
Role of Microstructure in Drug Release from Chitosan Amorphous Solid Dispersions

Abstract
:1. Introduction
2. Results and Discussion
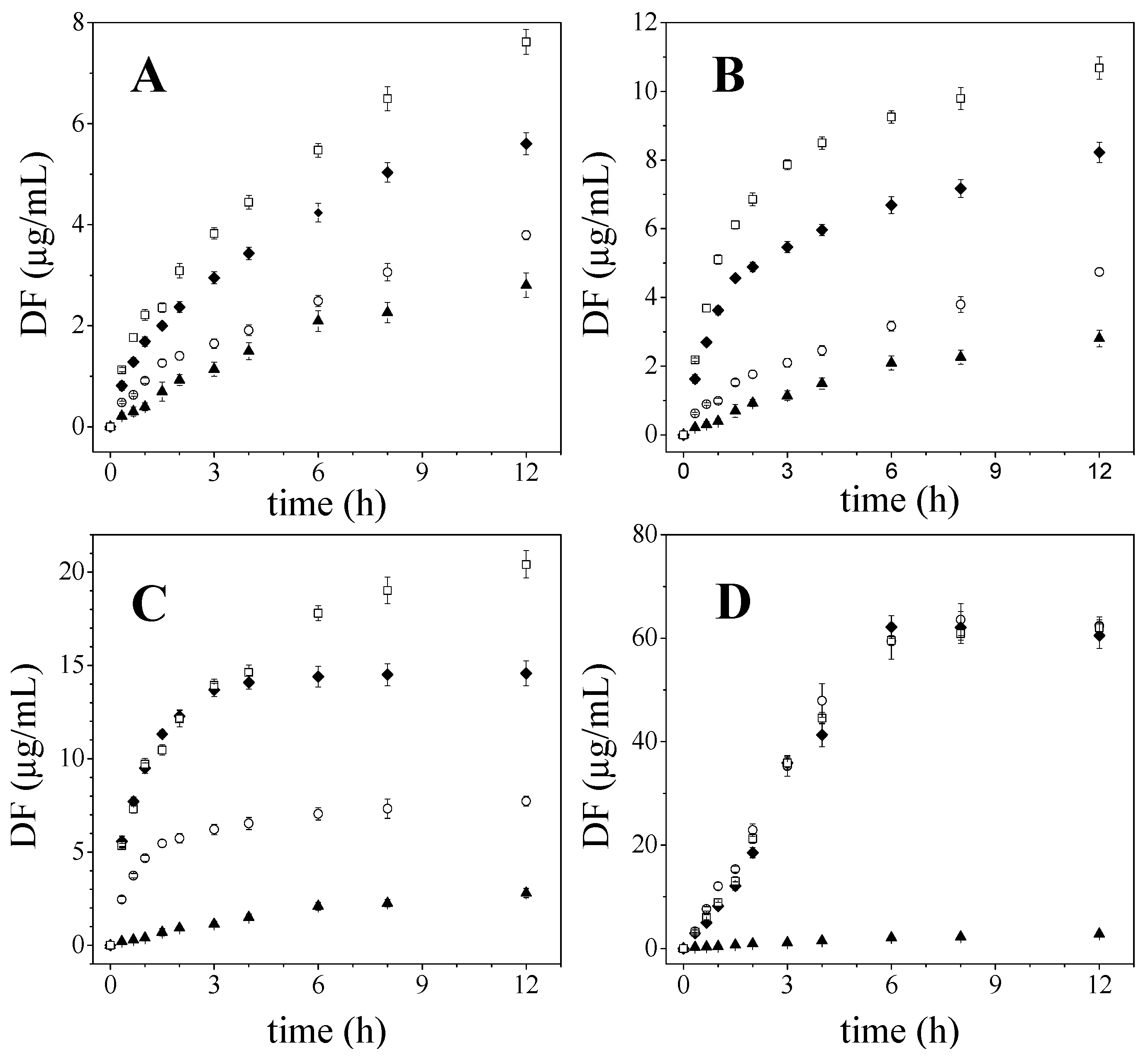
2.1. Diflunisal Release
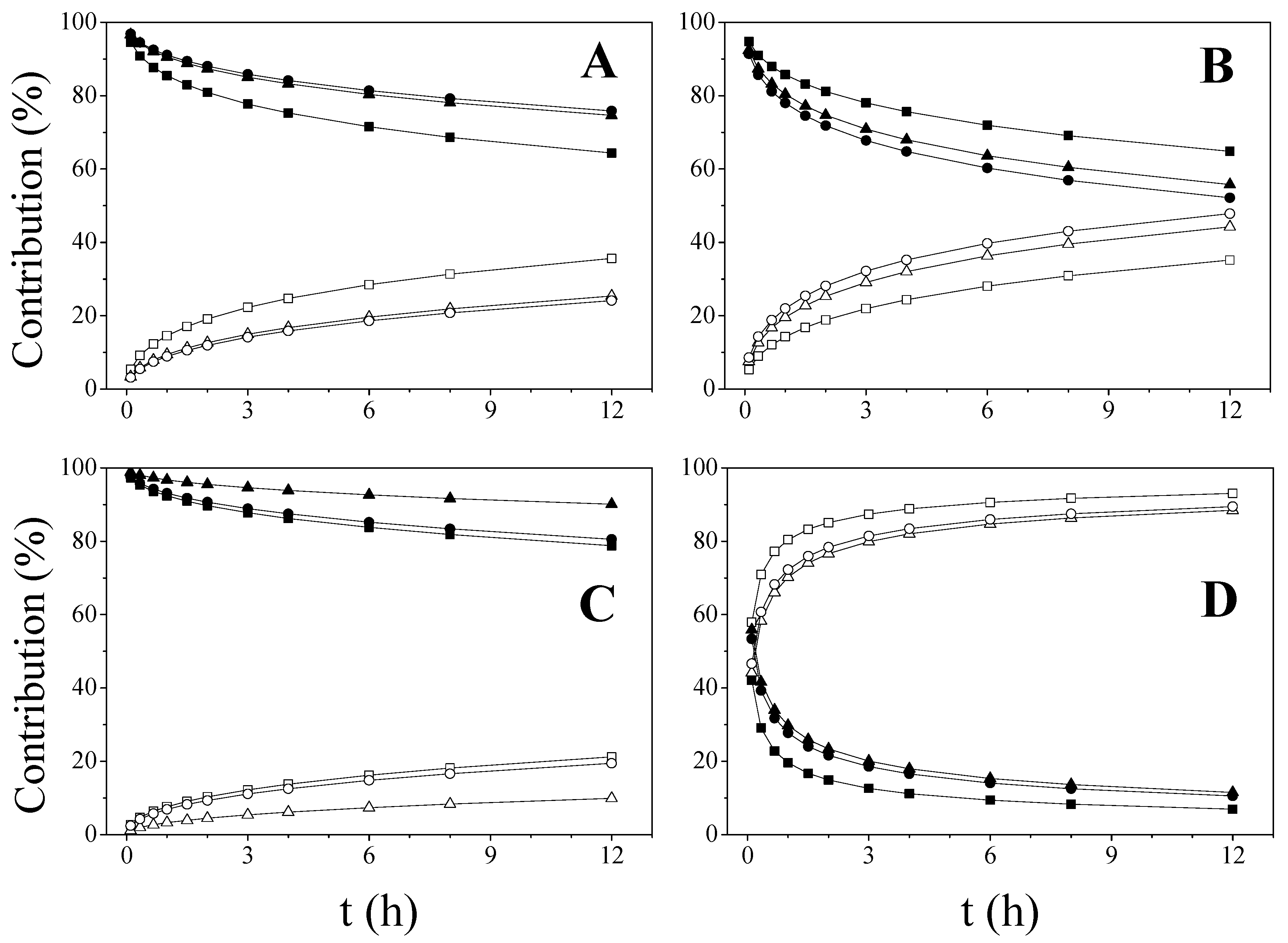
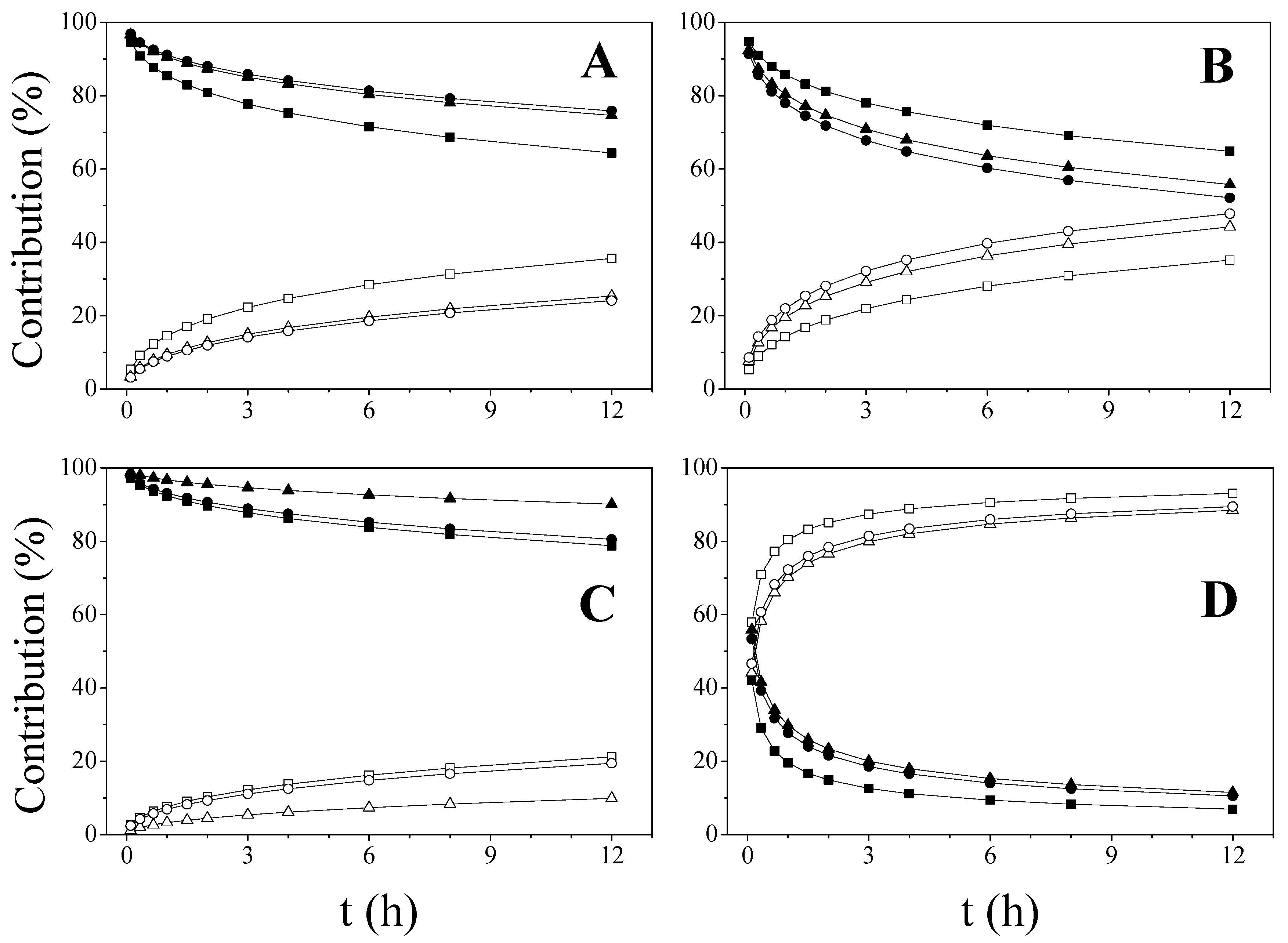
2.2. Influence of Microstructure on Drug Release
2.3. Mathematical Models
3. Materials and Methods
3.1. Materials
3.2. Preparation of Solid Dispersions
3.3. Dissolution Rate Studies
3.3.1. Release Parameters
3.3.2. Mathematical Models
3.4. Mercury Intrusion Porosimetry
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jia, W.; Yawman, P.D.; Pandya, K.M.; Sluga, K.; Ng, T.; Kou, D.; Nagapudi, K.; Luner, P.; Zhu, A.; Zhang, S.; et al. Assessing the Interrelationship of Microstructure, Properties, Drug Release Performance, and Preparation Process for Amorphous Solid Dispersions via Noninvasive Imaging Analytics and Material Characterization. Pharm. Res. 2022, 1–18. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef]
- Everaerts, M.; Cools, L.; Adriaensens, P.; Reekmans, G.; Baatsen, P.; van den Mooter, G. Investigating the Potential of Ethyl Cellulose and a Porosity-Increasing Agent as a Carrier System for the Formulation of Amorphous Solid Dispersions. Mol. Pharm. 2022, 19, 2712–2724. [Google Scholar] [CrossRef] [PubMed]
- Hamedi, H.; Moradi, S.; Hudson, S.M.; Tonelli, A.E.; King, M.W. Chitosan based bioadhesives for biomedical applications: A review. Carbohyd. Polym. 2022, 282, 119100. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An update on potential biomedical and pharmaceutical applications. Mar. Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef] [PubMed]
