

Type 2 Diabetes Mellitus and Alzheimer’s Disease: Shared Molecular Mechanisms and Potential Common Therapeutic Targets

 ,
, 
 ,
, 
Abstract
1. Introduction
2. Insulin and IGF-1: Physiological Role in the Brain
2.1. The Origin of Insulin in the Brain
2.2. Expression of Insulin and IGF Receptors in the Brain
2.3. Insulin and IGF Signaling and Actions in the Brain
3. Molecular Mechanisms Linking T2D to AD
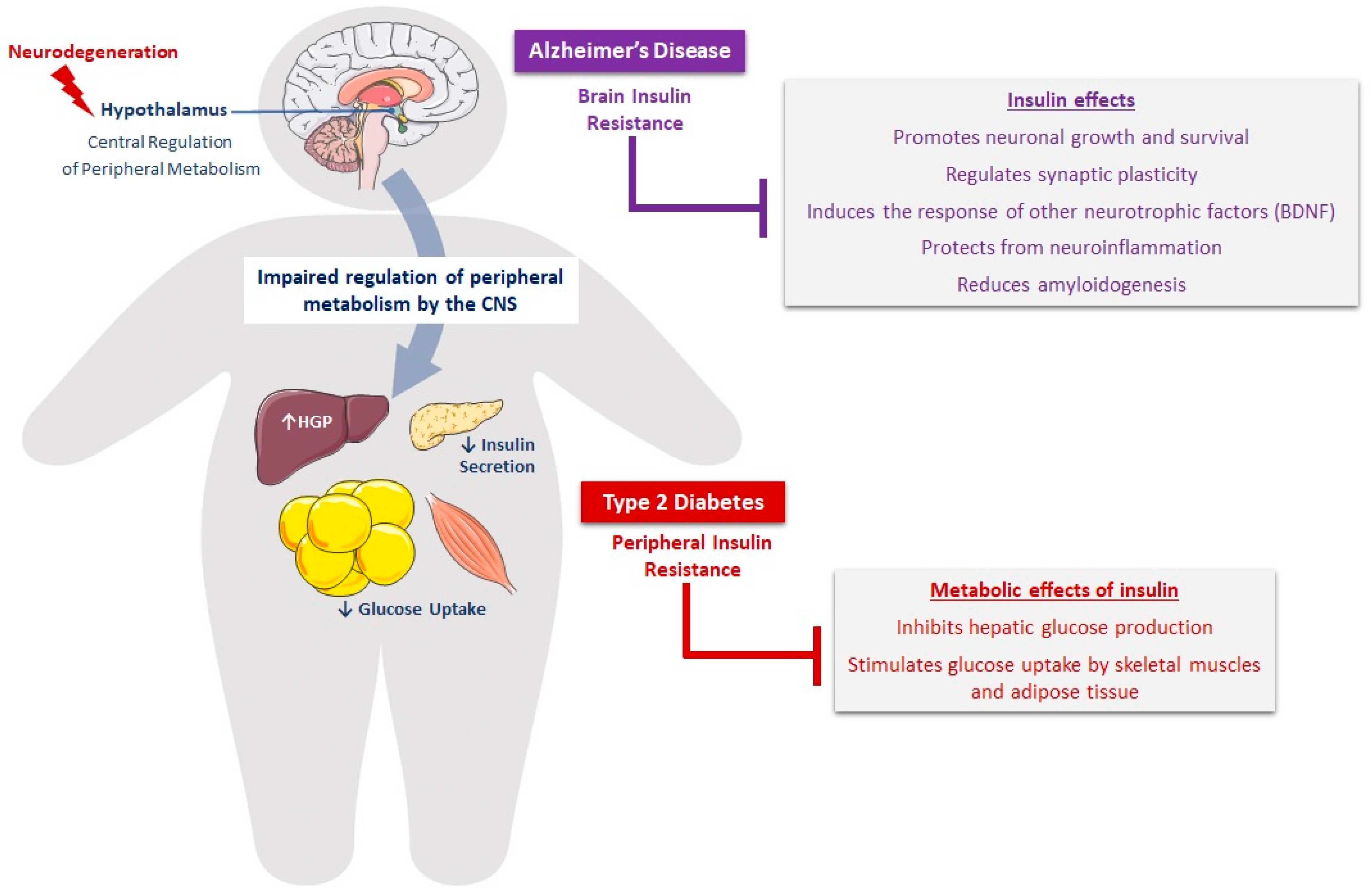
3.1. Cerebrovascular Abnormalities in Diabetes and AD
3.2. Alteration of Insulin and IGF-1 Signaling in the Brain
3.2.1. Insulin/IGF-1 Resistance, Neurodegeneration, and Cognition
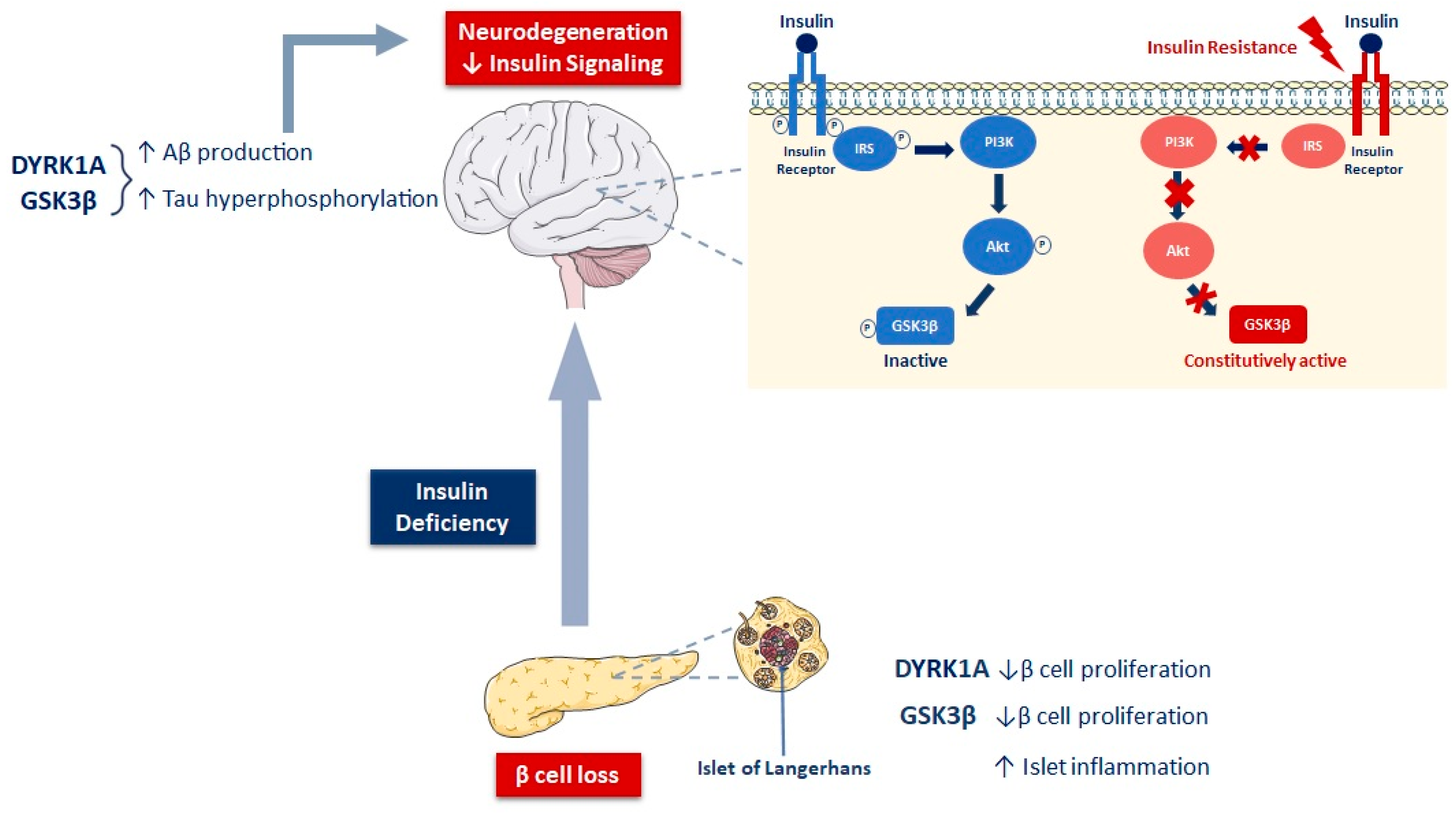
3.2.2. Bidirectional Relationship between Insulin/IGF-1 Resistance and Amyloidogenesis in T2D and AD
3.2.3. Insulin and IGF-1 Resistance, GSK3β, and Tauopathy in T2D and AD
3.3. Involvement of DYRK1A in AD and Diabetes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| [18F]-FDG | 18-fluorodeoxyglucose |
| ABCB1 | ATP-binding cassette subfamily B member 1 |
| AD | Alzheimer’s disease |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ApoE | apolipoprotein E |
| APP | amyloid precursor protein |
| ASF | alternative splicing factor |
| Aβ | amyloid beta peptide |
| BACE-1 | β-site APP-cleaving enzyme 1 |
| BBB | blood-brain barrier |
| BDNF | brain-derived neurotrophic factor |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DPP4 | dipeptidyl peptidase 4 |
| DS | Down syndrome |
| DYRK1A | dual-specificity tyrosine phosphorylation-regulated kinase 1A |
