

The PI3K/Akt Pathway in Meta-Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The PI3K/Akt Signaling Pathway, An Overview
3. The Role of Meta-Inflammation in the Development of Insulin Resistance
4. The Cellular Players of Meta-Inflammation: Adipocytes and Macrophages
4.1. Lean Versus Obese Adipose Tissue
Inflammation-Mediated Impairment of PI3K/Akt Signaling in Adipocytes
4.2. Macrophage Phenotypic Dynamics in Response to the Microenvironment
4.2.1. PI3K/Akt in Metabolic-Activated Macrophages
4.2.2. The Metabolic Reprogramming of Macrophage Polarization
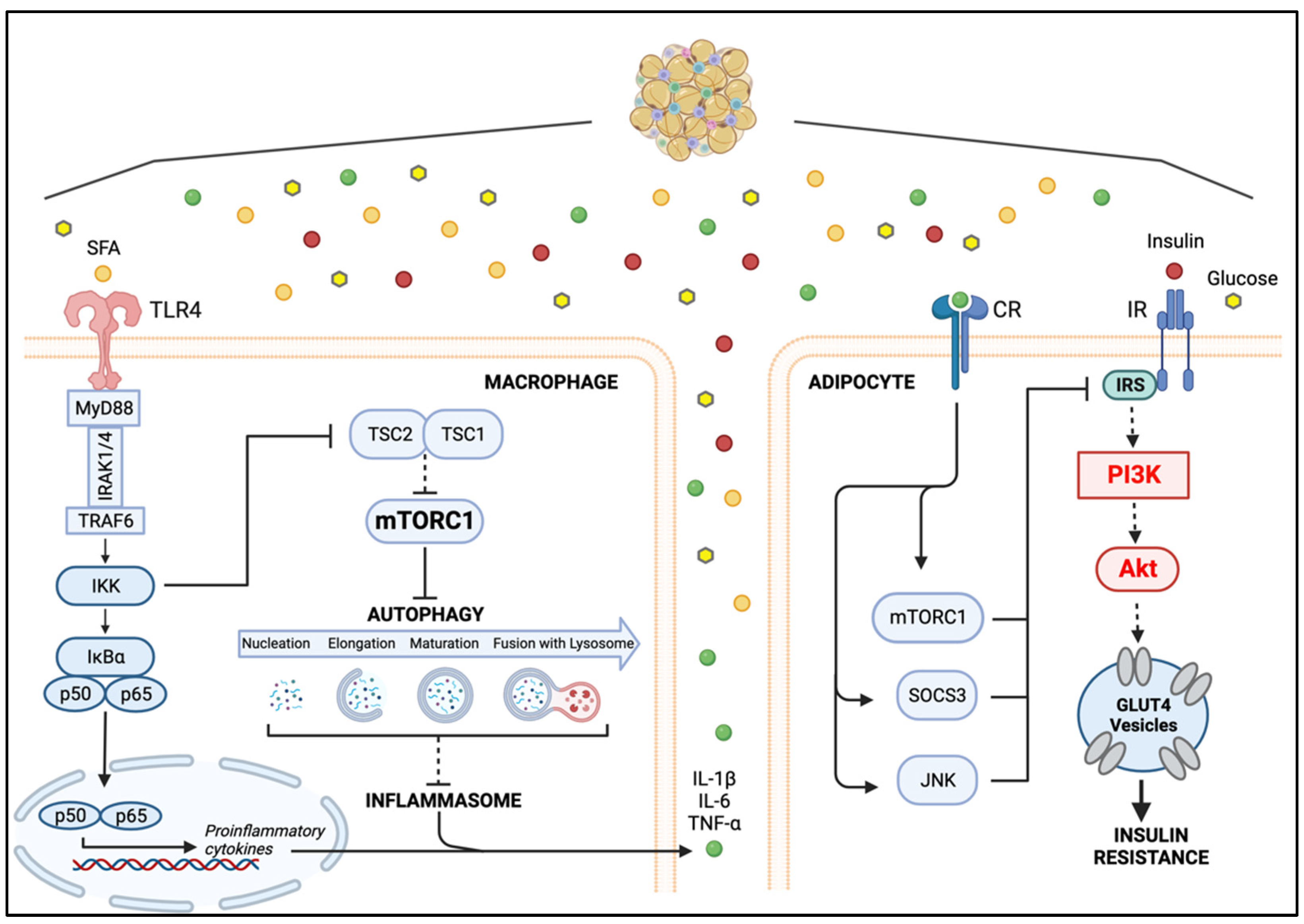
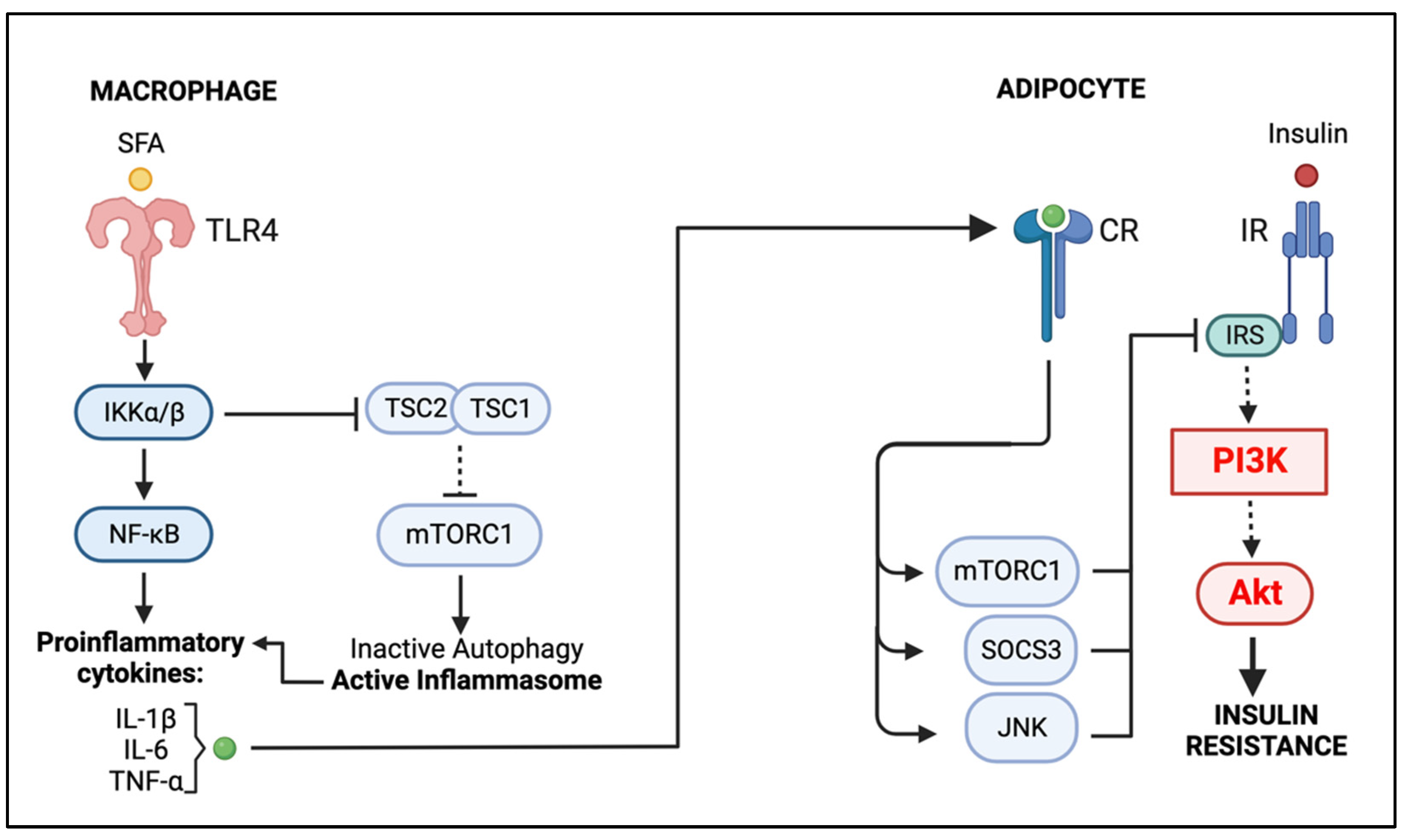
5. Macrophage–Adipocyte Crosstalk: Contribution of Impaired Autophagy to Adipose Tissue Insulin Resistance
6. Conclusions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- James, W.P.T. WHO recognition of the global obesity epidemic. Int. J. Obes. 2008, 32, S120–S126. [Google Scholar] [CrossRef] [PubMed]
- Caballero, B. The Global Epidemic of Obesity: An Overview. Epidemiol. Rev. 2007, 29, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, E.A.; Trogdon, J.G.; Cohen, J.W.; Dietz, W. Annual Medical Spending Attributable to Obesity: Payer-and Service-Specific Estimates. Health Aff. 2009, 28, w822–w831. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.; Dixit, V. Mechanisms of disease: Inflammasome activation and the development of type 2 diabetes. Front. Immunol. 2013, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Stevens, G.A.; Singh, G.M.; Lu, Y.; Danaei, G.; Lin, J.K.; Finucane, M.M.; Bahalim, A.N.; McIntire, R.K.; Gutierrez, H.R.; Cowan, M.; et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul. Health Metr. 2012, 10, 22. [Google Scholar] [CrossRef]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [PubMed]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage polarization and meta-inflammation. Transl. Res. 2018, 191, 29–44. [Google Scholar] [CrossRef]
- Charles-Messance, H.; Mitchelson, K.A.J.; De Marco Castro, E.; Sheedy, F.J.; Roche, H.M. Regulating metabolic inflammation by nutritional modulation. J. Allergy Clin. Immunol. 2020, 146, 706–720. [Google Scholar] [CrossRef]
- Engin, A.B.; Engin, A.; Gonul, II. The effect of adipocyte-macrophage crosstalk in obesity-related breast cancer. J. Mol. Endocrinol. 2019, 62, R201–R222. [Google Scholar] [CrossRef] [PubMed]
- Nitta, C.F.; Orlando, R.A. Crosstalk between immune cells and adipocytes requires both paracrine factors and cell contact to modify cytokine secretion. PLoS ONE 2013, 8, e77306. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Nishida, J.; Ogawa, Y. A Paracrine Loop Between Adipocytes and Macrophages Aggravates Inflammatory Changes. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D.; Shoelson, S.E. Immunometabolism: An emerging frontier. Nat. Rev. Immunol. 2011, 11, 81. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 2017, 127, 83–93. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Klemm, D.J. Adipose tissue insulin sensitivity and macrophage recruitment: Does PI3K pick the pathway? Adipocyte 2013, 2, 135–142. [Google Scholar] [CrossRef]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2015, 1851, 882–897. [Google Scholar] [CrossRef]
