
Impact of IBD-Associated Dysbiosis on Bacterial Quorum Sensing Mediated by Acyl-Homoserine Lactone in Human Gut Microbiota
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Presence of AHL in the Gut Microbiota of Controls and IBD Patients
2.2. QS-Related Genes in Human Gut Microbiota
2.2.1. Patients’ Characteristics of the in Silico Study
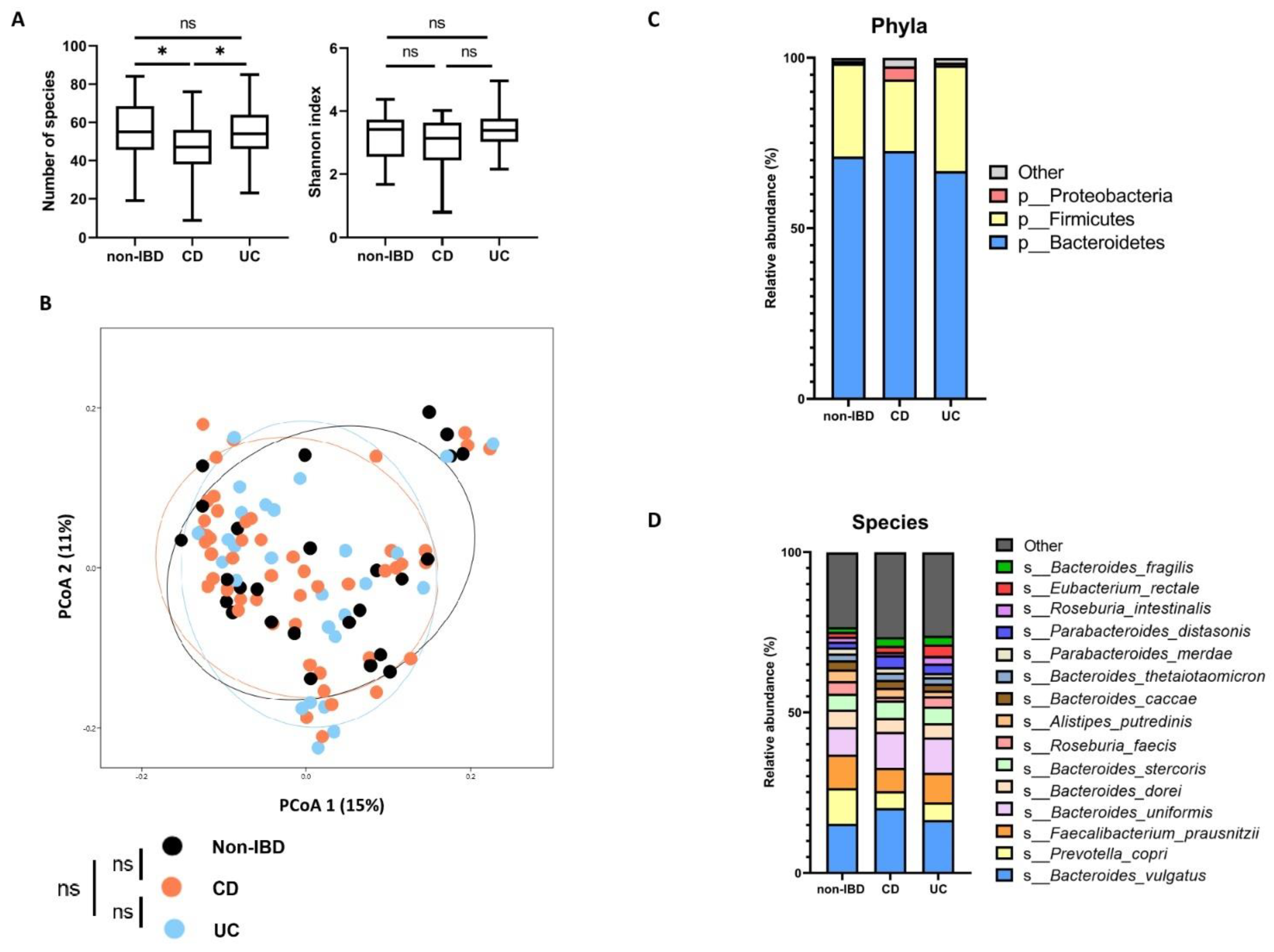
2.2.2. Microbiota Composition According to Disease Phenotypes
2.2.3. QS-Related Genes
2.2.4. Presence of Homologous QS-Related Genes
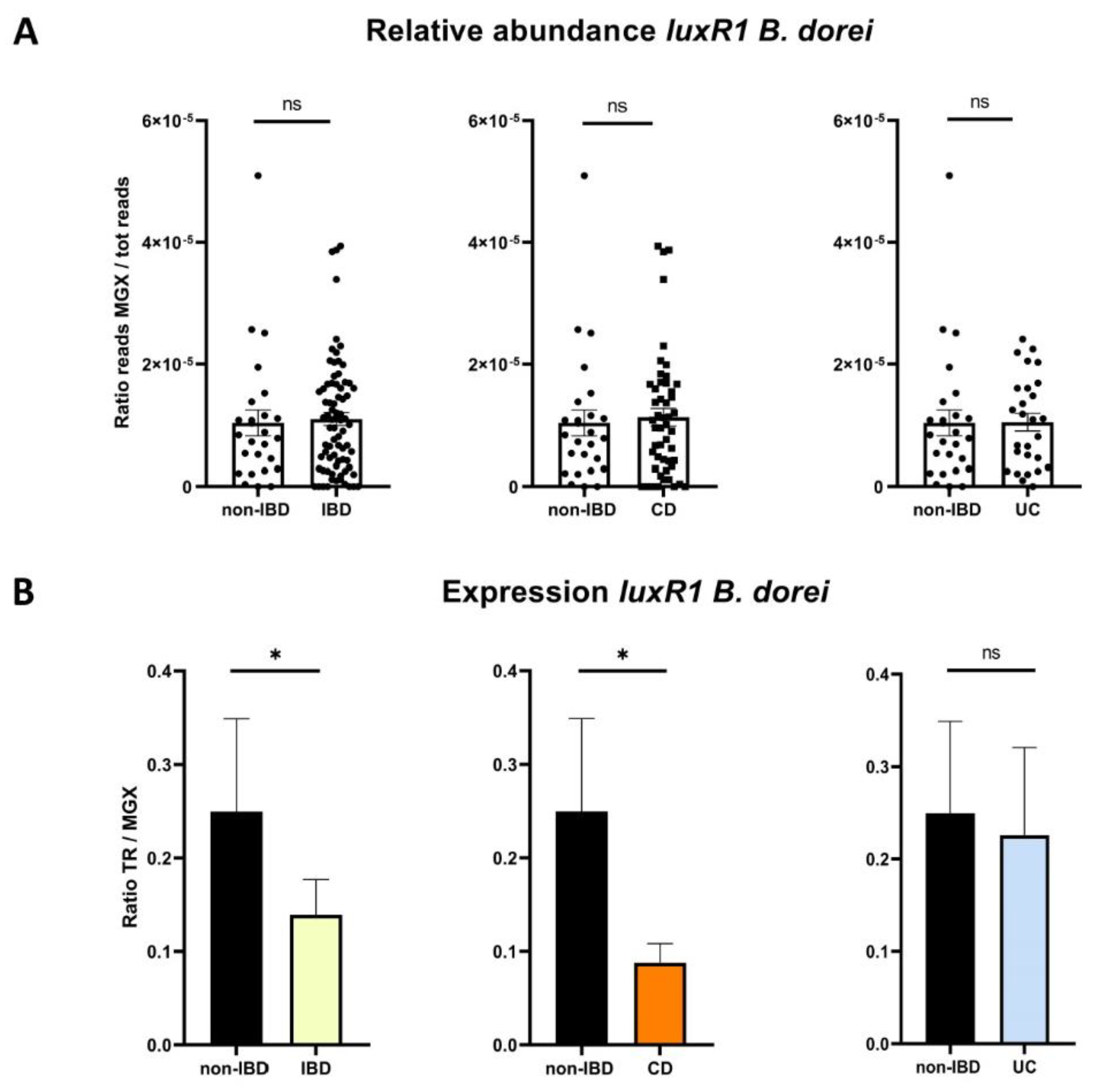
2.2.5. AHL Receptor Genes According to Disease Phenotypes
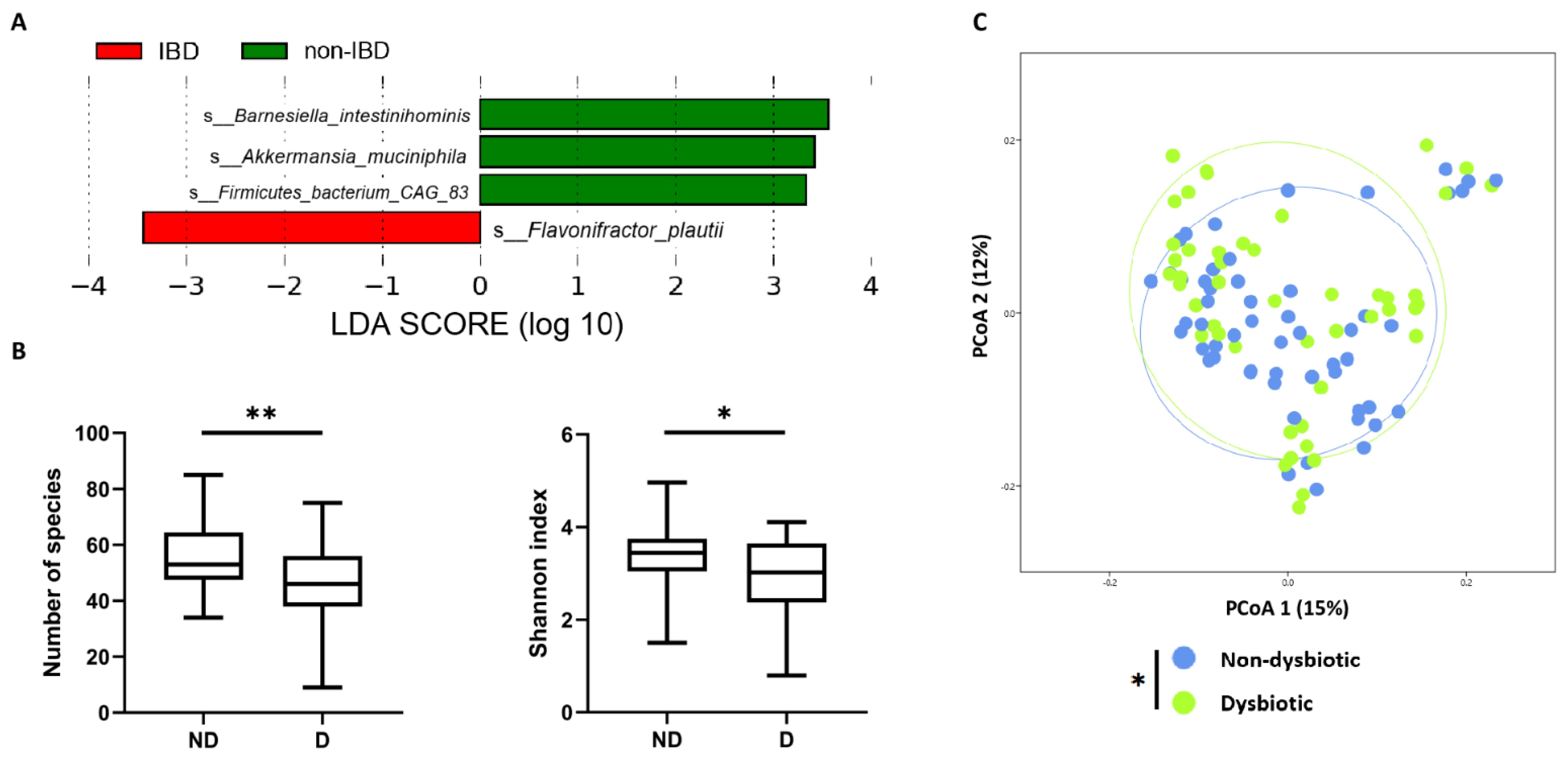
2.2.6. QS-Related Genes According to IBD-Associated Dysbiosis Status
3. Discussion
4. Materials and Methods
4.1. HPLC-MS/MS Samples Pre-Treatment and AHL Extraction
4.2. AHL Detection with HPLC-MS/MS
4.3. Inflammatory Bowel Disease Multi’omics Database and Samples
4.4. Dysbiosis Definitions
4.5. QS-Related Genes
4.6. Sequences Alignment
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Hardt, W.-D. The role of microbiota in infectious disease. Trends Microbiol. 2008, 16, 107–114. [Google Scholar] [CrossRef]
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef]
- Smith, M.B.; Lashner, B.A.; Hanauer, S.B. Smoking and inflammatory bowel disease in families. Am. J. Gastroenterol. 1988, 83, 407–409. [Google Scholar] [PubMed]
- Seksik, P. Alterations of the dominant faecal bacterial groups in patients with Crohns disease of the colon. Gut 2003, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; DA Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.-A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Aviello, G.; Singh, A.K.; O’Neill, S.; Conroy, E.; Gallagher, W.; D'Agostino, G.; Walker, A.; Bourke, B.; Scholz, D.; Knaus, U.G. Colitis susceptibility in mice with reactive oxygen species deficiency is mediated by mucus barrier and immune defense defects. Mucosal Immunol. 