| GABA | gamma-aminobutyric acid |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GLP-1 | glucagon-like peptide-1 |
| GLUT | glucose transporter |
| GSK3β | glycogen synthase kinase 3β |
| HFD | high-fat diet |
| i.c.v | intracerebroventricular |
| IAPP | islet amyloid polypeptide |
| IDE | insulin-degrading enzyme |
| IDF | International Diabetes Federation |
| IGF | insulin-like growth factor |
| IRS | insulin receptor substrate |
| ISF | interstitial fluid |
| JNK | c-Jun N-terminal kinase |
| LRP1 | low-density lipoprotein receptor-related protein 1 |
| NFTs | neurofibrillary tangles |
| NMDA | N-methyl-D-aspartate |
| PET | positron emission tomography |
| PHF | paired helical filaments |
| PP2A | protein phosphatase 2A |
| PPAR | peroxisome proliferator-activated receptor |
| PSD95 | postsynaptic density protein 95 |
| PSEN1 | presenilin 1 |
| PSEN2 | presenilin 2 |
| ROS | reactive oxygen species |
| sAPPα | soluble APPα fragment |
| T2D | type 2 diabetes |
| Tau | tubulin-associated unit |
| Thr | threonin |
| TNF-α | tumor necrosis factor alpha |
| TrkB | tropomyosin receptor kinase B |
| WHO | World Health Organization |
References
- Baglietto-Vargas, D.; Shi, J.; Yaeger, D.M.; Ager, R.; LaFerla, F.M. Diabetes and Alzheimer’s Disease Crosstalk. Neurosci. Biobehav. Rev. 2016, 64, 272–287. [Google Scholar] [CrossRef]
- Centre Européen D’Étude Du Diabète. Les Chiffres du Diabète. Le diabète. Available online: http://ceed-diabete.org/fr/le-diabete/les-chiffres/ (accessed on 18 August 2022).