- Kim, S.-P.; Ha, J.M.; Yun, S.J.; Kim, E.K.; Chung, S.W.; Hong, K.W.; Kim, C.D.; Bae, S.S. Transcriptional activation of peroxisome proliferator-activated receptor-γ requires activation of both protein kinase A and Akt during adipocyte differentiation. Biochem. Biophys. Res. Commun. 2010, 399, 55–59. [Google Scholar] [CrossRef]
- Whitman, M.; Kaplan, D.R.; Schaffhausen, B.; Cantley, L.; Roberts, T.M. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 1985, 315, 239–242. [Google Scholar] [CrossRef]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef]
- Okkenhaug, K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol. 2013, 31, 675–704. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Chen, J.Z. Obesity, the PI3K/Akt signal pathway and colon cancer. Obes. Rev. 2009, 10, 610–616. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef]
- Tung, O.C.; Susan, E.; Rittenhouse, A.; Tsichlis, P.N. AKT/PKB and Other D3 Phosphoinositide-Regulated Kinases: Kinase Activation by Phosphoinositide-Dependent Phosphorylation. Annu. Rev. Biochem. 1999, 68, 965–1014. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Ieronymaki, E.; Daskalaki, M.G.; Lyroni, K.; Tsatsanis, C. Insulin Signaling and Insulin Resistance Facilitate Trained Immunity in Macrophages Through Metabolic and Epigenetic Changes. Front. Immunol. 2019, 10, 1330. [Google Scholar] [CrossRef]
- Cabail, M.Z.; Li, S.; Lemmon, E.; Bowen, M.E.; Hubbard, S.R.; Miller, W.T. The insulin and IGF1 receptor kinase domains are functional dimers in the activated state. Nat. Commun. 2015, 6, 6406. [Google Scholar] [CrossRef]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef]
- Lauterbach, M.A.R.; Wunderlich, F.T. Macrophage function in obesity-induced inflammation and insulin resistance. Pflügers Arch.-Eur. J. Physiol. 2017, 469, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Solis-Herrera, C.; Triplitt, C.; Cersosimo, E.; DeFronzo, R.A. Pathogenesis of Type 2 Diabetes Mellitus. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Blank, A.; Cui, C.; Schoenfelt, K.Q.; Zhou, G.; Xu, Y.; Khramtsova, G.; Olopade, F.; Shah, A.M.; Khan, S.A.; et al. Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. J. Exp. Med. 2019, 216, 1345–1358. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.-i.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Wu, D.; Qiu, Y. Adipose tissue macrophag.ge in obesity-associated metabolic diseases. Front. Immunol. 2022, 13, 977485. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- De Boer, A.A.; Monk, J.M.; Liddle, D.M.; Hutchinson, A.L.; Power, K.A.; Ma, D.W.L.; Robinson, L.E. Fish-oil-derived n-3 polyunsaturated fatty acids reduce NLRP3 inflammasome activity and obesity-related inflammatory cross-talk between adipocytes and CD11b+ macrophages. J. Nutr. Biochem. 2016, 34, 61–72. [Google Scholar] [CrossRef]