2019, 12, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Bassler, B.L. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Engebrecht, J.; Silverman, M. Identification of genes and gene products necessary for bacterial bioluminescence. Proc. Natl. Acad. Sci. USA 1984, 81, 4154–4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambello, M.J.; Iglewski, B.H. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J. Bacteriol. 1991, 173, 3000–3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, M.M.; Sperandio, V. Quorum sensing by enteric pathogens. Curr. Opin. Gastroenterol. 2007, 23, 10–15. [Google Scholar] [CrossRef]
- Swearingen, M.C.; Sabag-Daigle, A.; Ahmer, B.M.M. Are there acyl-homoserine lactones within mammalian intestines? J. Bacteriol. 2013, 195, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Dyszel, J.L.; Soares, J.A.; Swearingen, M.C.; Lindsay, A.; Smith, J.N.; Ahmer, B.M.M. E. coli K-12 and EHEC genes regulated by SdiA. PLoS ONE 2010, 5, e8946. [Google Scholar] [CrossRef] [Green Version]
- Michael, B.; Smith, J.N.; Swift, S.; Heffron, F.; Ahmer, B.M. SdiA of Salmonella enterica is a LuxR homolog that detects mixed microbial communities. J. Bacteriol. 2001, 183, 5733–5742. [Google Scholar] [CrossRef] [Green Version]
- Landman, C.; Grill, J.-P.; Mallet, J.-M.; Marteau, P.; Humbert, L.; Le Balc’H, E.; Maubert, M.-A.; Perez, K.; Chaara, W.; Brot, L.; et al. Inter-kingdom effect on epithelial cells of the N-Acyl homoserine lactone 3-oxo-C12:2, a major quorum-sensing molecule from gut microbiota. PLoS ONE 2018, 13, e0202587. [Google Scholar] [CrossRef]
