
Discovery of a Flavonoid FM04 as a Potent Inhibitor to Reverse P-Glycoprotein-Mediated Drug Resistance in Xenografts and Improve Oral Bioavailability of Paclitaxel

 , ,
, ,
Abstract
:1. Introduction
2. Results
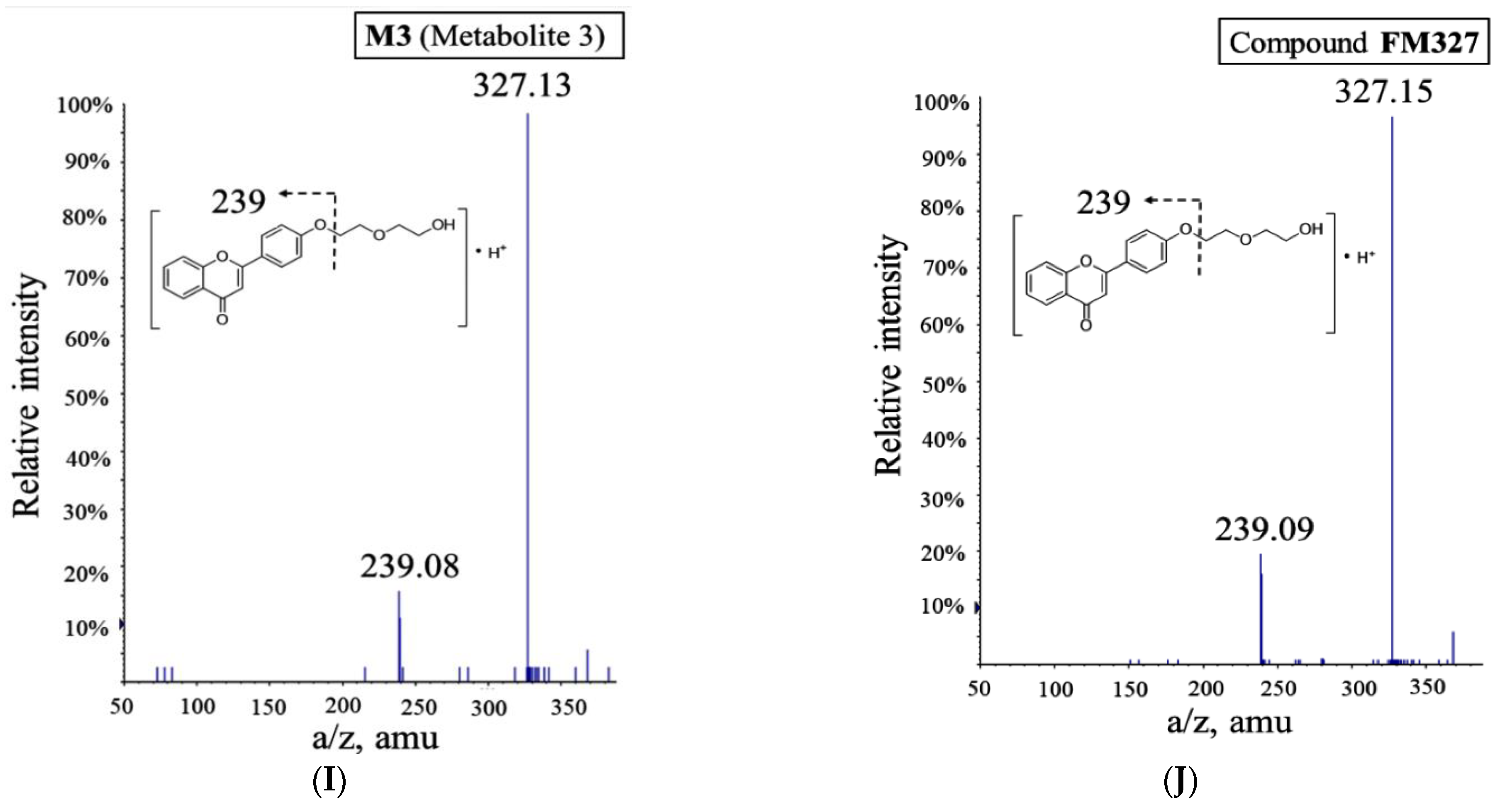
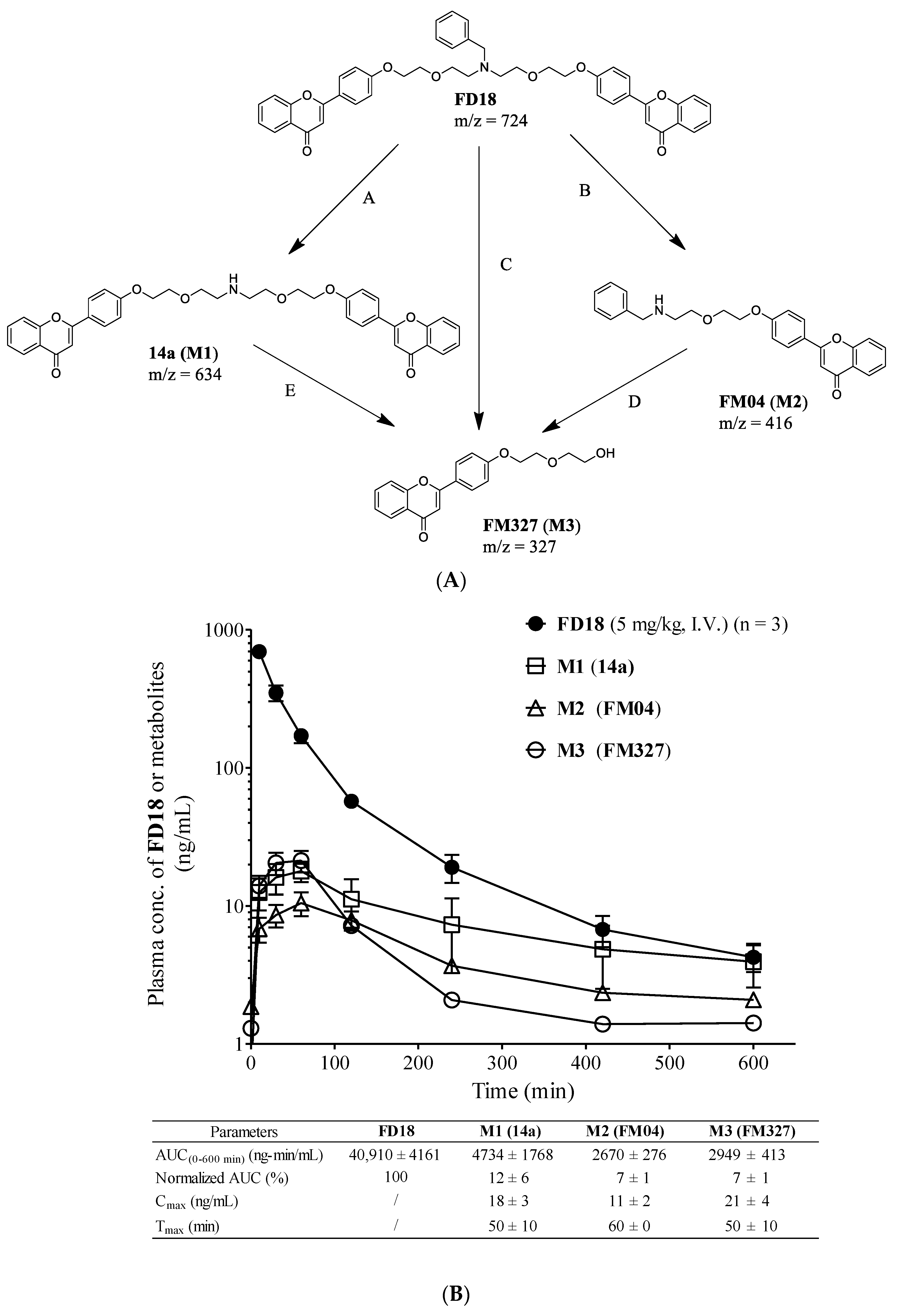
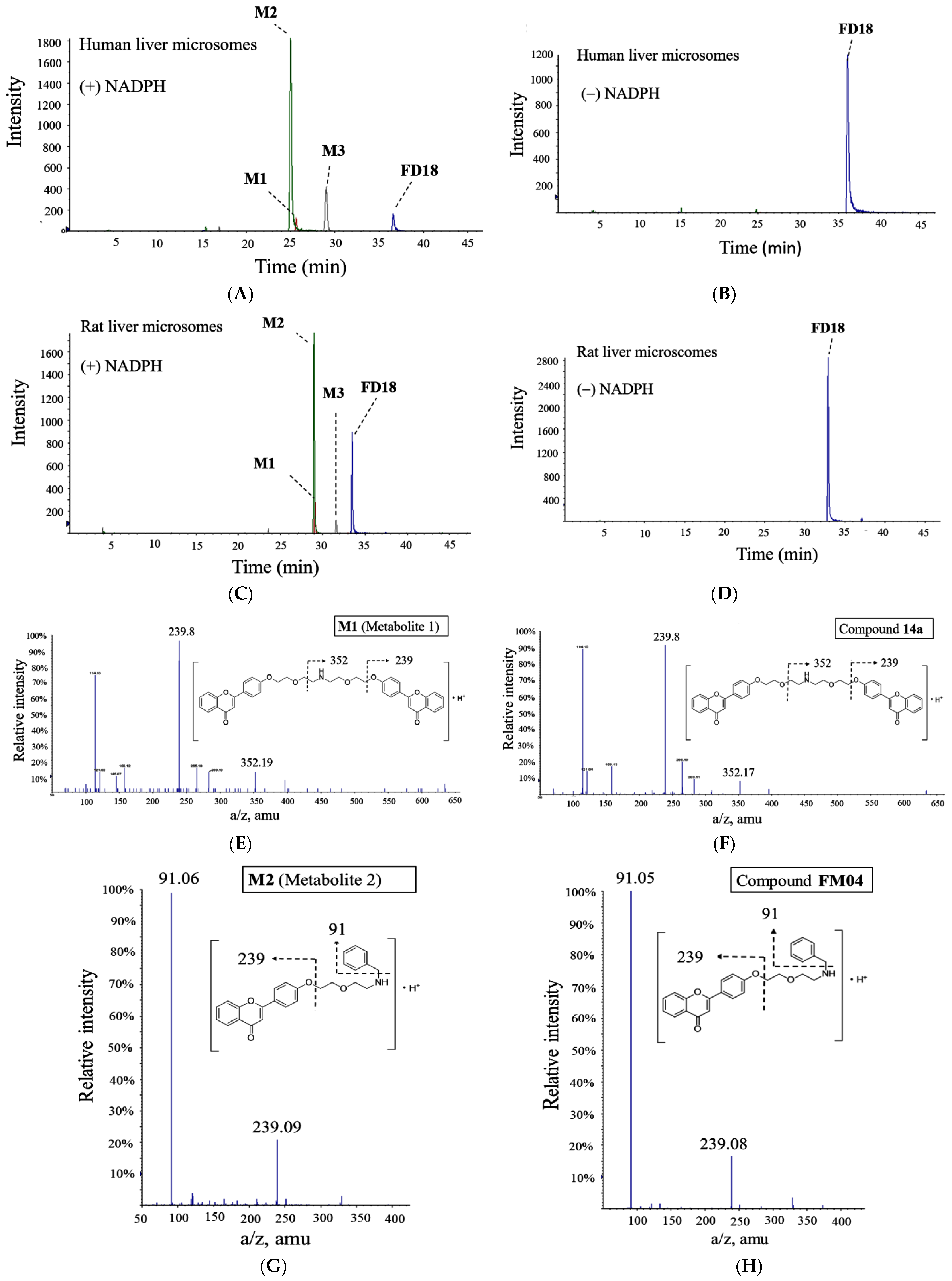
2.1. Metabolism of FD18 and Metabolites Identification in Rat and Human Liver Microsomes
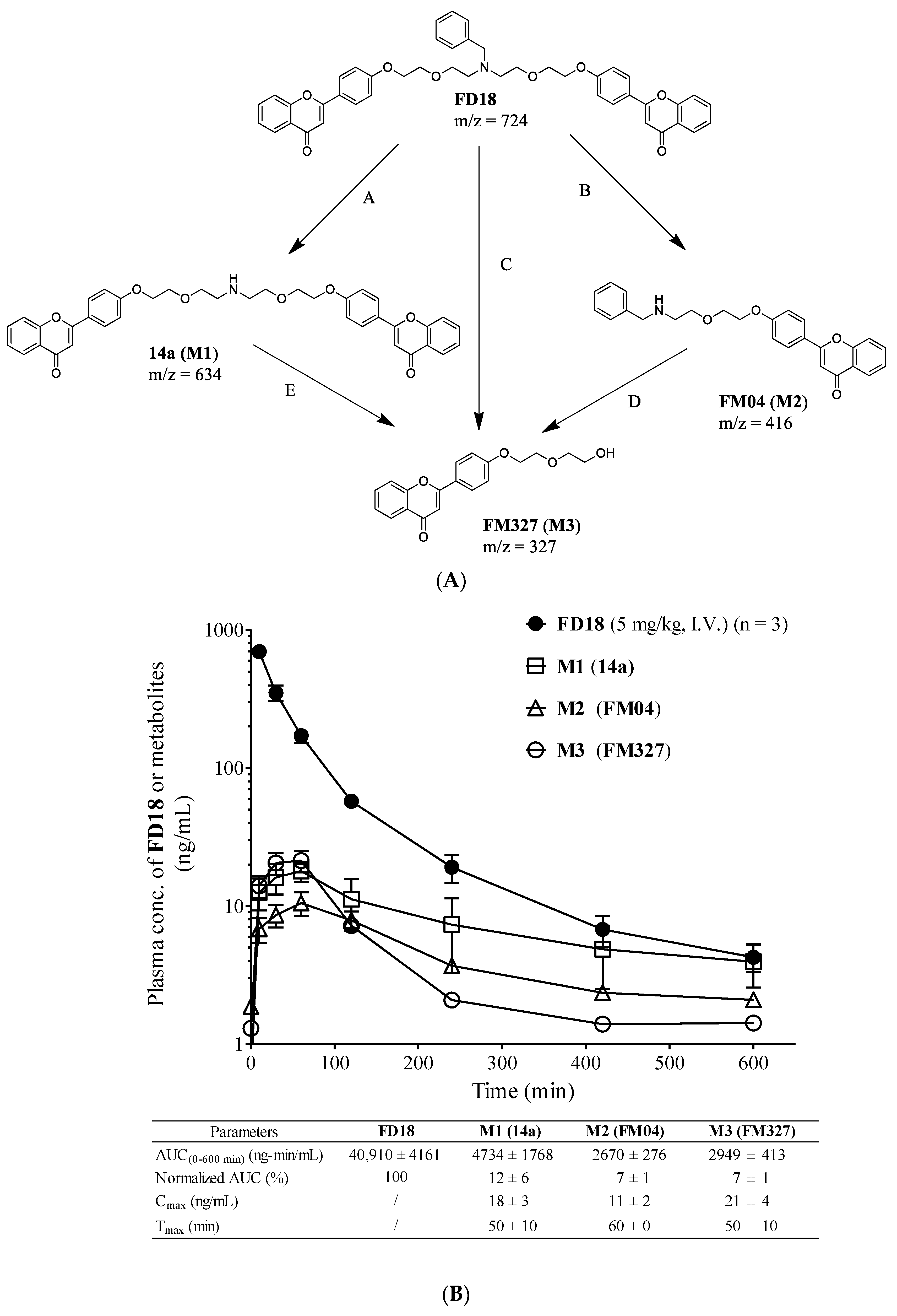
2.2. In Vivo Monitoring of Three Metabolites M1, M2, M3 Formation
2.3. In Vitro P-gp-Modulating Activity of Metabolites 14a (M1), FM04 (M2) and FM327 (M3)
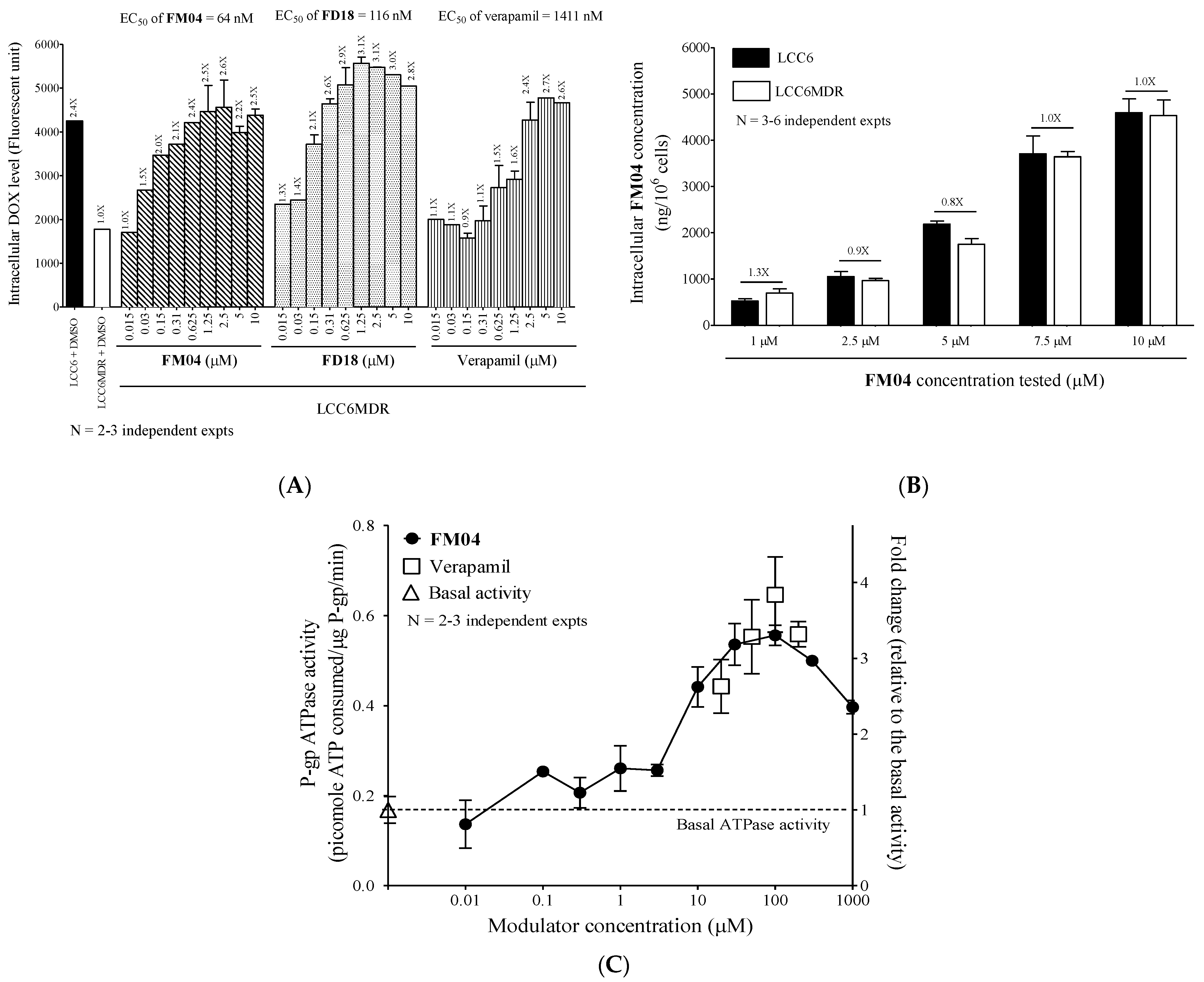
2.4. Selectivity of FM04 towards P-gp, BCRP and MRP1 Transporters
2.5. FM04 Increases Intracellular DOX Accumulation in LCC6MDR Cells
2.6. FM04 Is Not a Transport Substrate of P-gp
2.7. FM04 Does Not Inhibit P-gp ATPase Activity
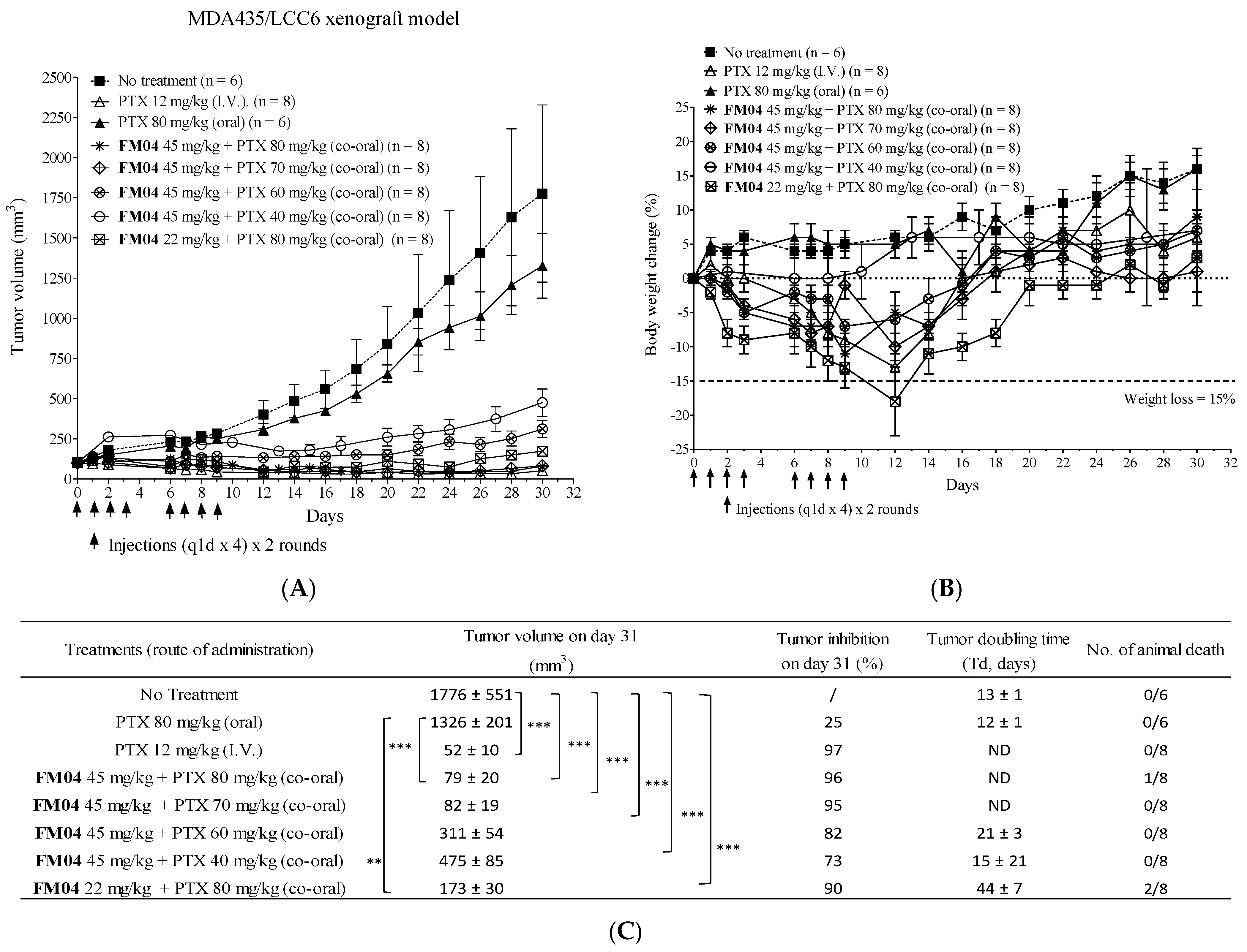
2.8. FM04 Reverses P-gp-Mediated PTX Drug Resistance in Human Melanoma MDA435/LCC6MDR Mice Xenograft Model
2.9. Pharmacokinetics Study of PTX or FM04 after Their Oral Co-Administration
2.10. Mechanistic Studies of FM04 on the Enhancement of Oral Bioavailability of PTX In Vitro
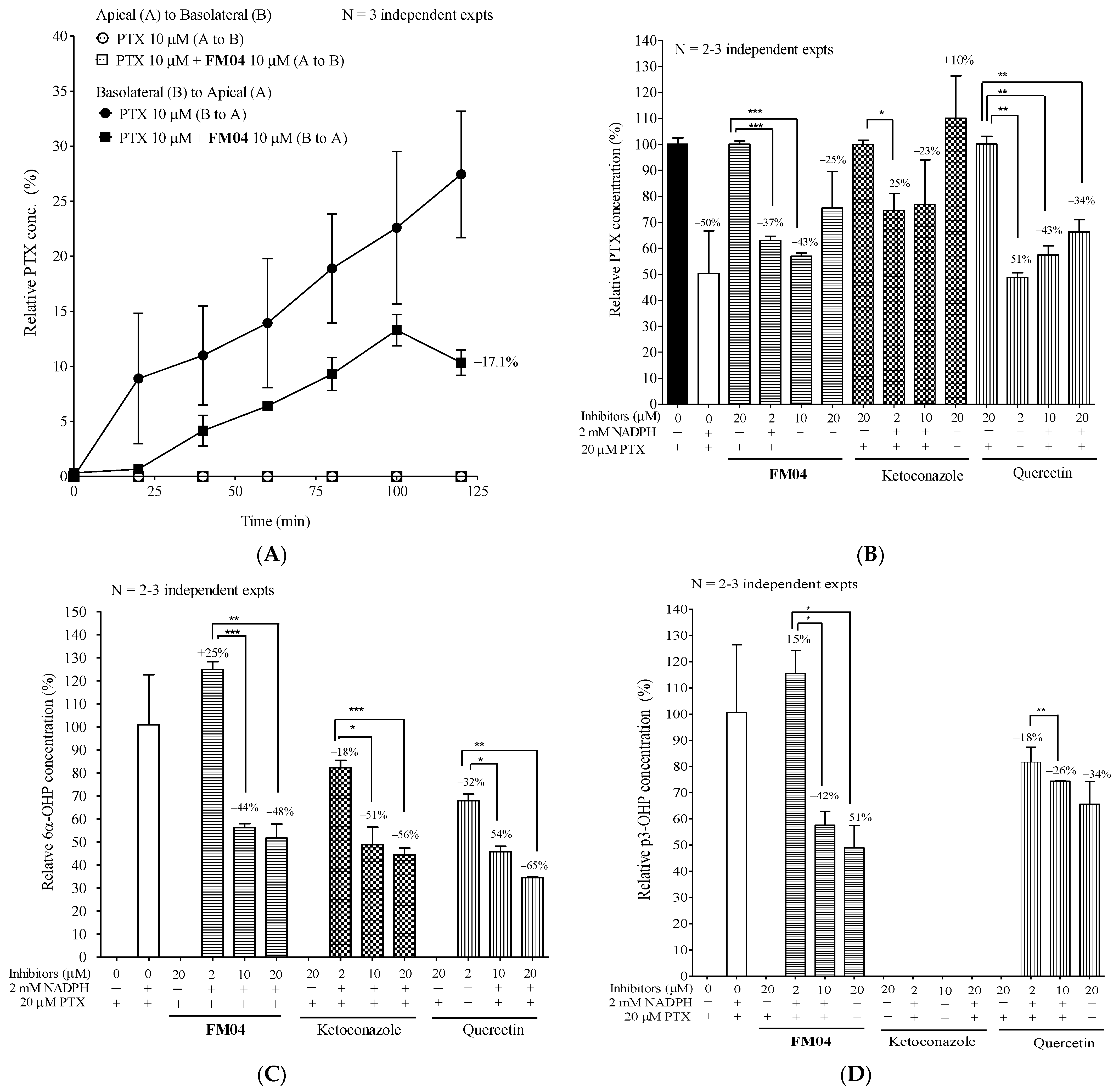
2.10.1. Effect of FM04 on Transepithelial Transport of PTX Using Caco-2 Transwell Assay In Vitro
2.10.2. Effect of FM04 on PTX Metabolism Using Human Liver Microsomes Assay In Vitro
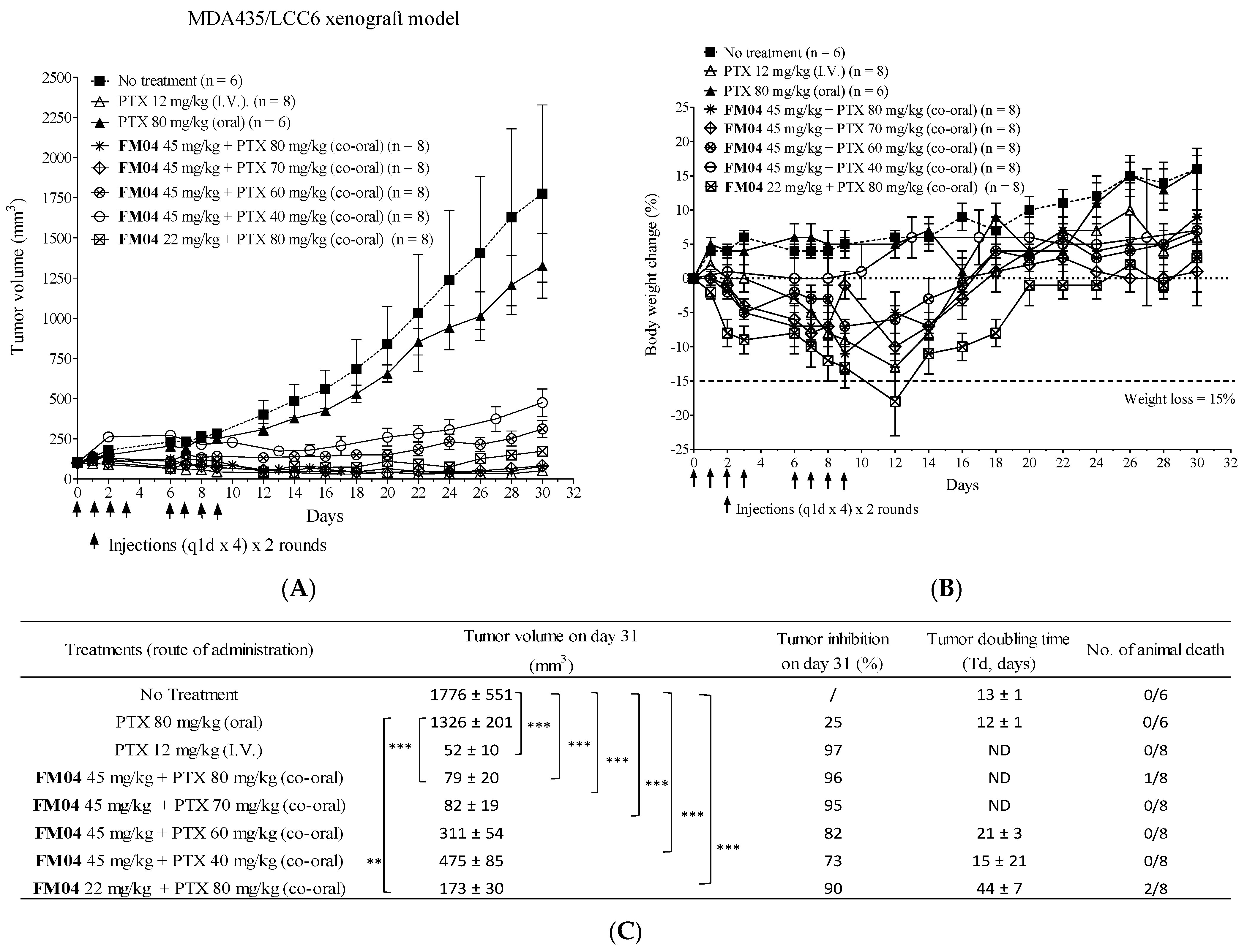
2.11. Efficacy of Orally Co-Administered FM04 and PTX in Treating the Human Melanoma MDA435/LCC6 Xenograft Model In Vivo
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Liver Microsomes Metabolism Study of FD18
4.3. HPLC-MS/MS Quadrupole-Time of Flight Analysis for Metabolite Identification
4.4. Synthesis of 1,13-Bis[4′-(4H-chromen-4-on-2-yl)phenyl]-1,4,10,13-tetraoxa-7-azatridecane (14a)
4.5. Synthesis of 2-(4-(2-(2-(benzylamino)ethoxy)ethoxy)phenyl)-4H-chromen-4-one (FM04)
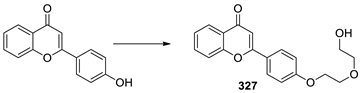
4.6. Synthesis of 2-(4-(2-(2-hydroxyethoxy)ethoxy)phenyl)-4H-chromen-4-one (FM327)
4.7. Cell Culture
4.8. In Vitro P-gp-Modulating Activity of FD18, 14a (M1), FM04 (M2) and FM327 (M3)