- Ardean, C.; Davidescu, C.M.; Nemeş, N.S.; Negrea, A.; Ciopec, M.; Duteanu, N.; Negrea, P.; Duda-Seiman, D.; Musta, V. Factors influencing the antibacterial activity of chitosan and chitosan modified by functionalization. Int. J. Mol. Sci. 2021, 22, 7449. [Google Scholar] [CrossRef] [PubMed]
- Fatullayeva, S.; Tagiyev, D.; Zeynalov, N.; Mammadova, S.; Aliyeva, E. Recent advances of chitosan-based polymers in biomedical applications and environmental protection. J. Polym. Res. 2022, 29, 259. [Google Scholar] [CrossRef]
- Kurakula, M.; Raghavendra, N. Prospection of recent chitosan biomedical trends: Evidence from patent analysis (2009–2020). Int. J. Biol. Macromol. 2020, 165, 1924–1938. [Google Scholar] [CrossRef]
- Shariatinia, Z. Carboxymethyl chitosan: Properties and biomedical applications. Int. J. Biol. Macromol. 2018, 120, 1406–1419. [Google Scholar] [CrossRef]
- Wang, W.; Meng, Q.; Li, Q.; Liu, J.; Zhou, M.; Jin, Z.; Zhao, K. Chitosan derivatives and their application in biomedicine. Int. J. Mol. Sci. 2020, 21, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S.; Singireddy, A.; Krishnamoorthy, K.; Rajappan, M. Nanosponges: A Novel Class of Drug Delivery System-Review. J. Pharm. Pharmaceut. Sci. 2012, 15, 103–111. [Google Scholar]
- Shirakawa, K.; Wang, L.; Man, N.; Maksimoska, J.; Sorum, A.W.; Lim, H.W.; Lee, I.S.; Shimazu, T.; Newman, J.C.; Schröder, S.; et al. Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity. eLife 2016, 5, e11156. [Google Scholar] [CrossRef] [PubMed]
- Snetkov, P.; Morozkina, S.; Olekhnovich, R.; Uspenskaya, M. Diflunisal targeted delivery systems: A review. Materials 2021, 14, 6687. [Google Scholar] [CrossRef] [PubMed]