- International Diabetes Federation. IDF Diabetes Atlas|Tenth Edition. Diabetes around the World in 2021. Available online: https://diabetesatlas.org/ (accessed on 18 August 2022).
- Hamed, S.A. Brain Injury with Diabetes Mellitus: Evidence, Mechanisms and Treatment Implications. Expert Rev. Clin. Pharmacol. 2017, 10, 409–428. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Quesada, I.; Nadal, A. Endocrine Disruptors in the Etiology of Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2011, 7, 346–353. [Google Scholar] [CrossRef]
- Lin, J.-Y.; Yin, R.-X. Exposure to Endocrine-Disrupting Chemicals and Type 2 Diabetes Mellitus in Later Life. Expo. Health 2022. [Google Scholar] [CrossRef]
- Paul, K.C.; Jerrett, M.; Ritz, B. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Overlapping Biologic Mechanisms and Environmental Risk Factors. Curr. Environ. Health Rep. 2018, 5, 44–58. [Google Scholar] [CrossRef]
- Cavaghan, M.K.; Ehrmann, D.A.; Polonsky, K.S. Interactions between Insulin Resistance and Insulin Secretion in the Development of Glucose Intolerance. J. Clin. Investig. 2000, 106, 329–333. [Google Scholar] [CrossRef]
- Muoio, D.M.; Newgard, C.B. Molecular and Metabolic Mechanisms of Insulin Resistance and β-Cell Failure in Type 2 Diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef]
- Wijesekara, N.; Gonçalves, R.A.; De Felice, F.G.; Fraser, P.E. Impaired Peripheral Glucose Homeostasis and Alzheimer’s Disease. Neuropharmacology 2018, 136, 172–181. [Google Scholar] [CrossRef]
- Umegaki, H. Neurodegeneration in Diabetes Mellitus. In Neurodegenerative Diseases; Ahmad, S.I., Ed.; Advances in Experimental Medicine and Biology; Springer US: New York, NY, USA, 2012; pp. 258–265. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes Mellitus and the Risk of Dementia: The Rotterdam Study. Neurology 1999, 53, 1937. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An Updated Meta-Analysis of Cohort Studies: Diabetes and Risk of Alzheimer’s Disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47. [Google Scholar] [CrossRef]
- Huang, C.-C.; Chung, C.-M.; Leu, H.-B.; Lin, L.-Y.; Chiu, C.-C.; Hsu, C.-Y.; Chiang, C.-H.; Huang, P.-H.; Chen, T.-J.; Lin, S.-J.; et al. Diabetes Mellitus and the Risk of Alzheimer’s Disease: A Nationwide Population-Based Study. PLoS ONE 2014, 9, e87095. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.N.N.; Lima-Filho, R.A.S.; De Felice, F.G. Connecting Alzheimer’s Disease to Diabetes: Underlying Mechanisms and Potential Therapeutic Targets. Neuropharmacology 2018, 136, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes Mellitus and Risk of Alzheimer Disease and Decline in Cognitive Function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, G.D.; Bild, V.; Ababei, D.C.; Rusu, R.N.; Cobzaru, A.; Paduraru, L.; Bulea, D. Link between Diabetes and Alzheimer’s Disease Due to the Shared Amyloid Aggregation and Deposition Involving Both Neurodegenerative Changes and Neurovascular Damages. J. Clin. Med. 2020, 9, 1713. [Google Scholar] [CrossRef] [PubMed]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased Risk of Type 2 Diabetes in Alzheimer Disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef]
- Jiménez-Palomares, M.; Ramos-Rodríguez, J.J.; López-Acosta, J.F.; Pacheco-Herrero, M.; Lechuga-Sancho, A.M.; Perdomo, G.; García-Alloza, M.; Cózar-Castellano, I. Increased Aβ Production Prompts the Onset of Glucose Intolerance and Insulin Resistance. Am. J. Physiol.-Endocrinol. Metab. 2012, 302, E1373–E1380. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, H.H.; Chi, T.; Shin, A.C.; Lindtner, C.; Hsieh, W.; Ehrlich, M.; Gandy, S.; Buettner, C. Increased Susceptibility to Metabolic Dysregulation in a Mouse Model of Alzheimer’s Disease Is Associated with Impaired Hypothalamic Insulin Signaling and Elevated BCAA Levels. Alzheimers Dement. J. Alzheimers Assoc. 2016, 12, 851–861. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Wirths, O.; Zampar, S. Neuron Loss in Alzheimer’s Disease: Translation in Transgenic Mouse Models. Int. J. Mol. Sci. 2020, 21, 8144. [Google Scholar] [CrossRef]
- World Health Organization. Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 18 August 2022).
