

Glycosphingolipids in Diabetes, Oxidative Stress, and Cardiovascular Disease: Prevention in Experimental Animal Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
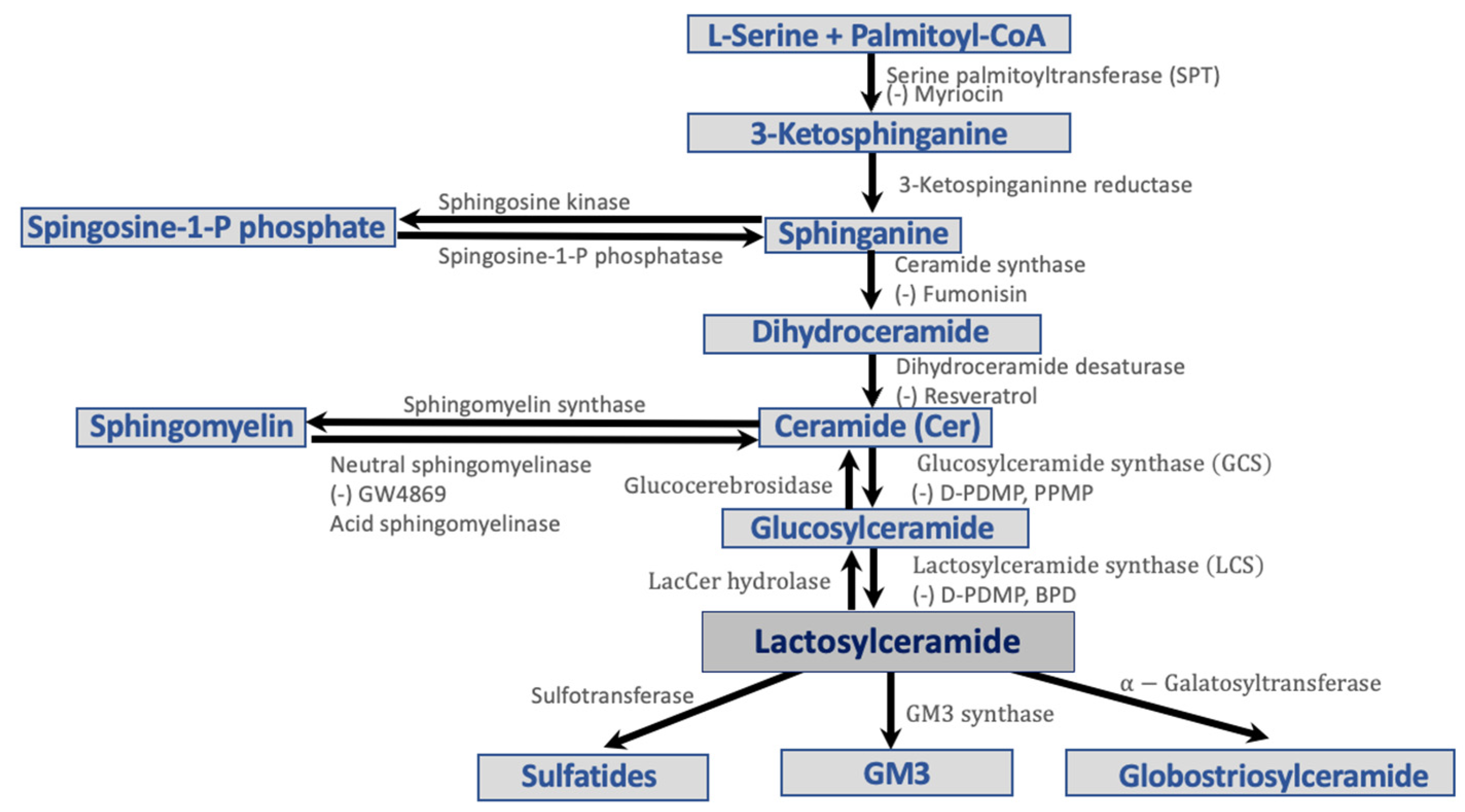
2. Lipoproteins and Sphingolipids
3. Sphingolipid Pathways in Diabetes
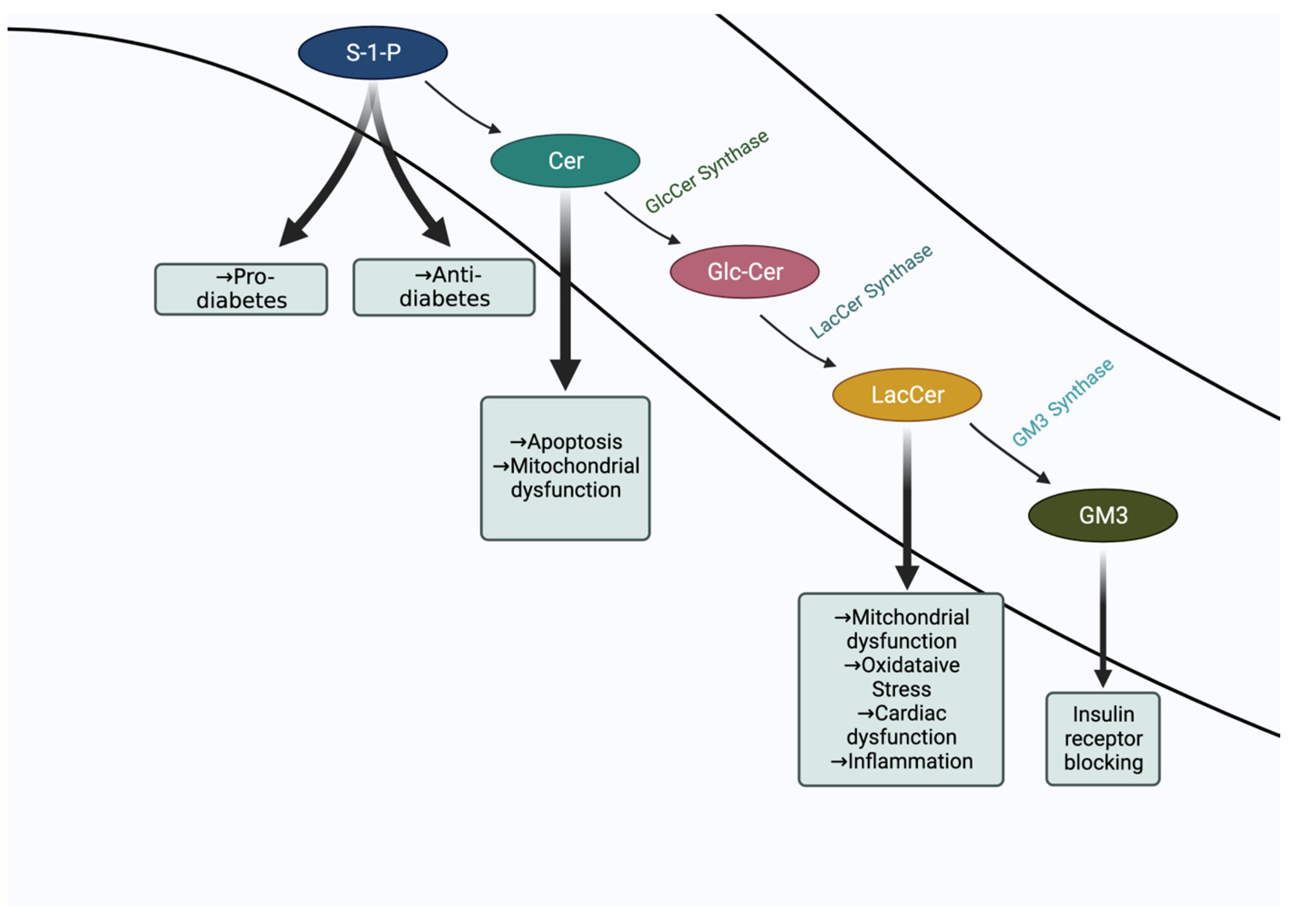
- Diabetes and Ceramide

- Diabetes and GM3 Ganglioside
- Diabetes and Sphingosine-1-Phosphate
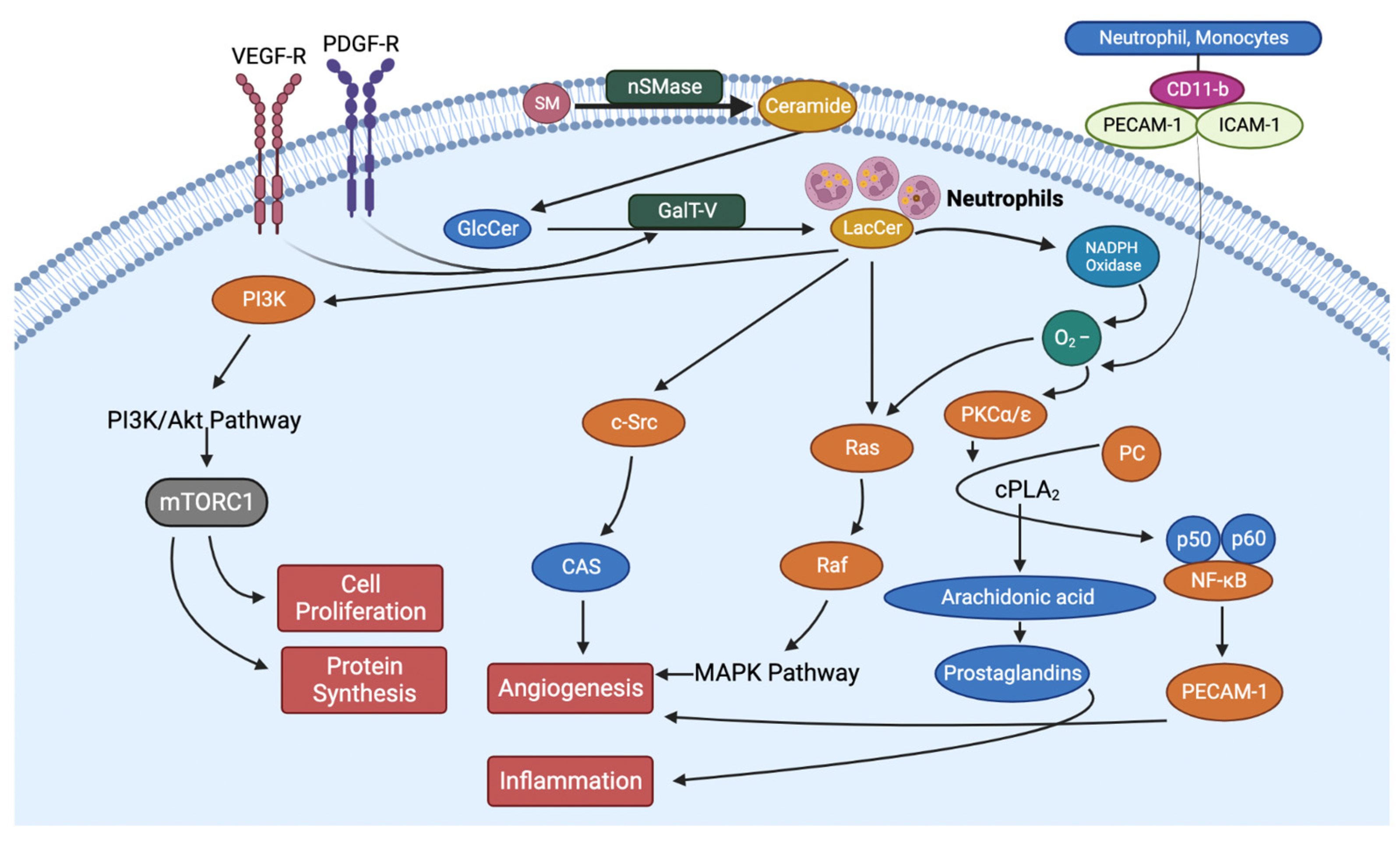
- Diabetes and Lactosylceramide
- Diabetes and Sphingomyelin
4. Sphingolipid Inhibitors Mitigate the Pathology of Insulin Resistance
5. Inhibition of LacCer Synthesis Can Mitigate the Pathology of Type II Diabetes by Reducing Blood Glucose, Body Weight, and Inflammation
6. Sphingolipidomics Helps Diagnose and Raise the Predictive Value in Diabetes
7. Conclusions
8. Perspectives
- Although experiments in vitro and in mouse models of diabetes suggest a potential role of GM3 in competing with the insulin–insulin receptor interaction and to contribute to T2D, robust evidence using GM3 synthase specific inhibitors is needed.
- Hyperglycemia and increased levels of LacCer collectively can increase oxidative stress and the generation of advanced glycation end (AGE) products, which is a formidable force in cardiovascular complications and may well adversely affect kidney function in T2D [80].