- Lucio, D.; Zornoza, A.; Martínez-Ohárriz, M.C. Influence of chitosan and carboxymethylchitosan on the polymorphism and solubilisation of diflunisal. Int. J. Pharm. 2014, 467, 19–26. [Google Scholar] [CrossRef]
- Sapkal, S.; Babhulkar, M.; Rathi, A.; Mehetre, G.; Narkhede, M.B. An overview on the mechanisms of solubility and dissolution rate enhancement in solid dispersion. Int. J. Pharmtech. Res. 2013, 5, 31–39. [Google Scholar]
- Markl, D.; Strobel, A.; Schlossnikl, R.; Bøtker, J.; Bawuah, P.; Ridgway, C.; Rantanen, J.; Rades, T.; Gane, P.; Peiponen, K.E.; et al. Characterisation of pore structures of pharmaceutical tablets: A review. Int. J. Pharm. 2018, 538, 188–214. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.C. Microstructure of Tablet—Pharmaceutical Significance, Assessment, and Engineering. Pharm. Res. 2017, 34, 918–928. [Google Scholar] [CrossRef]
- McKeen, L.W. Permeability Properties of Plastics and Elastomers. In Permeation Reference Guide and Databook; Elsevier: Cambridge, CA, USA, 2012. [Google Scholar]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug dissolution. Int. J. Pharm. 2013, 453, 12–24. [Google Scholar] [CrossRef]
- Weinhold, M.X.; Thöming, J. On conformational analysis of chitosan. Carbohydr. Polym. 2011, 84, 1237–1243. [Google Scholar] [CrossRef]
- Chen, X.G.; Park, H.J. Chemical characteristics of O-carboxymethyl chitosans related to the preparation conditions. Carbohydr. Polym. 2003, 53, 355–359. [Google Scholar] [CrossRef]
- Ferrero, C.; Muñoz-Ruiz, A.; Jiménez-Castellanos, M.R. Fronts movement as a useful tool for hydrophilic matrix release mechanism elucidation. Int. J. Pharm. 2000, 202, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Crowley, M.M.; Schroeder, B.; Fredersdorf, A.; Obara, S.; Talarico, M.; Kucera, S.; McGinity, J.W. Physicochemical properties and mechanism of drug release from ethyl cellulose matrix tablets prepared by direct compression and hot-melt extrusion. Int. J. Pharm. 