- Lindhorst, A.; Raulien, N.; Wieghofer, P.; Eilers, J.; Rossi, F.M.V.; Bechmann, I.; Gericke, M. Adipocyte death triggers a pro-inflammatory response and induces metabolic activation of resident macrophages. Cell Death Dis. 2021, 12, 579. [Google Scholar] [CrossRef]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef]
- Nelson, V.L.; Jiang, Y.P.; Dickman, K.G.; Ballou, L.M.; Lin, R.Z. Adipose tissue insulin resistance due to loss of PI3K p110α leads to decreased energy expenditure and obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1205–E1216. [Google Scholar] [CrossRef] [PubMed]
- Wibmer, A.G.; Becher, T.; Eljalby, M.; Crane, A.; Andrieu, P.C.; Jiang, C.S.; Vaughan, R.; Schöder, H.; Cohen, P. Brown adipose tissue is associated with healthier body fat distribution and metabolic benefits independent of regional adiposity. Cell Rep. Med. 2021, 2, 100332. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Martínez, M. PI3K: An Attractive Candidate for the Central Integration of Metabolism and Reproduction. Front. Endocrinol. 2011, 2, 110. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E. Adipose tissue: From lipid storage compartment to endocrine organ. Diabetes 2006, 55, 1537–1545. [Google Scholar] [CrossRef]
- Kiernan, K.; MacIver, N.J. The Role of the Adipokine Leptin in Immune Cell Function in Health and Disease. Front. Immunol. 2020, 11, 622468. [Google Scholar] [CrossRef]
- Iikuni, N.; Lam, Q.L.; Lu, L.; Matarese, G.; La Cava, A. Leptin and Inflammation. Curr. Immunol. Rev. 2008, 4, 70–79. [Google Scholar] [CrossRef]
- Nguyen, T.M.D. Adiponectin: Role in Physiology and Pathophysiology. Int. J. Prev. Med. 2020, 11, 136. [Google Scholar] [CrossRef]
- McLaughlin, T.; Craig, C.; Liu, L.F.; Perelman, D.; Allister, C.; Spielman, D.; Cushman, S.W. Adipose Cell Size and Regional Fat Deposition as Predictors of Metabolic Response to Overfeeding in Insulin-Resistant and Insulin-Sensitive Humans. Diabetes 2016, 65, 1245–1254. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef]
- Alkhouri, N.; Gornicka, A.; Berk, M.P.; Thapaliya, S.; Dixon, L.J.; Kashyap, S.; Schauer, P.R.; Feldstein, A.E. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J. Biol. Chem. 2010, 285, 3428–3438. [Google Scholar] [CrossRef]
- Johnson, A.M.; Olefsky, J.M. The origins and drivers of insulin resistance. Cell 2013, 152, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, M.J. Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell 1989, 57, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Bogan, J.S.; Kandror, K.V. Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr. Opin. Cell Biol. 2010, 22, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Belman, J.P.; Habtemichael, E.N.; Bogan, J.S. A proteolytic pathway that controls glucose uptake in fat and muscle. Rev. Endocr. Metab. Disord. 2014, 15, 55–66. [Google Scholar] [CrossRef]