4.9. In Vitro BCRP- and MRP-1-Modulating Activity of FM04
4.10. Cytotoxicity Assay of FM04
4.11. Intracellular DOX Accumulation Assay
4.12. P-gp-ATPase Activity
4.13. Animal Study
4.14. In Vivo Metabolism Monitoring of FD18 and Its Three Metabolites M1 (14a), M2 (FM04), M3 (FM327)
4.15. In Vivo Efficacy of FM04 in Reversing PTX Resistance in the Human Melanoma MDA435/LCC6MDR Xenograft Model
4.16. Pharmacokinetic (PK) Study of FM04 and Its Effect on PK of PTX in Mice after Oral Co-administration
4.17. UPLC-MSMS Quantification of FM04 and Paclitaxel (PTX)
4.18. Effect of FM04 on Transepithelial Transport of PTX Using Caco-2 Transwell Assay In Vitro
4.19. Effect of FM04 on PTX Metabolism Using Human Liver Microsomes (HLM) Assay In Vitro
4.20. UPLC-MSMS Quantification of PTX and Its Metabolites
4.21. Efficacy of Orally Co-Administered PTX with FM04 in Treating the Human Melanoma MDA435/LCC6 Xenograft Model In Vivo
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| P-gp | P-glycoprotein |
| MDR | multidrug resistance |
| PTX | paclitaxel |
| DOX | doxorubicin |
| AUC | area under the curve |
| I.P. | intraperitoneal |
| I.V. | Intravenous |
| BCRP | breast cancer resistance protein |
| MRP1 | multidrug resistance associated protein 1 |
| GI | gastrointestinal |
References
- Abraham, J.; Salama, N.N.; Azab, A.K. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk. Lymphoma 2015, 56, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Amawi, H.; Sim, H.-M.; Tiwari, A.K.; Ambudkar, S.V.; Shukla, S. ABC Transporter-Mediated Multidrug-Resistant Cancer. Adv. Exp. Med. Biol. 2019, 1141, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehne, G. P-glycoprotein as a Drug Target in the Treatment of Multidrug Resistant Cancer. Curr. Drug Targets 2000, 1, 85–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981, 41, 1967–1972. [Google Scholar]
- Mickisch, G.H.; Noordzij, M.A.; vd Gaast, A.; Gebreamlack, P.; Köhrmann, K.U.; Mogler-Drautz, E.; Kupper, H.; Schröder, F.H. Dexverapamil to modulate vinblastine resistance in metastatic renal cell carcinoma. J. Cancer Res. Clin. Oncol. 