2004, 269, 509–522. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.E.; Rekhi, G.S.; Augsburger, L.L.; Shah, V.P. Methods to Compare Dissolution Profiles and a Rationale for Wide Dissolution Specifications for Metoprolol Tartrate Tablets. J. Pharm. Sci. 1997, 86, 690–700. [Google Scholar] [CrossRef]
- Muselík, J.; Komersová, A.; Kubová, K.; Matzick, K.; Skalická, B. A critical overview of FDA and EMA statistical methods to compare in vitro drug dissolution profiles of pharmaceutical products. Pharmaceutics 2021, 13, 1073. [Google Scholar] [CrossRef]
- Rescigno, A. Bioequivalence. Pharm. Res. 1992, 9, 925–928. [Google Scholar] [CrossRef]
- Laracuente, M.L.; Yu, M.H.; McHugh, K.J. Zero-order drug delivery: State of the art and future prospects. J. Control. Release 2020, 327, 834–856. [Google Scholar] [CrossRef]
- Higuchi, T.; York, N. Mechanism of Sustained-Action Medication Theoretical Analysis of Rate of Release of Solid Drugs Dispersed in Solid Matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Sujja-Areevath, J.; Munday, D.L.; Cox, P.J.; Khan, K.A. Relationship between swelling, erosion and drug release in hydrophillic natural gum mini-matrix formulations. Eur. J. Pharm. Sci. 1998, 6, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Giesche, H. Mercury porosimetry: A general (practical) overview. Part. Part. Syst. Charact. 2006, 23, 9–19. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef]
- Grund, J.; Körber, M.; Bodmeier, R. Predictability of drug release from water-insoluble polymeric matrix tablets. Eur. J. Pharm. Biopharm. 2013, 85, 650–655. [Google Scholar] [CrossRef]