- Zhong, X.; Ke, C.; Cai, Z.; Wu, H.; Ye, Y.; Liang, X.; Yu, L.; Jiang, S.; Shen, J.; Wang, L.; et al. LNK deficiency decreases obesity-induced insulin resistance by regulating GLUT4 through the PI3K-Akt-AS160 pathway in adipose tissue. Aging 2020, 12, 17150–17166. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Li, Y.; Matye, D.J.; Wang, Y.; Ding, W.X.; Li, T. Inhibition of insulin/PI3K/AKT signaling decreases adipose Sortilin 1 in mice and 3T3-L1 adipocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2924–2933. [Google Scholar] [CrossRef]
- Emanuelli, B.; Peraldi, P.; Filloux, C.; Chavey, C.; Freidinger, K.; Hilton, D.J.; Hotamisligil, G.S.; Van Obberghen, E. SOCS-3 Inhibits Insulin Signaling and Is Up-regulated in Response to Tumor Necrosis Factor-α in the Adipose Tissue of Obese Mice. J. Biol. Chem. 2001, 276, 47944–47949. [Google Scholar] [CrossRef]
- Shi, H.; Tzameli, I.; Bjørbaek, C.; Flier, J.S. Suppressor of cytokine signaling 3 is a physiological regulator of adipocyte insulin signaling. J. Biol. Chem. 2004, 279, 34733–34740. [Google Scholar] [CrossRef]
- Yoshimura, A.; Ito, M.; Chikuma, S.; Akanuma, T.; Nakatsukasa, H. Negative Regulation of Cytokine Signaling in Immunity. Cold Spring Harb. Perspect. Biol. 2018, 10, a028571. [Google Scholar] [CrossRef]
- Rui, L.; Yuan, M.; Frantz, D.; Shoelson, S.; White, M.F. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 2002, 277, 42394–42398. [Google Scholar] [CrossRef]
- Shi, H.; Cave, B.; Inouye, K.; Bjørbæk, C.; Flier, J.S. Overexpression of Suppressor of Cytokine Signaling 3 in Adipose Tissue Causes Local but Not Systemic Insulin Resistanc.c.ce. Diabetes 2006, 55, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Reyes, J.A.; Arellano-Plancarte, A.; Castillo-Hernandez, J.R. Angiotensin II and the development of insulin resistance: Implications for diabetes. Mol. Cell Endocrinol. 2009, 302, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Morisco, C.; Lembo, G.; Trimarco, B. Insulin Resistance and Cardiovascular Risk: New Insights From Molecular and Cellular Biology. Trends Cardiovasc. Med. 2006, 16, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Valente, V.; Izzo, R.; Manzi, M.V.; De Luca, M.R.; Barbato, E.; Morisco, C. Modulation of insulin resistance by renin angiotensin system inhibitors: Implications for cardiovascular prevention. Monaldi Arch. Chest Dis. 2021, 91, 1602. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.C.; Giani, J.F.; Burghi, V.; Mayer, M.A.; Carranza, A.; Taira, C.A.; Dominici, F.P. The Mas receptor mediates modulation of insulin signaling by angiotensin-(1-7). Regul. Pept. 2012, 177, 1–11. [Google Scholar] [CrossRef]