- To further understand the complexities in T2D and to develop drugs, a systems-based biology approach is needed that could integrate molecular markers including sphingolipids, genomics, and physiology.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGE | Advanced glycation end |
| AOIMT | Aortic media-intima media thickening |
| BDP | Biopolymer-encapsulated D-PDMP |
| BMI | Body mass index |
| Cav-1 | Caveolin-1 |
| Cer | Ceramide |
| CRC | Calcium retention capacity |
| DAGS | Diacylglycerols |
| DM | Diabetes mellitus |
| D-PDMP | D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol |
| D-EtDO-P4 | D-thero-1-(3,4,-ethylenedioxy) phenyl-2-palmitoylamino-1-pyrrolidino-1propanol |
| GlcCer | Glucosylceramide |
| GCase | Glucosylceramidase |
| GLUT4 | Glucose transporter type 4 |
| GM3 | Monosialodihexosylceramide |
| GSL | Glycosphingolipidids |
| HDL | High-density lipoprotein |
| HFD | High fat diet |
| ICAM-1 | Intercellular cell adhesion molecule |
| IR | Insulin receptor |
| IRS-1 | Insulin receptor substrate 1 |
| IRS | Insulin receptor substrate |
| LDL | Low-density lipoprotein |
| NAFLD | Non-alcoholic fatty liver disease |
| NPC-1 | Niemann-Pick C1 |
| Ox-LDL | Oxidized low-density lipoprotein |
| PECAM-1 | Platelet cell adhesion molecule |
| PDGF | Platelet derived growth factor |
| PDGF | Platelet derived growth factor receptor |
| P13K | Phosphoinositide 3-kinases |
| PKB | Protein kinase B |
| PKC | Protein kinase C |
| PSL | Phosphosphingolipids |
| PWV | Pulse wave velocity |
| S1P | Sphingosine-1-phosphate |
| S1PR | Spingosine-1-phosphate receptor |
| SLs | Sphingolipids |
| SM | Sphingomyelin |
| SMase | Sphingomyelinases |
| SphK1 | Sphingosine kinases 1 |
| Sphk2 | Sphingosine kinases 2 |
| SPT | Serine palmitoyltransferase |
| T1D | Type-I diabetes |
| T2D | Type-ll diabetes |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor-alpha |
| UDP-galactose | Uridine-diphosphate galactose |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
| VLDL | Very-low-density-lipoprotein |
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [Green Version]
- Maddatu, J.; Anderson-Baucum, E.; Evans-Molina, C. Smoking and the risk of type 2 diabetes. Transl. Res. 2017, 184, 101–107. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45 (Suppl. 1), S17–S38. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Mu, J.; Kim, J.K.; Thorvaldsen, J.L.; Chu, Q.; Crenshaw, E.B.; Kaestner, K.H.; Bartolomei, M.S.; Shulman, G.I.; Birnbaum, M.J. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001, 292, 1728–1731. [Google Scholar] [CrossRef]
- Whitehead, J.P.; Humphreys, P.; Krook, A.; Jackson, R.; Hayward, A.; Lewis, H.; Siddle, K.; O’Rahilly, S. Molecular scanning of the insulin receptor substrate 1 gene in subjects with severe insulin resistance: Detection and functional analysis of a naturally occurring mutation in a YMXM motif. Diabetes 1998, 47, 837–839. [Google Scholar] [CrossRef]
- Yamauchi, T.; Tobe, K.; Tamemoto, H.; Ueki, K.; Kaburagi, Y.; Yamamoto-Honda, R.; Takahashi, Y.; Yoshizawa, F.; Aizawa, S.; Akanuma, Y.; et al. Insulin signalling and insulin actions in the muscles and livers of insulin-resistant, insulin receptor substrate 1-deficient mice. Mol. Cell. Biol. 1996, 16, 3074–3084. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef]
- Kim, J.K.; Fillmore, J.J.; Sunshine, M.J.; Albrecht, B.; Higashimori, T.; Kim., D.W.; Liu, Z.X.; Soos, J.; Cline, G.W.; O’Brien, W.R.; et al. PKC-theta knockout mice are protected from fat-induced insulin resistance. J. Clin. Investig. 2004, 114, 823–827. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef]
- Furukawa, N.; Ongusaha, P.; Jahng, W.J.; Araki, K.; Choi, C.S.; Kim, H.J.; Lee, Y.H.; Kaibuchi, K.; Kahn, B.B.; Masuzaki, H.; et al. Role of Rho-kinase in regulation of insulin action and glucose homeostasis. Cell Metab. 2005, 2, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Hardy, O.T.; Czech, M.P.; Corvera, S. What causes the insulin resistance underlying obesity? Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Shulman, G.I. Etiology of insulin resistance. Am J Med. 2006, 119 (Suppl. 1), S10–S16. [Google Scholar] [CrossRef] [Green Version]
