CrossRef][Green Version]
- Ahlskog, J.E.; Waring, S.C.; Petersen, R.C.; Esteban-Santillan, C.; Craig, U.-K.; O’Brien, P.C.; Plevak, M.F.; Kurland, L.T. Olfactory dysfunction in Guamanian ALS, parkinsonism, and dementia. Neurology 1998, 51, 1672–1677. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Shephard, B.C.; Geddes, J.F.; Body, G.D.; Martin, J.E. Olfactory disorder in motor neuron disease. Exp. Neurol. 1998, 150, 248–253. [Google Scholar] [CrossRef]
- Takeda, T.; Iijima, M.; Uchihara, T.; Ohashi, T.; Seilhean, D.; Duyckaerts, C.; Uchiyama, S. TDP-43 pathology progression along the olfactory pathway as a possible substrate for olfactory impairment in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2015, 74, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Viguera, C.; Wang, J.; Mosmiller, E.; Cerezo, A.; Maragakis, N.J. Olfactory dysfunction in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Watanabe, H.; Ogura, A.; Ohdake, R.; Kato, T.; Kawabata, K.; Hara, K.; Nakamura, R.; Atsuta, N.; Epifanio, B.; et al. Clinicoradiological features in amyotrophic lateral sclerosis patients with olfactory dysfunction. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Pilotto, A.; Rossi, F.; Rinaldi, F.; Compostella, S.; Cosseddu, M.; Borroni, B.; Filosto, M.; Padovani, A. Exploring olfactory function and its relation with behavioral and cognitive impairment in amyotrophic lateral sclerosis patients: A cross-sectional study. Neurodegener. Dis. 2016, 16, 411–416. [Google Scholar] [CrossRef]
- Günther, R.; Schrempf, W.; Hähner, A.; Hummel, T.; Wolz, M.; Storch, A.; Hermann, A. Impairment in respiratory function contributes to olfactory impairment in amyotrophic lateral sclerosis. Front. Neurol. 2018, 9, 79. [Google Scholar] [CrossRef]
- Lang, C.J.; Schwandner, K.; Hecht, M. Do patients with motor neuron disease suffer from disorders of taste or smell? Amyotroph. Lateral Scler. 2011, 12, 368–371. [Google Scholar] [CrossRef]
- Machts, J.; Loewe, K.; Kaufmann, J.; Jakubiczka, S.; Abdulla, S.; Petri, S.; Dengler, R.; Heinze, H.-J.; Vielhaber, S.; Schoenfeld, M.A.; et al. Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology 2015, 85, 1301–1309. [Google Scholar] [CrossRef]
- Pinkhardt, E.H.; van Elst, L.T.; Ludolph, A.C.; Kassubek, J. Amygdala size in amyotrophic lateral sclerosis without dementia: An in vivo study using MRI volumetry. BMC Neurol. 2006, 6, 48. [Google Scholar] [CrossRef]
- Takeda, T.; Uchihara, T.; Kawamura, S.; Ohashi, T. Olfactory dysfunction related to TDP-43 pathology in amyotrophic lateral sclerosis. Clin. Neuropathol. 2014, 33, 65–67. [Google Scholar] [CrossRef]
- Eisen, A.; Braak, H.; Del Tredici, K.; Lemon, R.; Ludolph, A.C.; Kiernan, M.C. Cortical influences drive amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 917–924. [Google Scholar] [CrossRef]
- Rey, N.L.; Wesson, D.W.; Brundin, P. The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol. Dis. 2018, 109 Pt B, 226–248. [Google Scholar] [CrossRef]
- Brozzetti, L.; Sacchetto, L.; Cecchini, M.P.; Avesani, A.; Perra, D.; Bongianni, M.; Portioli, C.; Scupoli, M.; Ghetti, B.; Monaco, S.; et al. Neurodegeneration-associated proteins in human olfactory neurons collected by nasal brushing. Front. Neurosci. 2020, 14, 145. [Google Scholar] [CrossRef] [PubMed]
- Lachén-Montes, M.; Mendizuri, N.; Ausin, K.; Andrés-Benito, P.; Ferrer, I.; Fernández-Irigoyen, J.; Santamaría, E. Amyotrophic lateral sclerosis is accompanied by protein derangements in the olfactory bulb-tract axis. Int. J. Mol. Sci. 2020, 21, 8311. [Google Scholar] [CrossRef] [PubMed]