- Gutierrez-Rodelo, C.; Arellano-Plancarte, A.; Hernandez-Aranda, J.; Landa-Galvan, H.V.; Parra-Mercado, G.K.; Moreno-Licona, N.J.; Hernandez-Gonzalez, K.D.; Catt, K.J.; Villalobos-Molina, R.; Olivares-Reyes, J.A. Angiotensin II Inhibits Insulin Receptor Signaling in Adipose Cells. Int. J. Mol. Sci. 2022, 23, 6048. [Google Scholar] [CrossRef]
- Kolliniati, O.; Ieronymaki, E.; Vergadi, E.; Tsatsanis, C. Metabolic Regulation of Macrophage Activation. J. Innate Immun. 2022, 14, 51–68. [Google Scholar] [CrossRef]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Wentworth, J.M.; Naselli, G.; Brown, W.A.; Doyle, L.; Phipson, B.; Smyth, G.K.; Wabitsch, M.; O’Brien, P.E.; Harrison, L.C. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 2010, 59, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, Z.; Gomez-Salazar, M.; Alexaki, V.I. Innate Immune Cells in the Adipose Tissue in Health and Metabolic Disease. J. Innate Immun. 2022, 14, 4–30. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Xu, L.; Dong, N.; Li, F. NLRP3 inflammasome: The rising star in cardiovascular diseases. Front. Cardiovasc. Med. 2022, 9, 927061. [Google Scholar] [CrossRef]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef]
- Yang, Z.; Ming, X.F. Functions of arginase isoforms in macrophage inflammatory responses: Impact on cardiovascular diseases and metabolic disorders. Front. Immunol. 2014, 5, 533. [Google Scholar] [CrossRef]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef]
- Skuratovskaia, D.; Vulf, M.; Khaziakhmatova, O.; Malashchenko, V.; Komar, A.; Shunkin, E.; Shupletsova, V.; Goncharov, A.; Urazova, O.; Litvinova, L. Tissue-Specific Role of Macrophages in Noninfectious Inflammatory Disorders. Biomedicines 2020, 8, 400. [Google Scholar] [CrossRef]
- Xu, Z.J.; Gu, Y.; Wang, C.Z.; Jin, Y.; Wen, X.M.; Ma, J.C.; Tang, L.J.; Mao, Z.W.; Qian, J.; Lin, J. The M2 macrophage marker CD206: A novel prognostic indicator for acute myeloid leukemia. Oncoimmunology 2020, 9, 1683347. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.; Kwiatkowski, M.; Govorukhina, N.; Bischoff, R.; Melgert, B.N. Meta-Inflammation and Metabolic Reprogramming of Macrophages in Diabetes and Obesity: The Importance of Metabolites. Front. Immunol. 2021, 12, 746151. [Google Scholar] [CrossRef] [PubMed]
- Muir, L.A.; Cho, K.W.; Geletka, L.M.; Baker, N.A.; Flesher, C.G.; Ehlers, A.P.; Kaciroti, N.; Lindsly, S.; Ronquist, S.; Rajapakse, I.; et al. Human CD206+ macrophages associate with diabetes and adipose tissue lymphoid clusters. JCI Insight 2022, 7, e146563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kalkum, M.; Chait, B.T.; Roeder, R.G. The N-CoR-HDAC3 Nuclear Receptor Corepressor Complex Inhibits the JNK Pathway through the Integral Subunit GPS2. Mol. Cell 2002, 9, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Toubal, A.; Goñi, S.; Drareni, K.; Huang, Z.; Alzaid, F.; Ballaire, R.; Ancel, P.; Liang, N.; Damdimopoulos, A.; et al. Loss of the co-repressor GPS2 sensitizes macrophage activation upon metabolic stress induced by obesity and type 2 diabetes. Nat. Med. 2016, 22, 780–791. [Google Scholar] [CrossRef]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006. [Google Scholar] [CrossRef]