- Heng, P.W.S.; Chan, L.W.; Easterbrook, M.G.; Li, X. Investigation of the influence of mean HPMC particle size and number of polymer particles on the release of aspirin from swellable hydrophilic matrix tablets. J. Control. Release 2001, 76, 39–49. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tablet | Profiles Compared | f1 | Rescigno Index (ξ) |
|---|---|---|---|
| DF-CSH | KN vs. PM | 40.22 | 0.24 |
| PM vs. CO | 23.45 | 0.13 | |
| DF-CSM | KN vs. PM | 56.69 | 0.35 |
| PM vs. CO | 27.31 | 0.16 | |
| DF-CSL | KN vs. PM | 51.64 | 0.34 |
| PM vs. CO | 10.03 | 0.09 |
| Sample | Total Intrusion Volume | Porosity | Permeability | Tortuosity Factor | |
|---|---|---|---|---|---|
| (mL/g) | (%) | (MDarcy) | |||
| DF-CSH | KN | 0.083 | 09.12 | 0.44 | 2.17 |
| DF-CSH | PM | 0.097 | 13.10 | 7.98 | 2.16 |
| DF-CSH | CO | 0.109 | 13.59 | 17.07 | 2.11 |
| DF-CSM | KN | 0.090 | 12.62 | 0.61 | 2.16 |
| DF-CSM | PM | 0.115 | 15.05 | 7.00 | 2.14 |
| DF-CSM | CO | 0.187 | 20.28 | 27.89 | 2.09 |
| DF-CSL | KN | 0.119 | 15.08 | 1.87 | 2.14 |
| DF-CSL | PM | 0.172 | 19.78 | 13.79 | 2.10 |
| DF-CSL | CO | 0.291 | 30.51 | 87.22 | 2.01 |
| DF-CMCS | KN | 0.083 | 10.37 | 0.49 | 2.21 |
| DF-CMCS | PM | 0.067 | 08.81 | 0.54 | 2.15 |
| DF-CMCS | CO | 0.057 | 07.83 | 0.52 | 2.18 |
| Tablets | Korsmeyer-Peppas | Peppas-Sahlin (m = 0.475) | |||||
|---|---|---|---|---|---|---|---|
| n | kKP × 102 (h−n) | R2 | kD × 102 (h−m) | kE × 102 (h−2m) | R2 | ||
| DF-CSH | KN | 0.58 (±0.02) | 17.0 (±0.6) | 0.994 | 14.7 (±1.0) | 2.5 (±0.5) | 0.994 |
| PM | 0.54 (±0.01) | 23.2 (±0.3) | 0.999 | 21.1 (±0.7) | 2.2 (±0.4) | 0.998 | |
| CO | 0.54 (±0.02) | 23.6 (±0.5) | 0.996 | 21.5 (±1.1) | 2.1 (±0.6) | 0.996 | |