- Freeman, A.M.; Pennings, N. Insulin Resistance. [Updated 2021 Jul 10]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Saini, V. Molecular mechanisms of insulin resistance in type 2 diabetes mellitus. World J. Diabetes 2010, 1, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Balram, A.; Li, W. Convergence: Lactosylceramide-Centric Signaling Pathways Induce Inflammation, Oxidative Stress, and Other Phenotypic Outcomes. Int. J. Mol. Sci. 2021, 22, 1816. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids—and their metabolism in physiology and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Chatterjee, S.; Kwiterovich, P.O. Glycosphingolipids of human plasma lipoproteins. Lipids 1976, 11, 462–466. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sekerke, S.C.; Kwiterovich, P.O., Jr. Increased urinary excretion of glycosphingolipids in familial hypercholesterolemia. J. Lipid Res. 1982, 23, 513–522. [Google Scholar] [CrossRef]
- Chatterjee, S.; Clarke, K.S.; Kwiterovich, P.O., Jr. Regulation of synthesis of lactosylceramide and long chain bases in normal and familial hypercholesterolemic cultured proximal tubular cells. J. Biol. Chem. 1986, 261, 13474–13479. [Google Scholar] [CrossRef]
- Kwok, B.C.; Dawson, G.; Ritter, M.C. Stimulation of glycolipid synthesis and exchange by human serum high density lipoprotein-3 in human fibroblasts and leukocytes. J. Biol. Chem. 1981, 256, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Snider, A. Sphingolipids in High Fat Diet and Obesity-Related Diseases. Mediat. Inflamm. 2015, 2015, 520618. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S. Sphingosine-1-phosphate: From insipid lipid to a key regulator. J. Biol. Chem. 2020, 295, 3371–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, M.B.; Prieto, M.; Silva, C.L. Ceramide: A simple sphingolipid with unique biophysical properties. Prog. Lipid Res. 2014, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Bedja, D.; Yan, W.; Lad, V.; Iocco, D.; Sivakumar, N.; Bandaru, V.V.R.; Chatterjee, S. Inhibition of glycosphingolipid synthesis reverses skin inflammation and hair loss in ApoE−/− mice fed western diet. Sci. Rep. 2018, 8, 11463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, C.B.; Gordillo, R.; Scherer, E.P. The Role of Ceramides in Diabetes and Cardiovascular Disease Regulation of Ceramides by Adipokines. Front. Endocrinol. 2020, 11, 569250. [Google Scholar] [CrossRef]
- Coen, P.M.; Dubé, J.J.; Amati, F.; Stefanovic-Racic, M.; Ferrell, R.E.; Toledo, F.G.; Goodpaster, B.H. Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes 2010, 59, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Bikman, B.T.; Wang, L.-P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid–induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Saberi, M.; Woods, N.B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Summers, S.A.; Garza, L.A.; Zhou, H.; Birnbaum, M.J. Regulation of insulin-stimulated glucose transporter GLUT4 translocation and Akt kinase activity by ceramide. Mol. Cell. Biol. 1998, 18, 5457–5464. [Google Scholar] [CrossRef]
- Powell, D.J.; Hajduch, E.; Kular, G.; Hundal, H.S. Ceramide Disables 3-Phosphoinositide Binding to the Pleckstrin Homology Domain of Protein Kinase B (PKB)/Akt by a PKCζ-Dependent Mechanism. Mol. Cell. Biol. 2003, 23, 7794–7808. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Bo, S.; Ruscica, M.; Sahebkar, A. Ceramides and diabetes mellitus: An update on the potential molecular relationships. Diabet. Med. 2020, 37, 742–3071. [Google Scholar] [CrossRef] [PubMed]