1995, 121, R11–R16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pea, F.; Damiani, D.; Michieli, M.; Ermacora, A.; Baraldo, M.; Russo, D.; Fanin, R.; Baccarani, M.; Furlanut, M. Multidrug resistance modulation in vivo: The effect of cyclosporin A alone or with dexverapamil on idarubicin pharmacokinetics in acute leukemia. Eur. J. Clin. Pharmacol. 1999, 55, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Warner, E.; Hedley, D.; Andrulis, I.; Myers, R.; Trudeau, M.; Warr, D.; Pritchard, K.I.; Blackstein, M.; Goss, P.E.; Franssen, E.; et al. Phase II study of dexverapamil plus anthracycline in patients with metastatic breast cancer who have progressed on the same anthracycline regimen. Clin. Cancer Res. 1998, 4, 1451–1457. [Google Scholar]
- Chen, H.; Shien, K.; Suzawa, K.; Tsukuda, K.; Tomida, S.; Sato, H.; Torigoe, H.; Watanabe, M.; Namba, K.; Yamamoto, H.; et al. Elacridar, a third-generation ABCB1 inhibitor, overcomes resistance to docetaxel in non-small cell lung cancer. Oncol. Lett. 2017, 14, 4349–4354. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Son, J.-Y.; Lee, B.-M.; Kim, H.S.; Yoon, S. Highly Eribulin-resistant KBV20C Oral Cancer Cells Can Be Sensitized by Co-treatment with the Third-generation P-Glycoprotein Inhibitor, Elacridar, at a Low Dose. Anticancer Res. 2017, 37, 4139–4146. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Stewart, A.J.; Dangerfield, W.; Okiji, S.; Liddle, C.; Bootle, D.; Plumb, J.A.; Templeton, D.; Charlton, P. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res. 2001, 61, 749–758. [Google Scholar] [PubMed]
- Dantzig, A.H.; Shepard, R.L.; Cao, J.; Law, K.L.; Ehlhardt, W.J.; Baughman, T.M.; Bumol, T.F.; Starling, J.J. Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979. Cancer Res. 1996, 56, 4171–4179. [Google Scholar] [PubMed]
- Shepard, R.L.; Cao, J.; Starling, J.J.; Dantzig, A.H. Modulation of P-glycoprotein but not MRP1- or BCRP-mediated drug resistance by LY335979. Int. J. Cancer 2002, 103, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Edgerly, M.; Wilson, R.; Chen, C.; Rutt, A.; Bakke, S.; Robey, R.; Dwyer, A.; Goldspiel, B.; Balis, F.; et al. A Phase I Study of the P-Glycoprotein Antagonist Tariquidar in Combination with Vinorelbine. Clin. Cancer Res. 2009, 15, 