- García-Escudero, V.; Rosales, M.; Muñoz, J.L.; Scola, E.; Medina, J.; Khalique, H.; Garaulet, G.; Rodriguez, A.; Lim, F. Patient-derived olfactory mucosa for study of the non-neuronal contribution to amyotrophic lateral sclerosis pathology. J. Cell Mol. Med. 2015, 19, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Stifani, S. Roles of Runx genes in nervous system development. Adv. Exp. Med. Biol. 2017, 962, 103–116. [Google Scholar] [PubMed]
- Steinbach, S.; Hundt, W.; Vaitl, A.; Heinrich, P.; Förster, S.; Bürger, K.; Zahnert, T. Taste in mild cognitive impairment and Alzheimer’s disease. J. Neurol. 2010, 257, 238–246. [Google Scholar] [CrossRef]
- Shah, M.; Deeb, J.; Fernando, M.; Noyce, A.; Visentin, E.; Findley, L.J.; Hawkes, C.H. Abnormality of taste and smell in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 232–237. [Google Scholar] [CrossRef]
- Heckmann, J.G.; Stössel, C.; Lang, C.J.; Neundörfer, B.; Tomandl, B.; Hummel, T. Taste disorders in acute stroke: A prospective observational study on taste disorders in 102 stroke patients. Stroke 2005, 36, 1690–1694. [Google Scholar] [CrossRef]
- Kim, J.S.; Choi-Kwon, S.; Kwon, S.U.; Kwon, J.H. Taste perception abnormalities after acute stroke in postmenopausal women. J. Clin. Neurosci. 2009, 16, 797–801. [Google Scholar] [CrossRef]
- Scinska, A.; Sienkiewicz-Jarosz, H.; Kuran, W.; Ryglewicz, D.; Rogowski, A.; Wrobel, E.; Korkosz, A.; Kukwa, A.; Kostowski, W.; Bienkowski, P. Depressive symptoms and taste reactivity in humans. Physiol. Behav. 2004, 82, 899–904. [Google Scholar] [CrossRef]
- Imoscopi, A.; Inelmen, E.M.; Sergi, G.; Miotto, F.; Manzato, E. Taste loss in the elderly: Epidemiology, causes and consequences. Aging Clin. Exp. Res. 2012, 24, 570–579. [Google Scholar] [PubMed]
- Tarlarini, C.; Greco, L.C.; Lizio, A.; Gerardi, F.; Sansone, V.A.; Lunetta, C. Taste changes in amyotrophic lateral sclerosis and effects on quality of life. Neurol. Sci. 2019, 40, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Ashary, A.A.; Patel, D.N.; Hirsch, A.R. 101 Amyotrophic lateral sclerosis (ALS)—Not just a motor disease? Isolated bitter and sweet taste loss in ALS. CNS Spectr. 2020, 25, 266. [Google Scholar] [CrossRef][Green Version]
- Nishihira, Y.; Tan, C.-F.; Onodera, O.; Toyoshima, Y.; Yamada, M.; Morita, T.; Nishizawa, M.; Kakita, A.; Takahashi, H. Sporadic amyotrophic lateral sclerosis: Two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol. 2008, 116, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, T.; Hokonohara, T.; Yamasaki, R.; Miura, S.; Kikuchi, H.; Iwaki, A.; Tashiro, H.; Furuya, H.; Nagara, Y.; Ohyagi, Y.; et al. Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol. 2010, 119, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, N.; Nonami, J.; Kodaira, M.; Yoshida, K.; Sekijima, Y. Taste disorder in facial onset sensory and motor neuronopathy: A case report. BMC Neurol. 2020, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, K.; Sasaki, K.; Tateishi, T.; Yamasaki, R.; Hayashi, S.; Sakae, N.; Ohyagi, Y.; Iwaki, T.; Kira, J.-I. TAR DNA-binding protein 43 pathology in a case clinically diagnosed with facial-onset sensory and motor neuronopathy syndrome: An autopsied case report and a review of the literature. J. Neurol. Sci. 2013, 332, 148–153. [Google Scholar] [CrossRef]
- Ziso, B.; Williams, T.L.; Walters, R.J.; Jaiser, S.R.; Attems, J.; Wieshmann, U.C.; Larner, A.J.; Jacob, A. Facial onset sensory and motor neuronopathy: Further evidence for a TDP-43 proteinopathy. Case Rep. Neurol. 2015, 7, 95–100. [Google Scholar] [CrossRef]