- Hage Hassan, R.; Pacheco de Sousa, A.C.; Mahfouz, R.; Hainault, I.; Blachnio-Zabielska, A.; Bourron, O.; Koskas, F.; Górski, J.; Ferré, P.; Foufelle, F.; et al. Sustained Action of Ceramide on the Insulin Signaling Pathway in Muscle Cells: IMPLICATION OF THE DOUBLE-STRANDED RNA-ACTIVATED PROTEIN KINASE. J. Biol. Chem. 2016, 291, 3019–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.D.; Pekala, P.H. Lipid mediators of insulin resistance: Ceramide signalling down-regulates GLUT4 gene transcription in 3T3-L1 adipocytes. Biochem. J. 1996, 319 Pt 1, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Li, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsayed, A.K.; Vimalraj, S.; Nandakumar, M.; Abdelalim, E.M. Insulin resistance in diabetes: The promise of using induced pluripotent stem cell technology. World J. Stem. Cells 2021, 13, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Tagami, S.; Inokuchi, J.; Kabayama, K.; Yoshimura, H.; Kitamura, F.; Uemura, S.; Ogawa, C.; Ishii, A.; Saito, A.; Ohtsuka, A.; et al. Ganglioside GM3 Participates in the Pathological Conditions of Insulin Resistance. J. Biol. Chem. 2002, 277, 3085–3092, ISSN 0021-9258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabayama, K.; Sato., T.; Kitamura, F.; Uemura, S.; Kang, B.W.; Igarashi, Y.; Inokuchi, J. TNFalpha-induced insulin resistance in adipocytes as a membrane microdomain disorder: Involvement of ganglioside GM3. Glycobiology 2005, 15, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Hashiramoto, A.; Haluzik, M.; Mizukami, H.; Beck, S.; Norton, A.; Kono, M.; Tsuji, S.; Daniotti, L.J.; Werth, N.; et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA 2003, 100, 3445–3449. [Google Scholar] [CrossRef]
- Kabayama, K.; Sato, T.; Saito, K.; Loberto, N.; Prinetti, A.; Sonnino, S.; Kinjo, M.; Igarashi, Y.; Inokuchi, J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 21, 13678–13683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekimoto, J.; Kabayama, K.; Gohara, K.; Inokuchi, J. Dissociation of the insulin receptor from caveolae during TNFα-induced insulin resistance and its recovery by d-PDMP. FEBS Lett. 2012, 586, 191–195, ISSN 0014-5793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitton, J.; Bandet, C.L.; Mariko, M.L.; Tan-Chen, S.; Bourron, O.; Benomar, Y.; Hajduch, E.; Le Stunff, H. Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes. Cells 2020, 9, 1682. [Google Scholar] [CrossRef]
- Wigger, D.; Schumacher, F.; Schneider-Schaulies, S.; Kleuser, B. Sphingosine 1-phosphate metabolism and insulin signaling. Cell. Signal. 2021, 82, 109959, ISSN 0898-6568. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Miller, R.A.; Wang, Z.V.; Sun, K.; Barth, B.M.; Bui, H.H.; Davis, K.E.; Bikman, B.T.; Halberg, N.; Rutkowski, J.M.; et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat. Med. 2010, 17, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, T.E.; Bewley, M.C.; Unrath, K.A.; Pedersen, M.M.; Anderson, R.E.; Jung, D.Y.; Jefferson, L.S.; Kim, J.K.; Bronson, S.K.; Flanagan, J.M.; et al. Circulating sphingolipid biomarkers in models of type 1 diabetes. J. Lipid Res. 2010, 52, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, J.C.; Morris, A.J.; Sunkara, M.; Popa, G.J.; Larson, K.L.; Özcan, S. Sphingosine 1-phosphate (S1P) regulates glucose-stimulated insulin secretion in pancreatic beta cells. J. Biol. Chem. 2012, 287, 13457–13464. [Google Scholar] [CrossRef] [Green Version]
- Novgorodov, A.S.; Riley, L.C.; Yu, J.; Keffler, A.J.; Clarke, J.C.; Van Laer, O.A.; Baicu, F.C.; Zile, R.M.; Gudz, I.T. Lactosylceramide Contributes to Mitochondrial Dysfunction in Diabetes. J. Lipid Res. 2016, 57, 546–562. [Google Scholar] [CrossRef] [Green Version]
- Schömel, N.; Gruber, L.; Alexopoulos, S.J.; Trautmann, S.; Olzomer, E.M.; Byrne, F.L.; Hoehn, K.L.; Gurke, R.; Thomas, D.; Ferreirós, N.; et al. UGCG overexpression leads to increased glycolysis and increased oxidative phosphorylation of breast cancer cells. Sci. Rep. 2020, 10, 8182. [Google Scholar] [CrossRef]
- Grösch, S.; Alessenko, V.A.; Albi, E. The Many Facets of Sphingolipids in the Specific Phases of Acure Inflammatory Response. Mediat. Inflamm. 2018, 2018, 5378284. [Google Scholar] [CrossRef]