3574–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planting, A.S.T.; Sonneveld, P.; Van Der Gaast, A.; Sparreboom, A.; Van Der Burg, M.E.L.; Luyten, G.P.M.; De Leeuw, K.; De Boer-Dennert, M.; Wissel, P.S.; Jewell, R.C.; et al. A phase I and pharmacologic study of the MDR converter GF120918 in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2005, 55, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Sandler, A.; Gordon, M.; de Alwis, D.P.; Pouliquen, I.; Green, L.; Marder, P.; Chaudhary, A.; Fife, K.; Battiato, L.; Sweeney, C.; et al. A Phase I Trial of a Potent P-Glycoprotein Inhibitor, Zosuquidar Trihydrochloride (LY335979), Administered Intravenously in Combination with Doxorubicin in Patients with Advanced Malignancy. Clin. Cancer Res. 2004, 10, 3265–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, Z.N.; Du, Y.J.; Li, X.Q.; Bao, Q.L.; Chen, P. Expression of MRP1, BCRP, LRP, and ERCC1 in advanced non-small-cell lung cancer: Correlation with response to chemotherapy and survival. Clin. Lung Cancer 2009, 10, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, R.; Nishiyama, Y.; Furuta, T.; Hatano, H.; Igarashi, Y.; Asakawa, N.; Kodaira, H.; Takahashi, H.; Aiyama, R.; Matsuzaki, T.; et al. Novel acrylonitrile derivatives, YHO-13177 and YHO-13351, reverse BCRP/ABCG2-mediated drug resistance in vitro and in vivo. Mol. Cancer Ther. 2011, 10, 1252–1263. [Google Scholar] [CrossRef] [Green Version]
- Chow, L.M.; Chan, T.H. ATP-Binding Cassette (ABC) Transporter Proteins, Multidrug Resistance and Novel Flavonoid Dimers as Potent, Nontoxic and Selective Inhibitors. Can. J. Chem. 2022, 100, 85–97. [Google Scholar] [CrossRef]
- Saeed, M.; Kadioglu, O.; Khalid, H.; Sugimoto, Y.; Efferth, T. Activity of the dietary flavonoid, apigenin, against multidrug-resistant tumor cells as determined by pharmacogenomics and molecular docking. J. Nutr. Biochem. 2015, 26, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.-F.; Zhao, Y.; Burkett, B.A.; Wong, I.L.K.; Chow, L.M.C.; Chan, T.H. Flavonoid Dimers as Bivalent Modulators for P-Glycoprotein-Based Multidrug Resistance: Synthetic Apigenin Homodimers Linked with Defined-Length Poly(ethylene glycol) Spacers Increase Drug Retention and Enhance Chemosensitivity in Resistant Cancer Cells. J. Med. Chem. 2006, 49, 6742–6759. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-F.; Zhao, Y.; Chow, T.W.S.; Yan, C.S.W.; Ma, D.L.; Burkett, B.A.; Wong, I.L.K.; Chow, L.M.C.; Chan, T.H. Flavonoid Dimers as Bivalent Modulators for P-Glycoprotein-Based Multidrug Resistance: Structure-Activity Relationships. ChemMedChem 2009, 4, 594–614. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-F.; Wong, I.L.K.; Kan, J.W.; Yan, C.S.W.; Chow, L.M.C.; Chan, T.H. Amine Linked Flavonoid Dimers as Modulators for P-Glycoprotein-Based Multidrug Resistance: Structure–Activity Relationship and Mechanism of Modulation. J. Med. Chem. 2012, 55, 1999–2014. [Google Scholar] [CrossRef]
- Yan, C.S.; Wong, I.L.; Chan, K.F.; Kan, J.W.; Chong, T.C.; Law, M.C.; Zhao, Y.; Chan, S.W.; Chan, T.H.; Chow, L.M. A New Class of Safe, Potent, and Specific P-gp Modulator: Flavonoid Dimer FD18 Reverses P-gp-Mediated Multidrug Resistance in Human Breast Xenograft in Vivo. Mol. Pharm. 2015, 12, 3507–3517. [Google Scholar] [CrossRef]
- Chan, C.-F.; Liu, Z.; Wong, I.L.K.; Zhao, X.; Yang, Z.; Zheng, J.; Lee, M.M.; Chan, M.K.; Chan, T.H.; Chow, L.M.C. Amine-Linked Flavonoids as Agents against Cutaneous Leishmaniasis. Antimicrob. Agents Chemother. 2021, 65, e02165-20. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.L.; Chan, K.F.; Tsang, K.H.; Lam, C.Y.; Zhao, Y.; Chan, T.H.; Chow, L.M. Modulation of multidrug resistance protein 1 (MRP1/ABCC1)-mediated multidrug resistance by bivalent apigenin homodimers and their derivatives. J. Med. Chem. 2009, 52, 5311–5322. [Google Scholar] [CrossRef] [PubMed]
- Cohn, D.E.; Sill, M.W.; Walker, J.L.; O′Malley, D.; Nagel, C.I.; Rutledge, T.L.; Bradley, W.; Richardson, D.L.; Moxley, K.M.; Aghajanian, C. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin®) in recurrent ovarian, tubal, or peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 146, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Morishige, K.; Enomoto, T.; Kimura, T. Carboplatin and paclitaxel as an initial treatment in patients with stage IVb cervical cancer: A report of 7 cases and a review of the literature. J. Gynecol. Oncol. 