- Troutman, T.D.; Bazan, J.F.; Pasare, C. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle 2012, 11, 3559–3567. [Google Scholar] [CrossRef]
- Tanti, J.F.; Ceppo, F.; Jager, J.; Berthou, F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front. Endocrinol. 2012, 3, 181. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Islam, R.; Zhang, S.; Fang, J. Metabolic reprogramming of macrophages and its involvement in inflammatory diseases. EXCLI J. 2021, 20, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [PubMed]
- Obaid, M.; Udden, S.M.N.; Alluri, P.; Mandal, S.S. LncRNA HOTAIR regulates glucose transporter Glut1 expression and glucose uptake in macrophages during inflammation. Sci. Rep. 2021, 11, 232. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef]
- Hutami, I.R.; Izawa, T.; Khurel-Ochir, T.; Sakamaki, T.; Iwasa, A.; Tanaka, E. Macrophage Motility in Wound Healing Is Regulated by HIF-1α via S1P Signaling. Int. J. Mol. Sci. 2021, 22, 8992. [Google Scholar] [CrossRef]
- Kim, S.Y.; Jeong, E.; Joung, S.M.; Lee, J.Y. PI3K/Akt contributes to increased expression of Toll-like receptor 4 in macrophages exposed to hypoxic stress. Biochem. Biophys. Res. Commun. 2012, 419, 466–471. [Google Scholar] [CrossRef]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.L.; Oh, M.H.; Sun, I.H.; Chan-Li, Y.; Zhao, L.; Powell, J.D.; Horton, M.R. mTORC1 Signaling Regulates Proinflammatory Macrophage Function and Metabolism. J. Immunol. 2021, 207, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, T.; Li, L.; Sun, L.; Hou, Y.; Hu, X.; Zhang, L.; Tian, H.; Zhao, Q.; Peng, J.; et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat. Commun. 2014, 5, 4696. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Adipose tissue dysfunction in obesity. Exp. Clin. Endocrinol. Diabetes 2009, 117, 241–250. [Google Scholar] [CrossRef]
- Graham, T.E.; Yang, Q.; Blüher, M.; Hammarstedt, A.; Ciaraldi, T.P.; Henry, R.R.; Wason, C.J.; Oberbach, A.; Jansson, P.A.; Smith, U.; et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med. 2006, 354, 2552–2563. [Google Scholar] [CrossRef]
- Norseen, J.; Hosooka, T.; Hammarstedt, A.; Yore, M.M.; Kant, S.; Aryal, P.; Kiernan, U.A.; Phillips, D.A.; Maruyama, H.; Kraus, B.J.; et al. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol. Cell Biol. 2012, 32, 2010–2019. [Google Scholar] [CrossRef]