- Deus, M.C.; Yambire, K.J.; Oliveira, J.P.; Raimundo, N. Mitochondria–LysosomeCrosstalk: FromPhysiologyto Neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, K.P.; Huynh, K.; Pernes, G.; Miotto, M.P.; Mellett, A.N.; Giles, C.; Meikle, J.P.; Murphy, J.A.; Lancaster, I.G. Macrophage polarization state affects lipid composition and the channeling of exgenous fatty acids into endogenous lipd pools. J. Biol. Chem. 2021, 297, 101341. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.S.; Torta, F.; Ji, S.; Choi, H.; Begum, H.; Sim, X.; Khoo, C.M.; Khoo, E.Y.H.; Ong, W.Y.; Van Dam, R.M.; et al. Large-scale lipidomics identifies associations between plasma sphingolipids and T2DM incidence. JCI Insight 2019, 5, e126925. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Kolmakova, A.; Miller, M. The role of the phospholipid sphingomyelin in heart disease. Curr. Opin. Investig. Drugs 2006, 7, 219–228. [Google Scholar]
- Yano, M.; Watanabe, K.; Yamamoto, T.; Taguchi, R.; Yamagata, K.; Oike, Y. Mitchordrial Dysfunction and Increased Reactive Oxygen Species Impair Insulin Secretion in Sphingomyelin Synthase 1-null Mice. J. Biol. Chem. 2011, 286, 3992–4002. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhang, H.; Liu, J.; Liang, C.; Li, Y.; Li, Y.; Teitelman, G.; Beyer, T.; Bui, H.H.; Peake, A.D.; et al. Reducing Plasam Membrane Shingomyelin Increases Insulin Sensitivity. Mol. Cell. Biol. 2011, 31, 4205–4218. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.C.; Jiang, X.C.; Tabas, I.; Tall, A.; Shea, S. Plasma sphingomyelin and subclinical atherosclerosis: Findings from the multi-ethnic study of atherosclerosis. Am. J. Epidemiol. 2006, 163, 903–912. [Google Scholar] [CrossRef]
- Hodson, A.; Tippetts, T.; Bikman, B. Insulin treatment increases myocardial ceramide accumulation and disrupts cardiometabolic function. Cardiovas. Diabetol. 2015, 14, 153. [Google Scholar] [CrossRef] [Green Version]
- Fedoryszak-Kuśka, N.; Panasiewicz, M.; Domek, H.; Pacuszka, T. Glucosylceramide synthase inhibitors D-PDMP and D-EtDO-P4 decrease the GM3 ganglioside level, differ in their effects on insulin receptor autophosphorylation but increase Akt1 kinase phosphorylation in human hepatoma HepG2 cells. Acta Biochim. Pol. 2016, 63, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Cleveland, T.; Shi, W.; Inokuchi, J.; Radin, S.N. Studies of the action of ceramide-like substances (d-andl-PDMP) on sphingolipid glycosyltransferases and purified lactosylceramide synthase. Glycoconj. J. 1996, 13, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi, J.; Kanoh, H. Pathophysiological Significance of Ganglioside Molecular Species With a Particular Attention to the Metabolic Syndrome Focusing on Toll-Like Receptor 4 Binding. Front. Mol. Biosci. 2022, 9, 918346. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Bedja, D.; Amuzie, C.; Foss, C.A.; Pomper, M.G.; Bhattacharya, R.; Yarema, K.J.; Chatterjee, S. Improved intervention of atherosclerosis and cardiac hypertrophy through biodegradable polymer-encapsulated delivery of glycosphingolipid inhibitor. Biomaterials 2015, 64, 125–135, ISSN 0142-9612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Ficicioglu, C. Review of miglustat for clinical management in Gaucher disease type 1. Ther. Clin. Risk Manag. 2008, 4, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, J.; Sun, Y.; Bangari, S.D.; Budman, E.; Park, H.; Nietupski, B.J.; Allaire, A.; Cromwell, A.M.; Wang, B.; Grabowski, A.G.; et al. CNS-accessible Inhibitor of Glucosylceramide Synthase for Substrate Reduction Therapy of Neuronopathic Gaucher Disease. Mol. Ther. 2016, 24, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Rojas, L.B.A.; Gomes, M.B. Metformin: An old but still the best treatment for type 2 diabetes. Diabetol. Metab. Syndr. 2013, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, L.S. Lactic Acidosis in a Patient with Type 2 Diabetes Mellitus. Clin. J. Am. Soc. Nephrol. 2015, 10, 1476–1483. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Jung, S.; Lee, S.H.; Lee, J.H. Association between arterial stiffness and serum L-octanoylcarnitine and lactosylceramide in overweight middle-aged subjects: 3-year follow-up study. PLoS ONE 2015, 10, e0119519. [Google Scholar] [CrossRef]