- Rossor, A.M.; Jaunmuktane, Z.; Rossor, M.N.; Hoti, G.; Reilly, M.M. TDP43 pathology in the brain, spinal cord, and dorsal root ganglia of a patient with FOSMN. Neurology 2019, 92, e951–e956, Correction in Neurology 2019, 93, e88. [Google Scholar]
- Low, P.A.; Ahlskog, J.E.; Petersen, R.C.; Waring, S.C.; Esteban-Santillan, C.; Kurland, L.T. Autonomic failure in Guamanian neurodegenerative disease. Neurology 1997, 49, 1031–1034. [Google Scholar] [CrossRef]
- Piccione, E.A.; Sletten, D.M.; Staff, N.P.; Low, P.A. Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve 2015, 51, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Hayashi, H.; Kato, S.; Hayashi, M.; Tanabe, H.; Oda, M. Circulatory collapse and sudden death in respirator-dependent amyotrophic lateral sclerosis. J. Neurol. Sci. 1994, 124, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Baltadzhieva, R.; Gurevich, T.; Korczyn, A.D. Autonomic impairment in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2005, 18, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, R.; Ferreira, C.; Valenti, V.; Garner, D.; Macedo, H.; Oliveira, A.; Leitão, F.; de Abreu, L. Complexity measures of heart-rate variability in amyotrophic lateral sclerosis with alternative pulmonary capacities. Entropy 2021, 23, 159. [Google Scholar] [CrossRef]
- Pimentel, R.M.M.; Macedo, H.; Valenti, V.; Rocha, F.O.; de Abreu, L.C.; Monteiro, C.B.D.M.; Ferreira, C. Decreased heart rate variability in individuals with amyotrophic lateral sclerosis. Respir. Care 2019, 64, 1088–1095. [Google Scholar] [CrossRef]
- Sachs, C.; Conradi, S.; Kaijser, L. Autonomic function in amyotrophic lateral sclerosis: A study of cardiovascular responses. Acta Neurol. Scand. 1985, 71, 373–378. [Google Scholar] [CrossRef]
- Kandinov, B.; Korczyn, A.D.; Rabinowitz, R.; Nefussy, B.; Drory, V.E. Autonomic impairment in a transgenic mouse model of amyotrophic lateral sclerosis. Auton. Neurosci. 2011, 159, 84–89. [Google Scholar] [CrossRef]
- Kandinov, B.; Drory, V.E.; Tordjman, K.; Korczyn, A.D. Blood pressure measurements in a transgenic SOD1-G93A mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 509–513. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yamada, M.; Koumura, A.; Sakurai, T.; Hayashi, Y.; Kimura, A.; Hozumi, I.; Inuzuka, T. Cardiac sympathetic function in the patients with amyotrophic lateral sclerosis: Analysis using cardiac [123I] MIBG scintigraphy. J. Neurol. 2013, 260, 2380–2386. [Google Scholar] [CrossRef]
- Beck, M.; Flachenecker, P.; Magnus, T.; Giess, R.; Reiners, K.; Toyka, K.V.; Naumann, M. Autonomic dysfunction in ALS: A preliminary study on the effects of intrathecal BDNF. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2005, 6, 100–103. [Google Scholar] [CrossRef]
- Vecchia, L.D.; De Maria, B.; Marinou, K.; Sideri, R.; Lucini, A.; Porta, A.; Mora, G. Cardiovascular neural regulation is impaired in amyotrophic lateral sclerosis patients. A study by spectral and complexity analysis of cardiovascular oscillations. Physiol. Meas. 2015, 36, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Tutaj, M.; Miller, M.; Tomik, B.; Golenia, A.; Stanuszek, A.; BŁońska, K.; SŁowik, A. Sympathetic vascular response to facial cooling is increased in flail phenotypes of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kato, S.; Hayashi, M.; Hayashi, H.; Tanabe, H. Amyotrophic lateral sclerosis with hypertensive attacks: Blood pressure changes in response to drug administration. Clin. Auton. Res. 1996, 6, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Hirano, M.; Udaka, F.; Shimada, K.; Oda, M.; Kubori, T.; Nishinaka, K.; Tsujimura, T.; Izumi, Y.; Konishi, N.; et al. Sympathetic disturbances increase risk of sudden cardiac arrest in sporadic ALS. J. Neurol. Sci. 2007, 254, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Drusehky, A.; Spitzer, A.; Platseh, G.; Claus, D.; Feistel, H.; Druschky, K.; Hilz, M.