- Aguanno, D.; Coquant, G.; Postal, B.G.; Osinski, C.; Wieckowski, M.; Stockholm, D.; Grill, J.-P.; Carrière, V.; Seksik, P.; Thenet, S. The intestinal quorum sensing 3-oxo-C12:2 Acyl homoserine lactone limits cytokine-induced tight junction disruption. Tissue Barriers 2020, 8, 1832877. [Google Scholar] [CrossRef] [PubMed]
- Coquant, G.; Aguanno, D.; Brot, L.; Belloir, C.; Delugeard, J.; Roger, N.; Pham, H.-P.; Briand, L.; Moreau, M.; de Sordi, L.; et al. 3-oxo-C12:2-HSL, quorum sensing molecule from human intestinal microbiota, inhibits pro-inflammatory pathways in immune cells via bitter taste receptors. Sci. Rep. 2022, 12, 9440. [Google Scholar] [CrossRef] [PubMed]
- Peyrottes, A.; Coquant, G.; Brot, L.; Rainteau, D.; Seksik, P.; Grill, J.-P.; Mallet, J.-M. Anti-Inflammatory Effects of Analogues of N-Acyl Homoserine Lactones on Eukaryotic Cells. Int. J. Mol. Sci. 2020, 21, 9448. [Google Scholar] [CrossRef]
- Cataldi, T.R.I.; Bianco, G.; Abate, S. Profiling of N-acyl-homoserine lactones by liquid chromatography coupled with electrospray ionization and a hybrid quadrupole linear ion-trap and Fourier-transform ion-cyclotron-resonance mass spec-trometry (LC-ESI-LTQ-FTICR-MS). J. Mass. Spectrom. 2008, 43, 82–96. [Google Scholar] [CrossRef] [PubMed]
- IBDMDB Investigators; Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Harvey, R.F.; Bradshaw, J.M. A simple index of Crohns-disease activity. Lancet 1980, 1, 514. [Google Scholar] [CrossRef]
- Walmsley, R.S.; Ayres, R.C.; Pounder, R.E.; Allan, R.N. A simple clinical colitis activity index. Gut 1998, 43, 29–32. [Google Scholar] [CrossRef] [Green Version]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal–response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef] [Green Version]