- Alshehry, Z.H.; Mundra, P.A.; Barlow, C.K.; Mellett, N.A.; Wong, G.; McConville, M.J.; Sime, J.; Tonkin, A.M.; Sullivan, D.R.; Barnes, E.H.; et al. Plasma Lipidomic Profiles Improve on Traditional Risk Factors for the Prediction of Cardiovascular Events in Type 2 Diabetes Mellitus. Circulation 2016, 134, 1637–1650. [Google Scholar] [CrossRef] [Green Version]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446.e1442. [Google Scholar] [CrossRef] [Green Version]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, K.W.; Gilijamse, P.W.; Versteeg, R.I.; Ackermans, M.T.; Nederveen, A.J.; la Fleur, S.E.; Romijn, J.A.; Nieuwdorp, M.; Zhang, D.; Samuel, V.T.; et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017, 19, 1997–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotronen, A.; Seppänen-Laakso, T.; Westerbacka, J.; Kiviluoto, T.; Arola, J.; Ruskeepaa, A.L.; Oresic, M.; Yki-Jarvinen, H. Hepatic stearoyl-CoA desaturase (SCD)-1 activity and diacylglycerol but not ceramide concentrations are increased in the nonalcoholic human fatty liver. Diabetes 2009, 58, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulou, M.; Gordillo, R.; Koliaki, C.; Gancheva, S.; Jelenik, T.; De Filippo, E.; Herder, C.; Markgraf, D.; Jankowiak, F.; Esposito, I.; et al. Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes Care 2018, 41, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, C.; Colell, A.; París, R.; Fernández-Checa, J.C. Direct interaction of GD3 ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial permeability transition; cytochrome c release; and caspase activation. FASEB J. 2000, 14, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Przybylska, M.; Wu, I.; Zhang, J.; Siegel, C.; Komarnitsky, S.; Yew, S.N.; Cheng., H.S. Inhibiting Glycosphingolipid Synthesis Improves Glycemic Control and Insulin Sensitivity in Animal Models of Type 2 Diabetes. Diabetes 2007, 56, 1210–1218. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Zheng, L.; Ma, S.; Bedja, D.; Bandaru, V.; Kim, G.; Rangecroft, A.B.; Iocco, D.; Campbell, S.A. Management of metabolic syndrome and reduction in body weight in type II diabetic mice by inhibiting glycosphingolipid synthesis. Biochem. Biophys. Res. Commun. 2020, 525, 455–461. [Google Scholar] [CrossRef]
- Conserva., F.; Gesualdo, L.; Papale, M. A Systems Biology Overview on Human Diabetic Nephropathy: From Genetic Susceptibility to Post-Transcriptional and Post-Translational Modifications. J. Diabetes Res. 2015, 2016, 7934504. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balram, A.; Thapa, S.; Chatterjee, S. Glycosphingolipids in Diabetes, Oxidative Stress, and Cardiovascular Disease: Prevention in Experimental Animal Models. Int. J. Mol. Sci. 2022, 23, 15442. https://doi.org/10.3390/ijms232315442
Balram A, Thapa S, Chatterjee S. Glycosphingolipids in Diabetes, Oxidative Stress, and Cardiovascular Disease: Prevention in Experimental Animal Models. International Journal of Molecular Sciences. 2022; 23(23):15442. https://doi.org/10.3390/ijms232315442
Chicago/Turabian StyleBalram, Amrita, Spriha Thapa, and Subroto Chatterjee. 2022. "Glycosphingolipids in Diabetes, Oxidative Stress, and Cardiovascular Disease: Prevention in Experimental Animal Models" International Journal of Molecular Sciences 23, no. 23: 15442. https://doi.org/10.3390/ijms232315442
APA StyleBalram, A., Thapa, S., & Chatterjee, S. (2022). Glycosphingolipids in Diabetes, Oxidative Stress, and Cardiovascular Disease: Prevention in Experimental Animal Models. International Journal of Molecular Sciences, 23(23), 15442. https://doi.org/10.3390/ijms232315442