-J.; Neundörfer, B. Cardiac sympathetic denervation in early stages of amyotrophic lateral sclerosis demonstrated by 123I-MIBG-SPECT. Acta Neurol. Scand. 1999, 99, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Karlsborg, M.; Andersen, E.B.; Wiinberg, N.; Gredal, O.; Jørgensen, L.; Mehlsen, J. Sympathetic dysfunction of central origin in patients with ALS. Eur. J. Neurol. 2003, 10, 229–234. [Google Scholar] [CrossRef]
- Provinciali, L.; Cangiotti, A.; Tulli, D.; Carboni, V.; Cinti, S. Skin abnormalities and autonomic involvement in the early stage of amyotrophic lateral sclerosis. J. Neurol. Sci. 1994, 126, 54–61. [Google Scholar] [CrossRef]
- Oey, P.L.; Vos, P.E.; Wieneke, G.H.; Wokke, J.H.; Blankestijn, P.J.; Karemaker, J.M. Subtle involvement of the sympathetic nervous system in amyotrophic lateral sclerosis. Muscle Nerve 2002, 25, 402–408. [Google Scholar] [CrossRef]
- Pavlovic, S.; Stevic, Z.; Milovanovic, B.; Milicic, B.; Rakocevic-Stojanovic, V.; Lavrnic, D.; Apostolski, S. Impairment of cardiac autonomic control in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 272–276. [Google Scholar] [CrossRef]
- Merico, A.; Cavinato, M. Autonomic dysfunction in the early stage of ALS with bulbar involvement. Amyotroph. Lateral Scler. 2011, 12, 363–367. [Google Scholar] [CrossRef]
- De Maria, B.; Bari, V.; Marchi, A.; Barbic, F.; Furlan, R.; Mora, G.; Dalla Vecchia, L.; Porta, A. Cardiovascular control indexes in amyotrophic lateral sclerosis patients and their relation with clinical markers. In Proceedings of the 2015 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Milan, Italy, 25–29 August 2015; Volume 2015, pp. 2055–2058. [Google Scholar]
- Pisano, F.; Miscio, G.; Mazzuero, G.; Lanfranchi, P.; Colombo, R.; Pinelli, P. Decreased heart rate variability in amyotrophic lateral sclerosis. Muscle Nerve 1995, 18, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.; Diehl, R.R.; Berlit, P. Reduced baroreflex sensitivity and cardiorespiratory transfer in amyotrophic lateral sclerosis. Electroencephalogr. Clin. Neurophysiol. 1998, 109, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Congiu, P.; Mariani, S.; Milioli, G.; Parrino, L.; Tamburrino, L.; Borghero, G.; Defazio, G.; Pereira, B.; Fantini, M.L.; Puligheddu, M. Sleep cardiac dysautonomia and EEG oscillations in amyotrophic lateral sclerosis. Sleep 2019, 42, zsz164. [Google Scholar] [CrossRef]
- Donadio, V.; Liguori, R. Microneurographic recording from unmyelinated nerve fibers in neurological disorders: An update. Clin. Neurophysiol. 2015, 126, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Shindo, K. [Sympathetic neurograms in patients with neurodegenerative disorders—An overview]. Brain Nerve 2009, 61, 263–269. [Google Scholar] [PubMed]
- Beck, M.; Giess, R.; Magnus, T.; Puls, I.; Reiners, K.; Toyka, K.V.; Naumann, M. Progressive sudomotor dysfunction in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2002, 73, 68–70. [Google Scholar] [CrossRef]
- Santos-Bento, M.; de Carvalho, M.; Evangelista, T.; Sales Luís, M.L. Sympathetic sudomotor function and amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2001, 2, 105–108. [Google Scholar] [CrossRef]