| DF-CSM | KN | 0.58 (±0.02) | 17.4 (±1.2) | 0.996 | 15.0 (±0.8) | 2.5 (±0.4) | 0.996 |
| PM | 0.58 (±0.05) | 40.0 (±1.2) | 0.987 | 32.0 (±5.6) | 7.8 (±4.8) | 0.985 | |
| CO | 0.59 (±0.05) | 40.7 (±0.8) | 0.989 | 31.6 (±5.4) | 8.9 (±4.6) | 0.986 | |
| DF-CSL | KN | 0.51 (±0.03) | 51.2 (±0.3) | 0.997 | 47.2 (±4.2) | 3.9 (±4.0) | 0.996 |
| PM | 0.49 (±0.01) | 64.2 (±0.9) | 0.999 | 62.1 (±1.4) | 2.1 (±1.6) | 0.999 | |
| CO | 0.44 (±0.04) | 41.0 (±0.6) | 0.991 | 44.5 (±3.8) | 3.3 (±3.2) | 0.992 | |
| DF-CMCS | KN | 1.06 (±0.03) | 17.0 (±1.1) | 0.998 | - | 22.6 (±1.1) | 0.997 |
| PM | 1.17 (±0.10) | 14.1 (±1.2) | 0.977 | - | 24.3 (±2.8) | 0.978 | |
| CO | 1.14 (±0.06) | 15.2 (±1.1) | 0.991 | - | 24.5 (±1.9) | 0.991 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucio, D.; Zornoza, A.; Martínez-Ohárriz, M.C. Role of Microstructure in Drug Release from Chitosan Amorphous Solid Dispersions. Int. J. Mol. Sci. 2022, 23, 15367. https://doi.org/10.3390/ijms232315367
Lucio D, Zornoza A, Martínez-Ohárriz MC. Role of Microstructure in Drug Release from Chitosan Amorphous Solid Dispersions. International Journal of Molecular Sciences. 2022; 23(23):15367. https://doi.org/10.3390/ijms232315367
Chicago/Turabian StyleLucio, David, Arantza Zornoza, and Maria Cristina Martínez-Ohárriz. 2022. "Role of Microstructure in Drug Release from Chitosan Amorphous Solid Dispersions" International Journal of Molecular Sciences 23, no. 23: 15367. https://doi.org/10.3390/ijms232315367
APA StyleLucio, D., Zornoza, A., & Martínez-Ohárriz, M. C. (2022). Role of Microstructure in Drug Release from Chitosan Amorphous Solid Dispersions. International Journal of Molecular Sciences, 23(23), 15367. https://doi.org/10.3390/ijms232315367