- Alzheimer’s Disease International. Dementia Statistics. Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 18 August 2022).
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s Disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Neth, B.J.; Craft, S. Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef]
- López, O.L.; DeKosky, S.T. Clinical Symptoms in Alzheimer’s Disease. In Handbook of Clinical Neurology; Dementias; Elsevier: Amsterdam, The Netherlands, 2008; Volume 89, pp. 207–216. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s Disease a Type 3 Diabetes? A Critical Appraisal. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Ingelsson, M.; Fukumoto, H.; Newell, K.L.; Growdon, J.H.; Hedley–Whyte, E.T.; Frosch, M.P.; Albert, M.S.; Hyman, B.T.; Irizarry, M.C. Early Aβ Accumulation and Progressive Synaptic Loss, Gliosis, and Tangle Formation in AD Brain. Neurology 2004, 62, 925–931. [Google Scholar] [CrossRef]
- Cole, G.M.; Frautschy, S.A. The Role of Insulin and Neurotrophic Factor Signaling in Brain Aging and Alzheimer’s Disease. Exp. Gerontol. 2007, 42, 10–21. [Google Scholar] [CrossRef]
- Huang, H.-C.; Jiang, Z.-F. Accumulated Amyloid-β Peptide and Hyperphosphorylated Tau Protein: Relationship and Links in Alzheimer’s Disease. J. Alzheimers Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Sen, N. Tauopathy: A Common Mechanism for Neurodegeneration and Brain Aging. Mech. Ageing Dev. 2019, 178, 72–79. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Li, L.; Hölscher, C. Common Pathological Processes in Alzheimer Disease and Type 2 Diabetes: A Review. Brain Res. Rev. 2007, 56, 384–402. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2015 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. J. Alzheimers Assoc. 2015, 11, 332–384. [Google Scholar] [CrossRef]
- Verghese, P.B.; Castellano, J.M.; Holtzman, D.M. Roles of Apolipoprotein E in Alzheimer’s Disease and Other Neurological Disorders. Lancet Neurol. 2011, 10, 241–252. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and Apolipoprotein E Receptors: Normal Biology and Roles in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward Defining the Preclinical Stages of Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. J. Alzheimers Assoc. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, E.; Hurtado-Carneiro, V.; LeBaut-Ayuso, Y.; Velázquez, E.; García-García, L.; Gómez-Oliver, F.; Ruiz-Albusac, J.M.; Ávila, J.; Pozo, M.Á. Significance of Brain Glucose Hypometabolism, Altered Insulin Signal Transduction, and Insulin Resistance in Several Neurological Diseases. Front. Endocrinol. 2022, 13, 873301. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Contributions of Brain Insulin Resistance and Deficiency in Amyloid-Related Neurodegeneration in Alzheimer’s Disease. Drugs 2012, 72, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Brain Glucose Transporters: Role in Pathogenesis and Potential Targets for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8142. [Google Scholar] [CrossRef] [PubMed]
- Roriz-Filho, J.S.; Sá-Roriz, T.M.; Rosset, I.; Camozzato, A.L.; Santos, A.C.; Chaves, M.L.F.; Moriguti, J.C.; Roriz-Cruz, M. (Pre)Diabetes, Brain Aging, and Cognition. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2009, 1792, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired Insulin and Insulin-like Growth Factor Expression and Signaling Mechanisms in Alzheimer’s Disease—Is This Type 3 Diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- de la Monte, S.M. Type 3 Diabetes Is Sporadic Alzheimer’s Disease: Mini-Review. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef]
- Mittal, K.; Katare, D.P. Shared Links between Type 2 Diabetes Mellitus and Alzheimer’s Disease: A Review. Diabetes Metab. Syndr. Clin. Res. Rev. 2016, 10 (Suppl. S1), S144–S149. [Google Scholar] [CrossRef] [PubMed]
- Bommer, C.; Sagalova, V.; Heesemann, E.; Manne-Goehler, J.; Atun, R.; Bärnighausen, T.; Davies, J.; Vollmer, S. Global Economic Burden of Diabetes in Adults: Projections From 2015 to 2030. Diabetes Care 2018, 41, 963–970. [Google Scholar] [CrossRef]
- Patterson, C.; Alzheimer’s Disease International. World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- Yang, Y.; Song, W. Molecular Links between Alzheimer’s Disease and Diabetes Mellitus. Neuroscience 2013, 250, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common Neurodegenerative Pathways in Obesity, Diabetes, and Alzheimer’s Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; De Felice, F.G. Insulin Resistance in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 830. [Google Scholar] [CrossRef]