- Shindo, K.; Tsunoda, S.; Shiozawa, Z. Increased sympathetic outflow to muscles in patients with amyotrophic lateral sclerosis: A comparison with other neuromuscular patients. J. Neurol. Sci. 1995, 134, 57–60. [Google Scholar] [CrossRef]
- Shindo, K.; Watanabe, H.; Ohta, E.; Nagasaka, T.; Shiozawa, Z.; Takiyama, Y. Sympathetic sudomotor neural function in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 39–44. [Google Scholar] [CrossRef]
- Hu, F.; Jin, J.; Qu, Q.; Dang, J. Sympathetic skin response in amyotrophic lateral sclerosis. J. Clin. Neurophysiol. 2016, 33, 60–65. [Google Scholar] [CrossRef]
- Dettmers, C.; Fatepour, D.; Faust, H.; Jerusalem, F. Sympathetic skin response abnormalities in amyotrophic lateral sclerosis. Muscle Nerve 1993, 16, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Barron, S.A.; Mazliah, J.; Bental, E. Sympathetic cholinergic dysfunction in amyotrophic lateral sclerosis. Acta Neurol. Scand. 1987, 75, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Masur, H.; Schulte-Oversohl, U.; Papke, K.; Oberwittler, C.; Vollmer, J. Sympathetic skin response in patients with amyotrophic lateral sclerosis. Funct. Neurol. 1995, 10, 131–135. [Google Scholar] [PubMed]
- Miscio, G.; Pisano, F. Sympathetic skin response in amyotrophic lateral sclerosis. Acta Neurol. Scand. 1998, 98, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Kihara, M.; Takahashi, A.; Sugenoya, J.; Kihara, Y.; Watanabe, H. Sudomotor dysfunction in amyotrophic lateral sclerosis. Funct. Neurol. 1994, 9, 193–197. [Google Scholar] [PubMed]
- Shindo, K.; Shimokawa, C.; Watanabe, H.; Iida, H.; Ohashi, K.; Nitta, K.; Nagasaka, T.; Tsunoda, S.; Shiozawa, Z. Chronological changes of sympathetic outflow to muscles in amyotrophic lateral sclerosis. J. Neurol. Sci. 2004, 227, 79–84. [Google Scholar] [CrossRef]
- Takeda, T.; Iijima, M.; Shimizu, Y.; Yoshizawa, H.; Miyashiro, M.; Onizuka, H.; Yamamoto, T.; Nishiyama, A.; Suzuki, N.; Aoki, M.; et al. p.N345K mutation in TARDBP in a patient with familial amyotrophic lateral sclerosis: An autopsy case. Neuropathology 2019, 39, 286–293. [Google Scholar] [CrossRef]
- Nübling, G.S.; Mie, E.; Bauer, R.M.; Hensler, M.; Lorenzl, S.; Hapfelmeier, A.; Irwin, D.E.; Borasio, G.D.; Winkler, A.S. Increased prevalence of bladder and intestinal dysfunction in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 174–179. [Google Scholar] [CrossRef]
- Charchaflie, R.J.; Bustos Fernandez, L.; Perec, C.J.; Gonzalez, E.; Marzi, A. Functional studies of the parotid and pancreas glands in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 1974, 37, 863–867. [Google Scholar] [CrossRef]
- Giess, R.; Werner, E.; Beck, M.; Reiners, C.; Toyka, K.V.; Naumann, M. Impaired salivary gland function reveals autonomic dysfunction in amyotrophic lateral sclerosis. J. Neurol. 2002, 249, 1246–1249. [Google Scholar] [CrossRef]
- Copperman, R. Letter: Decreased lacrimation in amyotrophic lateral sclerosis. JAMA 1974, 230, 536. [Google Scholar] [CrossRef]
- Toepfer, M.; Folwaczny, C.; Klauser, A.; Riepl, R.L.; Müller-Felber, W.; Pongratz, D. Gastrointestinal dysfunction in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 1999, 1, 15–19. [Google Scholar] [CrossRef]
- Toepfer, M.; Folwaczny, C.; Lochmüller, H.; Schroeder, M.; Riepl, R.; Pongratz, D.; Müller-Felber, W. Noninvasive (13)C-octanoic acid breath test shows delayed gastric emptying in patients with amyotrophic lateral sclerosis. Digestion 1999, 60, 567–571. [Google Scholar] [CrossRef]
- Toepfer, M.; Schroeder, M.; Klauser, A.; Lochmüller, H.; Hirschmann, M.; Riepl, R.L.; Pongratz, D.; Müller-Felber, W. Delayed colonic transit times in amyotrophic lateral sclerosis assessed with radio-opaque markers. Eur. J. Med. Res. 1997, 2, 473–476. [Google Scholar]