- Doberva, M.; Sanchez-Ferandin, S.; Toulza, E.; Lebaron, P.; Lami, R. Diversity of quorum sensing autoinducer synthases in the Global Ocean Sampling metagenomic database. Aquat. Microb. Ecol. 2015, 74, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Le Berre, R.; Nguyen, S.; Nowak, E.; Kipnis, E.; Pierre, M.; Ader, F.; Courcol, R.; Guery, B.; Faure, K. Quorum-sensing activity and related virulence factor expression in clinically pathogenic isolates of Pseudomonas aeruginosa. Clin. Microbiol. Infect. 2008, 14, 337–343. [Google Scholar] [CrossRef]
- Brint, J.M.; Ohman, D.E. Synthesis of multiple exoproducts in Pseudomonas aeruginosa is under the control of RhlR-RhlI, another set of regulators in strain PAO1 with homology to the autoinducer-responsive LuxR-LuxI family. J. Bacteriol. 1995, 177, 7155–7163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandner-Miranda, L.; Vinuesa, P.; Soberón-Chávez, G.; Morales-Espinosa, R. Complete Genome Sequence of Serratia marcescens SmUNAM836, a Nonpigmented Multidrug-Resistant Strain Isolated from a Mexican Patient with Obstructive Pulmonary Disease. Genome Announc. 2016, 4, e01417-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, N.K.; Bulgin, R.; Crepin, V.F.; Cerdeño-Tárraga, A.M.; Schroeder, G.N.; Quail, M.A.; Lennard, N.; Corton, C.; Barron, A.; Clark, L.; et al. The Citrobacter rodentium genome sequence reveals convergent evolution with human pathogenic Escherichia coli. J. Bacteriol. 2010, 192, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Pumbwe, L.; Skilbeck, C.A.; Wexler, H.M. Presence of quorum-sensing systems associated with multidrug resistance and biofilm formation in Bacteroides fragilis. Microb. Ecol. 2008, 56, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Coquant, G.; Aguanno, D.; Pham, S.; Grellier, N.; Thenet, S.; Carrière, V.; Grill, J.-P.; Seksik, P. Gossip in the gut: Quorum sensing, a new player in the host-microbiota interactions. World J. Gastroenterol. 2021, 27, 7247–7270. [Google Scholar] [CrossRef]