- Milstein, J.L.; Ferris, H.A. The Brain as an Insulin-Sensitive Metabolic Organ. Mol. Metab. 2021, 52, 101234. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the Brain: The Role of Glucose in Physiological and Pathological Brain Function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef]
- Salas, I.H.; De Strooper, B. Diabetes and Alzheimer’s Disease: A Link Not as Simple as It Seems. Neurochem. Res. 2019, 44, 1271–1278. [Google Scholar] [CrossRef]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline. Front. Neurosci. 2019, 13, 788. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; DeSilva, S.; Abbruscato, T. The Role of Glucose Transporters in Brain Disease: Diabetes and Alzheimer’s Disease. Int. J. Mol. Sci. 2012, 13, 12629–12655. [Google Scholar] [CrossRef]
- Sankar, R.; Thamotharan, S.; Shin, D.; Moley, K.H.; Devaskar, S.U. Insulin-Responsive Glucose Transporters-GLUT8 and GLUT4 Are Expressed in the Developing Mammalian Brain. Brain Res. Mol. Brain Res. 2002, 107, 157–165. [Google Scholar] [CrossRef]
- McEwen, B.S.; Reagan, L.P. Glucose Transporter Expression in the Central Nervous System: Relationship to Synaptic Function. Eur. J. Pharmacol. 2004, 490, 13–24. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain Insulin Resistance in Type 2 Diabetes and Alzheimer Disease: Concepts and Conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Reno, C.M.; Sharma, S.; Christensen, C.; Huang, Y.; Fisher, S.J. Insulin Action in the Brain Regulates Both Central and Peripheral Functions. Am. J. Physiol.-Endocrinol. Metab. 2021, 321, E156–E163. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.M.; Barrett, E.J. Insulin Transport into the Brain. Am. J. Physiol.-Cell Physiol. 2018, 315, C125–C136. [Google Scholar] [CrossRef]
- Heni, M.; Schöpfer, P.; Peter, A.; Sartorius, T.; Fritsche, A.; Synofzik, M.; Häring, H.-U.; Maetzler, W.; Hennige, A.M. Evidence for Altered Transport of Insulin across the Blood-Brain Barrier in Insulin-Resistant Humans. Acta Diabetol. 2014, 51, 679–681. [Google Scholar] [CrossRef]
- Sartorius, T.; Peter, A.; Heni, M.; Maetzler, W.; Fritsche, A.; Häring, H.-U.; Hennige, A.M. The Brain Response to Peripheral Insulin Declines with Age: A Contribution of the Blood-Brain Barrier? PLoS ONE 2015, 10, e0126804. [Google Scholar] [CrossRef]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in Insulin and Insulin Signaling in Alzheimer’s Disease: Cause or Consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef]
- Frölich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Türk, A.; Hoyer, S.; et al. Brain Insulin and Insulin Receptors in Aging and Sporadic Alzheimer’s Disease. J. Neural Transm. Vienna Austria 1996 1998, 105, 423–438. [Google Scholar] [CrossRef]
- Cole, A.R.; Astell, A.; Green, C.; Sutherland, C. Molecular Connexions between Dementia and Diabetes. Neurosci. Biobehav. Rev. 2007, 31, 1046–1063. [Google Scholar] [CrossRef]
- Movassat, J.; Delangre, E.; Liu, J.; Gu, Y.; Janel, N. Hypothesis and Theory: Circulating Alzheimer’s-Related Biomarkers in Type 2 Diabetes. Insight From the Goto-Kakizaki Rat. Front. Neurol. 2019, 10, 649. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; Chen, H.; Quon, M.J.; Alkon, D.L. Insulin and the Insulin Receptor in Experimental Models of Learning and Memory. Eur. J. Pharmacol. 2004, 490, 71–81. [Google Scholar] [CrossRef]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in Central Nervous System: More than Just a Peripheral Hormone. J. Aging Res. 2012, 2012, 384017. [Google Scholar] [CrossRef]
- Wrigley, S.; Arafa, D.; Tropea, D. Insulin-Like Growth Factor 1: At the Crossroads of Brain Development and Aging. Front. Cell. Neurosci. 2017, 11, 14. [Google Scholar] [CrossRef]
- Nieto-Estévez, V.; Defterali, Ç.; Vicario-Abejón, C. IGF-I: A Key Growth Factor That Regulates Neurogenesis and Synaptogenesis from Embryonic to Adult Stages of the Brain. Front. Neurosci. 2016, 10, 52. [Google Scholar] [CrossRef]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The Role of Insulin in Human Brain Glucose Metabolism: An 18Fluoro-Deoxyglucose Positron Emission Tomography Study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin, Cognition, and Dementia. Eur. J. Pharmacol. 2013, 719, 170–179. [Google Scholar] [CrossRef]