2010, 21, 93–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schettini, F.; Giuliano, M.; De Placido, S.; Arpino, G. Nab-paclitaxel for the treatment of triple-negative breast cancer: Rationale, clinical data and future perspectives. Cancer Treat. Rev. 2016, 50, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-H.; Mao, J.-W.; Tan, X.-L. Research progress on the source, production, and anti-cancer mechanisms of paclitaxel. Chin. J. Nat. Med. 2020, 18, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Kothandan, G.; Gadhe, C.G.; Madhavan, T.; Choi, C.H.; Cho, S.J. Docking and 3D-QSAR (quantitative structure activity relationship) studies of flavones, the potent inhibitors of p-glycoprotein targeting the nucleotide binding domain. Eur. J. Med. Chem. 2011, 46, 4078–4088. [Google Scholar] [CrossRef]
- Van Veen, H.W.; Margolles, A.; Müller, M.; Higgins, C.F.; Konings, W.N. The homodimeric ATP-binding cassette transporter LmrA mediates multidrug transport by an alternating two-site (two-cylinder engine) mechanism. EMBO J. 2000, 19, 2503–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Van Zuylen, L.; Gianni, L.; Verweij, J.; Mross, K.; Brouwer, E.; Loos, W.J.; Sparreboom, A. Inter-relationships of paclitaxel disposition, infusion duration and Cremophor EL kinetics in cancer patients. Anti-Cancer Drugs 2000, 11, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Wandel, C.; Kim, R.B.; Stein, C.M. “Inactive” excipients such as Cremophor can affect in vivo drug disposition. Clin. Pharmacol. Ther. 2003, 73, 394–396. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.T.; Misser, V.H.S.; Pieters, R.C.; Ten Bokkel Huinink, W.W.; Veenhof, C.H.N.; Vermorken, J.B.; Pinedo, H.M.; Beijnen, J.H. Taxanes: A New Class of Antitumor Agents. Cancer Investig. 1995, 13, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.-O.; Lee, S.H.; Lee, G.S.; Kim, M.S.; Ahn, Y.-G.; Lee, J.H.; Kim, S.W.; Kim, K.H.; Lee, M.G. Selective inhibition of MDR1 (ABCB1) by HM30181 increases oral bioavailability and therapeutic efficacy of paclitaxel. Eur. J. Pharmacol. 2010, 627, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Van Asperen, J.; Van Tellingen, O.; Sparreboom, A.; Schinkel, A.H.; Borst, P.; Nooijen, W.J.; Beijnen, J.H. Enhanced oral bioavailability of paclitaxel in mice treated with the P-glycoprotein blocker SDZ PSC 833. Br. J. Cancer 1997, 76, 1181–1183. [Google Scholar] [CrossRef] [Green Version]