- Smith, A.w.; Mulder, D.w.; Code, C.f. Esophageal motility in amyotrophic lateral sclerosis. Proc. Staff Meet Mayo Clin. 1957, 32, 438–441. [Google Scholar]
- Yamada, T.; Itoh, K.; Matsuo, K.; Yamamoto, Y.; Hosokawa, Y.; Koizumi, T.; Shiga, K.; Mizuno, T.; Nakagawa, M.; Fushiki, S. Concomitant alpha-synuclein pathology in an autopsy case of amyotrophic lateral sclerosis presenting with orthostatic hypotension and cardiac arrests. Neuropathology 2014, 34, 164–169. [Google Scholar] [CrossRef]
- Bogucki, A.; Salvesen, R. Sympathetic iris function in amyotrophic lateral sclerosis. J. Neurol. 1987, 234, 185–186. [Google Scholar] [CrossRef]
- Ohno, T.; Shimizu, T.; Kato, S.; Hayashi, H.; Hirai, S. Effect of tamsulosin hydrochloride on sympathetic hyperactivity in amyotrophic lateral sclerosis. Auton. Neurosci. 2001, 88, 94–98. [Google Scholar] [CrossRef]
- Nagai, M.; Hoshide, S.; Kario, K. The insular cortex and cardiovascular system: A new insight into the brain-heart axis. J. Am. Soc. Hypertens. 2010, 4, 174–182. [Google Scholar] [CrossRef]
- Hayashi, H.; Kato, S. Total manifestations of amyotrophic lateral sclerosis. ALS in the totally locked-in state. J. Neurol. Sci. 1989, 93, 19–35. [Google Scholar] [CrossRef]
- Kato, S.; Oda, M.; Tanabe, H. Diminution of dopaminergic neurons in the substantia nigra of sporadic amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 1993, 19, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kawata, A.; Kato, S.; Hayashi, M.; Takamoto, K.; Hirai, S.; Yamaguchi, S.; Komori, T.; Oda, M. Autonomic failure in ALS with a novel SOD1 gene mutation. Neurology 2000, 54, 1534–1537. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Mochizuki, Y.; Koide, R.; Kawata, A.; Homma, T.; Shimizu, T.; Komori, T.; Isozaki, E. A Japanese familial ALS patient with autonomic failure and a p.Cys146Arg mutation in the gene for SOD1 (SOD1). Neuropathology 2016, 36, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Hayashi, M.; Kawata, A.; Mizutani, T.; Watabe, K.; Matsubara, S. A morphometric study of the vagus nerve in amyotropic lateral sclerosis with circulatory collapse. Amyotroph. Lateral Scler. 2011, 12, 356–362. [Google Scholar] [CrossRef]
- Konno, H.; Yamamoto, T.; Iwasaki, Y.; Iizuka, H. Shy-Drager syndrome and amyotrophic lateral sclerosis. Cytoarchitectonic and morphometric studies of sacral autonomic neurons. J. Neurol. Sci. 1986, 73, 193–204. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Duchen, L.W. A quantitative study of intermediolateral column cells in motor neuron disease and the Shy-Drager syndrome. J. Neurol. Neurosurg. Psychiatry 1985, 48, 1103–1106. [Google Scholar] [CrossRef]
- Itoh, T.; Sobue, G.; Ken, E.; Mitsuma, T.; Takahashi, A.; Trojanowski, J.Q. Phosphorylated high molecular weight neurofilament protein in the peripheral motor, sensory and sympathetic neuronal perikarya: System-dependent normal variations and changes in amyotrophic lateral sclerosis and multiple system atrophy. Acta Neuropathol. 1992, 83, 240–245. [Google Scholar] [CrossRef]
- Takahashi, H.; Oyanagi, K.; Ikuta, F. The intermediolateral nucleus in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 1993, 86, 190–192. [Google Scholar] [CrossRef]
- Kandinov, B.; Grigoriadis, N.C.; Touloumi, O.; Drory, V.E.; Offen, D.; Korczyn, A.D. Immunohistochemical analysis of sympathetic involvement in the SOD1-G93A transgenic mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 424–433. [Google Scholar] [CrossRef]