- Piroli, G.G.; Grillo, C.A.; Reznikov, L.R.; Adams, S.; McEwen, B.S.; Charron, M.J.; Reagan, L.P. Corticosterone Impairs Insulin-Stimulated Translocation of GLUT4 in the Rat Hippocampus. Neuroendocrinology 2007, 85, 71–80. [Google Scholar] [CrossRef]
- Skeberdis, V.A.; Lan, J.; Zheng, X.; Zukin, R.S.; Bennett, M.V.L. Insulin Promotes Rapid Delivery of N-Methyl-d- Aspartate Receptors to the Cell Surface by Exocytosis. Proc. Natl. Acad. Sci. USA 2001, 98, 3561–3566. [Google Scholar] [CrossRef]
- Craft, S.; Watson, G.S. Insulin and Neurodegenerative Disease: Shared and Specific Mechanisms. Lancet Neurol. 2004, 3, 169–178. [Google Scholar] [CrossRef]
- Chen, T.-J.; Wang, D.-C.; Hung, H.-S.; Ho, H.-F. Insulin Can Induce the Expression of a Memory-Related Synaptic Protein through Facilitating AMPA Receptor Endocytosis in Rat Cortical Neurons. Cell. Mol. Life Sci. CMLS 2014, 71, 4069–4080. [Google Scholar] [CrossRef]
- Ge, Y.; Dong, Z.; Bagot, R.C.; Howland, J.G.; Phillips, A.G.; Wong, T.P.; Wang, Y.T. Hippocampal Long-Term Depression Is Required for the Consolidation of Spatial Memory. Proc. Natl. Acad. Sci. USA 2010, 107, 16697–16702. [Google Scholar] [CrossRef]
- Wan, Q.; Xiong, Z.G.; Man, H.Y.; Ackerley, C.A.; Braunton, J.; Lu, W.Y.; Becker, L.E.; MacDonald, J.F.; Wang, Y.T. Recruitment of Functional GABA(A) Receptors to Postsynaptic Domains by Insulin. Nature 1997, 388, 686–690. [Google Scholar] [CrossRef]
- Korol, S.V.; Jin, Z.; Babateen, O.; Birnir, B. GLP-1 and Exendin-4 Transiently Enhance GABAA Receptor–Mediated Synaptic and Tonic Currents in Rat Hippocampal CA3 Pyramidal Neurons. Diabetes 2014, 64, 79–89. [Google Scholar] [CrossRef]
- Limon, A.; Reyes-Ruiz, J.M.; Miledi, R. Loss of Functional GABAA Receptors in the Alzheimer Diseased Brain. Proc. Natl. Acad. Sci. USA 2012, 109, 10071–10076. [Google Scholar] [CrossRef]
- Hammoud, H.; Netsyk, O.; Tafreshiha, A.S.; Korol, S.V.; Jin, Z.; Li, J.-P.; Birnir, B. Insulin Differentially Modulates GABA Signalling in Hippocampal Neurons and, in an Age-Dependent Manner, Normalizes GABA-Activated Currents in the Tg-APPSwe Mouse Model of Alzheimer’s Disease. Acta Physiol. 2021, 232, e13623. [Google Scholar] [CrossRef]
- Chiu, S.-L.; Chen, C.-M.; Cline, H.T. Insulin Receptor Signaling Regulates Synapse Number, Dendritic Plasticity and Circuit Function in Vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef]
- Lee, C.-C.; Huang, C.-C.; Wu, M.-Y.; Hsu, K.-S. Insulin Stimulates Postsynaptic Density-95 Protein Translation via the Phosphoinositide 3-Kinase-Akt-Mammalian Target of Rapamycin Signaling Pathway. J. Biol. Chem. 2005, 280, 18543–18550. [Google Scholar] [CrossRef]
- González-García, I.; Gruber, T.; García-Cáceres, C. Insulin Action on Astrocytes: From Energy Homeostasis to Behaviour. J. Neuroendocrinol. 2021, 33, e12953. [Google Scholar] [CrossRef]
- Haas, C.B.; Kalinine, E.; Zimmer, E.R.; Hansel, G.; Brochier, A.W.; Oses, J.P.; Portela, L.V.; Muller, A.P. Brain Insulin Administration Triggers Distinct Cognitive and Neurotrophic Responses in Young and Aged Rats. Mol. Neurobiol. 2016, 53, 5807–5817. [Google Scholar] [CrossRef]
- Pezet, S.; Malcangio, M. Brain-Derived Neurotrophic Factor as a Drug Target for CNS Disorders. Expert Opin. Ther. Targets 2004, 8, 391–399. [Google Scholar] [CrossRef]
- Diniz, B.S.; Teixeira, A.L. Brain-Derived Neurotrophic Factor and Alzheimer’s Disease: Physiopathology and Beyond. Neuromolecular Med. 2011, 13, 217–222. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Canteiro, P.B.; Antero, D.C.; Tramontin, N.d.S.; Simon, K.U.; Mendes, C.; Anastácio Borges Correa, M.E.; Silveira, P.C.L.; Muller, A.P. Insulin Treatment Protects the Brain against Neuroinflammation by Reducing Cerebral Cytokines and Modulating Mitochondrial Function. Brain Res. Bull. 2019, 149, 120–128. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.-C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An Anti-Diabetes Agent Protects the Mouse Brain from Defective Insulin Signaling Caused by Alzheimer’s Disease–Associated Aβ Oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Lyra e Silva, N.D.M.; Gonçalves, R.A.; Boehnke, S.E.; Forny-Germano, L.; Munoz, D.P.; De Felice, F.G. Understanding the Link between Insulin Resistance and Alzheimer’s Disease: Insights from Animal Models. Exp. Neurol. 2019, 316, 1–11. [Google Scholar] [CrossRef]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal Insulin Improves Memory in Humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef]