- Thompson, J.A.; Oliveira, R.A.; Djukovic, A.; Ubeda, C.; Xavier, K.B. Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep. 2015, 10, 1861–1871. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Basset, C.; Holton, J.; Bazeos, A.; Vaira, D.; Bloom, S. Are Helicobacter species and enterotoxigenic Bacteroides fragilis involved in inflammatory bowel disease? Dig. Dis. Sci. 2004, 49, 1425–1432. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, Z.; Yan, Y.; Ji, L.; He, J.; Xuan, B.; Shen, C.; Ma, Y.; Jiang, S.; Ma, D.; et al. Enterotoxigenic Bacteroides fragilis Promotes Intestinal Inflammation and Malignancy by Inhibiting Exosome-Packaged miR-149-3p. Gastroenterology 2021, 161, 1552–1566.e12. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Lopez-Siles, M.; Enrich-Capó, N.; Aldeguer, X.; Sabat-Mir, M.; Duncan, S.; Garcia-Gil, L.J.; Martinez-Medina, M. Alterations in the Abundance and Co-occurrence of Akkermansia muciniphila and Faecalibacterium prausnitzii in the Colonic Mucosa of Inflammatory Bowel Disease Subjects. Front. Cell Infect. Microbiol. 2018, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. Available online: http://palaeo-electronica.org/2001_1/past/issue1_01.htm (accessed on 25 September 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-IBD n = 26 | CD n = 50 | UC n = 27 | |
|---|---|---|---|
| Sex (F/M) | 11/15 | 23/27 | 17/10 |
| Age (years) (mean +/− SEM) | 29.7 ± 4.0 | 25.9 ± 2.4 | 29.2 ± 3.5 |
| Paediatric * (n) | 12 | 26 | 12 |
| Disease duration (years) (mean +/− SEM) | 6.0 ± 2.8 | 5.7 ± 1.4 | |
| New diagnosis ** (n) | 35 | 14 | |
| Antibiotics | 0 | 8 | 1 |
| Steroids | 0 | 13 | 2 |
| History of surgery | 0 | 10 | 0 |
| Clinical activity score (mean +/− SEM) | HBI: 2.1 ± 0.3 Unknown: 4 patients | SCCAI: 1.6 ± 0.2 Unknown: 1 patient |
| Accession Number | Function | |
|---|---|---|
| luxR1 B. fragilis | BF9343_2602 | Putative DNA-binding response regulator |