- Van Asperen, J.; Van Tellingen, O.; Van Der Valk, M.A.; Rozenhart, M.; Beijnen, J.H. Enhanced oral absorption and decreased elimination of paclitaxel in mice cotreated with cyclosporin A. Clin. Cancer Res. 1998, 4, 2293–2297. [Google Scholar]
- Mei, L.; Zhang, Z.; Zhao, L.; Huang, L.; Yang, X.-L.; Tang, J.; Feng, S.-S. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv. Drug Deliv. Rev. 2013, 65, 880–890. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, J.J.M.A.; Lagas, J.S.; Wagenaar, E.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H.; Schinkel, A.H. Oral co-administration of elacridar and ritonavir enhances plasma levels of oral paclitaxel and docetaxel without affecting relative brain accumulation. Br. J. Cancer 2014, 110, 2669–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrikx, J.J.; Lagas, J.S.; Rosing, H.; Schellens, J.H.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein and cytochrome P450 3A act together in restricting the oral bioavailability of paclitaxel. Int. J. Cancer 2013, 132, 2439–2447. [Google Scholar] [CrossRef]
- König, J.; Müller, F.; Fromm, M.F. Transporters and Drug-Drug Interactions: Important Determinants of Drug Disposition and Effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauret, N.; Gauthier, A.; A. Nicoll-Griffith, D. Effect of common organic solvents on in vitro cytochrome P450-mediated metabolic activities in human liver microsomes. Drug Metab. Dispos. 1998, 26, 1–4. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | |||||
|---|---|---|---|---|---|
| FD18 | 14a (M1) | FM04 (M2) | FM327 (M3) | ||
| EC50 (nM) needed for reversing MDR in LCC6MDR cells | PTX | 148 ± 18 a | 305 ± 35 a | 83 ± 7 | >1000 |
| Vinblastine | 173 ± 27 a | ND | 61 ± 13 | ND | |
| Vincristine | 179 ± 32 a | ND | 83 ± 11 | ND | |
| DOX | 131 ± 13 a | ND | 153 ± 39 | ND | |
| Daunorubicin | 95 ± 25 a | ND | 88 ± 52 | ND | |
| Mitoxantrone | 90 ± 20 a | ND | 64 ± 27 | ND | |