- Adzovic, L.; Lynn, A.E.; D’Angelo, H.M.; Crockett, A.M.; Kaercher, R.M.; Royer, S.E.; Hopp, S.C.; Wenk, G.L. Insulin Improves Memory and Reduces Chronic Neuroinflammation in the Hippocampus of Young but Not Aged Brains. J. Neuroinflammation 2015, 12, 63. [Google Scholar] [CrossRef]
- Chapman, C.D.; Schiöth, H.B.; Grillo, C.A.; Benedict, C. Intranasal Insulin in Alzheimer’s Disease: Food for Thought. Neuropharmacology 2018, 136 Pt B, 196–201. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Rademakers, R.; Cruts, M.; Van Broeckhoven, C. Genetics of Early-Onset Alzheimer Dementia. Sci. World J. 2003, 3, 497–519. [Google Scholar] [CrossRef]
- Hoogmartens, J.; Cacace, R.; Van Broeckhoven, C. Insight into the Genetic Etiology of Alzheimer’s Disease: A Comprehensive Review of the Role of Rare Variants. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2021, 13, e12155. [Google Scholar] [CrossRef]
- Wildsmith, K.R.; Holley, M.; Savage, J.C.; Skerrett, R.; Landreth, G.E. Evidence for Impaired Amyloid β Clearance in Alzheimer’s Disease. Alzheimers Res. Ther. 2013, 5, 33. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS Amyloid-β in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Carare, R.O.; Aldea, R.; Agarwal, N.; Bacskai, B.J.; Bechman, I.; Boche, D.; Bu, G.; Bulters, D.; Clemens, A.; Counts, S.E.; et al. Clearance of Interstitial Fluid (ISF) and CSF (CLIC) Group—Part of Vascular Professional Interest Area (PIA). Alzheimers Dement. Diagn. Assess. Dis. Monit. 2020, 12, e12053. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, D.-Y.; Wang, Y.-J. Peripheral Clearance of Brain-Derived Aβ in Alzheimer’s Disease: Pathophysiology and Therapeutic Perspectives. Transl. Neurodegener. 2020, 9, 16. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s Amyloid-Β1-40 Peptide from Brain by LDL Receptor–Related Protein-1 at the Blood-Brain Barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.-M.; Sharom, F.J.; Reiner, P.B. β-Amyloid Efflux Mediated by p-Glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The Glymphatic Pathway in Neurological Disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.R.; Perry, V.H.; Weller, R.O. Solutes, but Not Cells, Drain from the Brain Parenchyma along Basement Membranes of Capillaries and Arteries: Significance for Cerebral Amyloid Angiopathy and Neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef]
- Weller, R.O.; Boche, D.; Nicoll, J.A.R. Microvasculature Changes and Cerebral Amyloid Angiopathy in Alzheimer’s Disease and Their Potential Impact on Therapy. Acta Neuropathol. 2009, 118, 87–102. [Google Scholar] [CrossRef]
- Aldea, R.; Weller, R.O.; Wilcock, D.M.; Carare, R.O.; Richardson, G. Cerebrovascular Smooth Muscle Cells as the Drivers of Intramural Periarterial Drainage of the Brain. Front. Aging Neurosci. 2019, 11, 1. [Google Scholar] [CrossRef]
- Farkas, E.; Luiten, P.G.M. Cerebral Microvascular Pathology in Aging and Alzheimer’s Disease. Prog. Neurobiol. 2001, 64, 575–611. [Google Scholar] [CrossRef]
- Brun, A.; Englund, E. A White Matter Disorder in Dementia of the Alzheimer Type: A Pathoanatomical Study. Ann. Neurol. 1986, 19, 253–262. [Google Scholar] [CrossRef]
- Lee, B.C.P.; Mintun, M.; Buckner, R.L.; Morris, J.C. Imaging of Alzheimer’s Disease. J. Neuroimaging 2003, 13, 199–214. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular Dysfunction and Faulty Amyloid β-Peptide Clearance in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef]
- Scheffer, S.; Hermkens, D.M.A.; van der Weerd, L.; de Vries, H.E.; Daemen, M.J.A.P. Vascular Hypothesis of Alzheimer Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283. [Google Scholar] [CrossRef]
- Chornenkyy, Y.; Wang, W.; Wei, A.; Nelson, P.T. Alzheimer’s Disease and Type 2 Diabetes Mellitus Are Distinct Diseases with Potential Overlapping Metabolic Dysfunction Upstream of Observed Cognitive Decline. Brain Pathol. 2018, 29, 3–17. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, X.; Lu, J. Progress on Diabetic Cerebrovascular Diseases. Bosn. J. Basic Med. Sci. 2014, 14, 185–190. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.-X.; Stern, Y.; Shea, S.; Mayeux, R. Diabetes Mellitus and Risk of Alzheimer’s Disease and Dementia with Stroke in a Multiethnic Cohort. Am. J. Epidemiol. 2001, 154, 635–641. [Google Scholar] [CrossRef]
- Roberts, R.O.; Kantarci, K.; Geda, Y.E.; Knopman, D.S.; Przybelski, S.A.; Weigand, S.D.; Petersen, R.C.; Jack, C.R. Untreated Type 2 Diabetes and Its Complications Are Associated With Subcortical Infarctions. Diabetes Care 2011, 34, 184–186. [Google Scholar] [CrossRef]