| luxR2 B. fragilis | BF9343_2858 | Putative sigma factor |
| luxR3 B. fragilis | BF9343_3797 | Putative LuxR-family regulatory protein |
| luxR4 B. fragilis | BF9343_4003 | Putative transcriptional regulator GerE |
| luxR1 B. dorei | NZ_LR699004.1:3959839-3960450 | Putative DNA-binding response regulator |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grellier, N.; Suzuki, M.T.; Brot, L.; Rodrigues, A.M.S.; Humbert, L.; Escoubeyrou, K.; Rainteau, D.; Grill, J.-P.; Lami, R.; Seksik, P. Impact of IBD-Associated Dysbiosis on Bacterial Quorum Sensing Mediated by Acyl-Homoserine Lactone in Human Gut Microbiota. Int. J. Mol. Sci. 2022, 23, 15404. https://doi.org/10.3390/ijms232315404
Grellier N, Suzuki MT, Brot L, Rodrigues AMS, Humbert L, Escoubeyrou K, Rainteau D, Grill J-P, Lami R, Seksik P. Impact of IBD-Associated Dysbiosis on Bacterial Quorum Sensing Mediated by Acyl-Homoserine Lactone in Human Gut Microbiota. International Journal of Molecular Sciences. 2022; 23(23):15404. https://doi.org/10.3390/ijms232315404
Chicago/Turabian StyleGrellier, Nathan, Marcelino T. Suzuki, Loic Brot, Alice M. S. Rodrigues, Lydie Humbert, Karine Escoubeyrou, Dominique Rainteau, Jean-Pierre Grill, Raphaël Lami, and Philippe Seksik. 2022. "Impact of IBD-Associated Dysbiosis on Bacterial Quorum Sensing Mediated by Acyl-Homoserine Lactone in Human Gut Microbiota" International Journal of Molecular Sciences 23, no. 23: 15404. https://doi.org/10.3390/ijms232315404
APA StyleGrellier, N., Suzuki, M. T., Brot, L., Rodrigues, A. M. S., Humbert, L., Escoubeyrou, K., Rainteau, D., Grill, J.-P., Lami, R., & Seksik, P. (2022). Impact of IBD-Associated Dysbiosis on Bacterial Quorum Sensing Mediated by Acyl-Homoserine Lactone in Human Gut Microbiota. International Journal of Molecular Sciences, 23(23), 15404. https://doi.org/10.3390/ijms232315404