- Pullen, A.H.; Martin, J.E. Ultrastructural abnormalities with inclusions in Onuf’s nucleus in motor neuron disease (amyotrophic lateral sclerosis). Neuropathol. Appl. Neurobiol. 1995, 21, 327–340. [Google Scholar] [CrossRef]
- Carvalho, M.; Schwartz, M.S.; Swash, M. Involvement of the external anal sphincter in amyotrophic lateral sclerosis. Muscle Nerve 1995, 18, 848–853. [Google Scholar] [CrossRef]
- Sasaki, S.; Maruyama, S. A fine structural study of Onuf’s nucleus in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1993, 119, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Manzano, R.; Sorarú, G.; Grunseich, C.; Fratta, P.; Zuccaro, E.; Pennuto, M.; Rinaldi, C. Beyond motor neurons: Expanding the clinical spectrum in Kennedy’s disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Powis, R.A.; Gillingwater, T.H. Selective loss of alpha motor neurons with sparing of gamma motor neurons and spinal cord cholinergic neurons in a mouse model of spinal muscular atrophy. J. Anat. 2016, 228, 443–451. [Google Scholar] [CrossRef]
- Anneser, J.M.; Borasio, G.D.; Berthele, A.; Zieglgänsberger, W.; Tölle, T.R. Differential expression of group I metabotropic glutamate receptors in rat spinal cord somatic and autonomic motoneurons: Possible implications for the pathogenesis of amyotrophic lateral sclerosis. Neurobiol. Dis. 1999, 6, 140–147. [Google Scholar] [CrossRef]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 252. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Xia, R.; Liu, Y.; Yang, L.; Gal, J.; Zhu, H.; Jia, J. Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol. Neurodegener. 2012, 7, 10. [Google Scholar] [CrossRef]
- Fischer, L.R.; Li, Y.; Asress, S.A.; Jones, D.P.; Glass, J.D. Absence of SOD1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol. 2012, 233, 163–171. [Google Scholar] [CrossRef]
- Jokic, N.; De Aguilar, J.-L.G.; Dimou, L.; Lin, S.; Fergani, A.; Ruegg, M.A.; Schwab, M.E.; Dupuis, L.; Loeffler, J.-P. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. 2006, 7, 1162–1167. [Google Scholar] [CrossRef]
- Teng, F.Y.; Tang, B.L. Nogo-A and Nogo-66 receptor in amyotrophic lateral sclerosis. J. Cell Mol. Med. 2008, 12, 1199–1204. [Google Scholar] [CrossRef]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part I, background and methods. Brain Behav. 2013, 3, 335–350. [Google Scholar] [CrossRef]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part II, results and discussion. Brain Behav. 2013, 3, 431–457. [Google Scholar] [CrossRef]
- Bruneteau, G.; Bauché, S.; de Aguilar, J.L.G.; Brochier, G.; Mandjee, N.; Tanguy, M.-L.; Hussain, G.; Behin, A.; Khiami, F.; Sariali, E.; et al. Endplate denervation correlates with Nogo-A muscle expression in amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2015, 2, 362–372. [Google Scholar] [CrossRef]
- De Winter, F.; Vo, T.; Stam, F.J.; Wisman, L.A.; Bär, P.R.; Niclou, S.; van Muiswinkel, F.L.; Verhaagen, J. The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell Neurosci. 2006, 32, 102–117. [Google Scholar] [CrossRef]
- Fanara, P.; Banerjee, J.; Hueck, R.V.; Harper, M.R.; Awada, M.; Turner, H.; Husted, K.H.; Brandt, R.; Hellerstein, M.K. Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J. Biol. Chem. 2007, 282, 23465–23472. [Google Scholar] [CrossRef]
- Bilsland, L.G.; Sahai, E.; Kelly, G.; Golding, M.; Greensmith, L.; Schiavo, G. Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 20523–20528. [Google Scholar] [CrossRef]
- Marinkovic, P.; Reuter, M.S.; Brill, M.S.; Godinho, L.; Kerschensteiner, M.; Misgeld, T. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 4296–4301. [Google Scholar] [CrossRef]