| Physicochemical properties | Molecular Weight | 724 | ND | 415 | ND |
| CLogP | 9.0 | ND | 4.9 | ND | |
| tPSA (Å2) | 92.8 | ND | 56.8 | ND | |
| Compounds | IC50 of Anticancer Drugs (nM) | |||||
|---|---|---|---|---|---|---|
| P-gp-Transfectant LCC6MDR | BCRP-Transfectant HEK293/R2 | MRP1-Transfectant 2008/MRP1 | ||||
| Paclitaxel (nM) | RF | Topotecan (nM) | RF | DOX (nM) | RF | |
| DMSO | 129.6 ± 7.9 | 1.0 | 224.8 ± 32.0 | 1.0 | 616.0 ± 54.2 | 1 |
| 1 µM FM04 | 4.6 ± 0.5 | 28.2 | 131.7 ± 14.5 | 1.7 | 522.2 ± 27.6 | 1.2 |
| 1 µM Cyclosporine A | 1.8 ± 0.5 | 72.0 | / | / | / | / |
| 1 µM Ko143 | / | / | 16.4 ± 1.2 | 13.7 | / | / |
| 1 µM FD-4e | / | / | / | / | 44.5 ± 10.1 | 13.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kan, J.W.Y.; Yan, C.S.W.; Wong, I.L.K.; Su, X.; Liu, Z.; Chan, T.H.; Chow, L.M.C. Discovery of a Flavonoid FM04 as a Potent Inhibitor to Reverse P-Glycoprotein-Mediated Drug Resistance in Xenografts and Improve Oral Bioavailability of Paclitaxel. Int. J. Mol. Sci. 2022, 23, 15299. https://doi.org/10.3390/ijms232315299
Kan JWY, Yan CSW, Wong ILK, Su X, Liu Z, Chan TH, Chow LMC. Discovery of a Flavonoid FM04 as a Potent Inhibitor to Reverse P-Glycoprotein-Mediated Drug Resistance in Xenografts and Improve Oral Bioavailability of Paclitaxel. International Journal of Molecular Sciences. 2022; 23(23):15299. https://doi.org/10.3390/ijms232315299
Chicago/Turabian StyleKan, Jason W. Y., Clare S. W. Yan, Iris L. K. Wong, Xiaochun Su, Zhen Liu, Tak Hang Chan, and Larry M. C. Chow. 2022. "Discovery of a Flavonoid FM04 as a Potent Inhibitor to Reverse P-Glycoprotein-Mediated Drug Resistance in Xenografts and Improve Oral Bioavailability of Paclitaxel" International Journal of Molecular Sciences 23, no. 23: 15299. https://doi.org/10.3390/ijms232315299
APA StyleKan, J. W. Y., Yan, C. S. W., Wong, I. L. K., Su, X., Liu, Z., Chan, T. H., & Chow, L. M. C. (2022). Discovery of a Flavonoid FM04 as a Potent Inhibitor to Reverse P-Glycoprotein-Mediated Drug Resistance in Xenografts and Improve Oral Bioavailability of Paclitaxel. International Journal of Molecular Sciences, 23(23), 15299. https://doi.org/10.3390/ijms232315299