- Fite, A.; Abou-Samra, A.B.; Seyoum, B. Macrophages inhibit insulin signalling in adipocytes: Role of inducible nitric oxide synthase and nitric oxide. Can. J. Diabetes 2015, 39, 36–43. [Google Scholar] [CrossRef][Green Version]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Behl, T.; Sehgal, A.; Bala, R.; Chadha, S. Understanding the molecular mechanisms and role of autophagy in obesity. Mol. Biol. Rep. 2021, 48, 2881–2895. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Takabatake, Y.; Takahashi, A.; Kimura, T.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Matsusaka, T.; et al. High-Fat Diet-Induced Lysosomal Dysfunction and Impaired Autophagic Flux Contribute to Lipotoxicity in the Kidney. J. Am. Soc. Nephrol. 2017, 28, 1534–1551. [Google Scholar] [CrossRef]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. Embo J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef]
- Fraiberg, M.; Elazar, Z. Genetic defects of autophagy linked to disease. Prog Mol. Biol. Transl. Sci. 2020, 172, 293–323. [Google Scholar] [CrossRef]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.S.; Peak, J.J.; Kang, S.W.; Yoon, S.Y.; Song, Y. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016, 7, 35577–35591. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT—Emerging Players in Metabolism. Trends Endocrinol. Metab. 2018, 29, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Urits, I.; Israel, J.; Hakobyan, H.; Yusin, G.; Lassiter, G.; Fackler, N.; Berger, A.A.; Kassem, H.; Kaye, A.; Viswanath, O. Baricitinib for the treatment of rheumatoid arthritis. Reumatologia 2020, 58, 407–415. [Google Scholar] [CrossRef]
- Collotta, D.; Hull, W.; Mastrocola, R.; Chiazza, F.; Cento, A.S.; Murphy, C.; Verta, R.; Alves, G.F.; Gaudioso, G.; Fava, F.; et al. Baricitinib counteracts metaflammation, thus protecting against diet-induced metabolic abnormalities in mice. Mol. Metab. 2020, 39, 101009. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, K.R.; Brosius, F.C., 3rd; Adler, S.G.; Kretzler, M.; Mehta, R.L.; Tumlin, J.A.; Tanaka, Y.; Haneda, M.; Liu, J.; Silk, M.E.; et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: Results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transpl. 2018, 33, 1950–1959. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acosta-Martinez, M.; Cabail, M.Z. The PI3K/Akt Pathway in Meta-Inflammation. Int. J. Mol. Sci. 2022, 23, 15330. https://doi.org/10.3390/ijms232315330
Acosta-Martinez M, Cabail MZ. The PI3K/Akt Pathway in Meta-Inflammation. International Journal of Molecular Sciences. 2022; 23(23):15330. https://doi.org/10.3390/ijms232315330
Chicago/Turabian StyleAcosta-Martinez, Maricedes, and Maria Zulema Cabail. 2022. "The PI3K/Akt Pathway in Meta-Inflammation" International Journal of Molecular Sciences 23, no. 23: 15330. https://doi.org/10.3390/ijms232315330
APA StyleAcosta-Martinez, M., & Cabail, M. Z. (2022). The PI3K/Akt Pathway in Meta-Inflammation. International Journal of Molecular Sciences, 23(23), 15330. https://doi.org/10.3390/ijms232315330