- Williamson, T.L.; Cleveland, D.W. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 1999, 2, 50–56. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef]
- Suzuki, N.; Akiyama, T.; Warita, H.; Aoki, M. Omics approach to axonal dysfunction of motor neurons in amyotrophic lateral sclerosis (ALS). Front. Neurosci. 2020, 14, 194. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; Lapointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef] [PubMed]
- Sau, D.; Rusmini, P.; Crippa, V.; Onesto, E.; Bolzoni, E.; Ratti, A.; Poletti, A. Dysregulation of axonal transport and motorneuron diseases. Biol. Cell 2011, 103, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Montiel, M.J.; Chaineau, M.; Durcan, T.M. The neglected genes of ALS: Cytoskeletal dynamics impact synaptic degeneration in ALS. Front. Cell Neurosci. 2020, 14, 594975. [Google Scholar] [CrossRef]
- Spaulding, E.L.; Burgess, R.W. Accumulating evidence for axonal translation in neuronal homeostasis. Front. Neurosci. 2017, 11, 312. [Google Scholar] [CrossRef]
- Nagano, S.; Jinno, J.; Abdelhamid, R.F.; Jin, Y.; Shibata, M.; Watanabe, S.; Hirokawa, S.; Nishizawa, M.; Sakimura, K.; Onodera, O.; et al. TDP-43 transports ribosomal protein mRNA to regulate axonal local translation in neuronal axons. Acta Neuropathol. 2020, 140, 695–713. [Google Scholar] [CrossRef]
- Fischer, L.R.; Glass, J.D. Axonal degeneration in motor neuron disease. Neurodegener. Dis. 2007, 4, 431–442. [Google Scholar] [CrossRef]
- Muller, F.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.-T.; Epstein, C.J.; Roberts, L.J.; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 function and its implications for amyotrophic lateral sclerosis pathology: New and renascent themes. Neuroscientist 2015, 21, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.; Fisher, E.M.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136 Pt 8, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef]
- Bidhendi, E.E.; Bergh, J.; Zetterström, P.; Forsberg, K.; Pakkenberg, B.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Mutant superoxide dismutase aggregates from human spinal cord transmit amyotrophic lateral sclerosis. Acta Neuropathol. 2018, 136, 939–953. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Hafezparast, M.; Klocke, R.; Ruhrberg, C.; Marquardt, A.; Ahmad-Annuar, A.; Bowen, S.; Lalli, G.; Witherden, A.S.; Hummerich, H.; Nicholson, S.; et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 2003, 300, 808–812. [Google Scholar] [CrossRef]
- Kieran, D.; Hafezparast, M.; Bohnert, S.; Dick, J.R.T.; Martin, J.; Schiavo, G.; Fisher, E.; Greensmith, L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J. Cell Biol. 2005, 169, 561–567. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio, M.A.; Herrando-Grabulosa, M.; Navarro, X. Sensory Involvement in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 15521. https://doi.org/10.3390/ijms232415521
Rubio MA, Herrando-Grabulosa M, Navarro X. Sensory Involvement in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2022; 23(24):15521. https://doi.org/10.3390/ijms232415521
Chicago/Turabian StyleRubio, Miguel A., Mireia Herrando-Grabulosa, and Xavier Navarro. 2022. "Sensory Involvement in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 23, no. 24: 15521. https://doi.org/10.3390/ijms232415521
APA StyleRubio, M. A., Herrando-Grabulosa, M., & Navarro, X. (2022). Sensory Involvement in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 23(24), 15521. https://doi.org/10.3390/ijms232415521