- Mukai, N.; Hori, S.; Pomeroy, M. Cerebral Lesions in Rats with Streptozotocin-Induced Diabetes. Acta Neuropathol. 1980, 51, 79–84. [Google Scholar] [CrossRef]
- Johnson, P.C.; Brendel, K.; Meezan, E. Thickened Cerebral Cortical Capillary Basement Membranes in Diabetics. Arch. Pathol. Lab. Med. 1982, 106, 214–217. [Google Scholar]
- Ergul, A.; Kelly-Cobbs, A.; Abdalla, M.; Fagan, S.C. Cerebrovascular Complications of Diabetes: Focus on Stroke. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 148–158. [Google Scholar] [CrossRef]
- McCuskey, P.A.; McCuskey, R.S. In Vivo and Electron Microscopic Study of the Development of Cerebral Diabetic Microangiography. Microcirc. Endothelium. Lymphat. 1984, 1, 221–244. [Google Scholar]
- Hou, Q.; Zuo, Z.; Michel, P.; Zhang, Y.; Eskandari, A.; Man, F.; Gao, Q.; Johnston, K.C.; Wintermark, M. Influence of Chronic Hyperglycemia on Cerebral Microvascular Remodeling. Stroke 2013, 44, 3557–3560. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 Diabetes, APOE Gene, and the Risk for Dementia and Related Pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef]
- Morris, J.K.; Vidoni, E.D.; Honea, R.A.; Burns, J.M. Impaired Glycemia Increases Disease Progression in Mild Cognitive Impairment. Neurobiol. Aging 2014, 35, 585–589. [Google Scholar] [CrossRef]
- Benedict, C.; Grillo, C.A. Insulin Resistance as a Therapeutic Target in the Treatment of Alzheimer’s Disease: A State-of-the-Art Review. Front. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef]
- Bruehl, H.; Sweat, V.; Hassenstab, J.; Polyakov, V.; Convit, A. Cognitive Impairment in Non-Diabetic Middle-Aged and Older Adults Is Associated with Insulin Resistance. J. Clin. Exp. Neuropsychol. 2010, 32, 487–493. [Google Scholar] [CrossRef]
- De Felice, F.G.; Ferreira, S.T. Inflammation, Defective Insulin Signaling, and Mitochondrial Dysfunction as Common Molecular Denominators Connecting Type 2 Diabetes to Alzheimer Disease. Diabetes 2014, 63, 2262–2272. [Google Scholar] [CrossRef]
- Biessels, G.J.; Reagan, L.P. Hippocampal Insulin Resistance and Cognitive Dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant Insulin Signaling in Alzheimer’s Disease: Current Knowledge. Front. Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated Brain Insulin Resistance in Alzheimer’s Disease Patients Is Associated with IGF-1 Resistance, IRS-1 Dysregulation, and Cognitive Decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. [Google Scholar] [CrossRef]
- Kumar, V.; Kim, S.-H.; Bishayee, K. Dysfunctional Glucose Metabolism in Alzheimer’s Disease Onset and Potential Pharmacological Interventions. Int. J. Mol. Sci. 2022, 23, 9540. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin Resistance and Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults With Prediabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Li, W.; Risacher, S.L.; Huang, E.; Saykin, A.J. Type 2 Diabetes Mellitus Is Associated with Brain Atrophy and Hypometabolism in the ADNI Cohort. Neurology 2016, 87, 595–600. [Google Scholar] [CrossRef]
- Shinohara, M.; Sato, N. Bidirectional Interactions between Diabetes and Alzheimer’s Disease. Neurochem. Int. 2017, 108, 296–302. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Review of Insulin and Insulin-like Growth Factor Expression, Signaling, and Malfunction in the Central Nervous System: Relevance to Alzheimer’s Disease. J. Alzheimers Dis. 2005, 7, 45–61. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.-X. Deficient Brain Insulin Signalling Pathway in Alzheimer’s Disease and Diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the Brain: There and Back Again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 Receptor, Insulin Receptor and IRS-1/2 in Alzheimer’s Disease Indicate Possible Resistance to IGF-1 and Insulin Signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef]
- Biessels, G.J.; Reijmer, Y.D. Brain Changes Underlying Cognitive Dysfunction in Diabetes: What Can We Learn From MRI? Diabetes 2014, 63, 2244–2252. [Google Scholar] [CrossRef]
- de la Monte, S.M. Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimer’s Disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef]
- Lester-Coll, N.; Rivera, E.J.; Soscia, S.J.; Doiron, K.; Wands, J.R.; de la Monte, S.M. Intracerebral Streptozotocin Model of Type 3 Diabetes: Relevance to Sporadic Alzheimer’s Disease. J. Alzheimers Dis. JAD 2006, 9, 13–33. [Google Scholar] [CrossRef]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-Accelerated Memory Dysfunction via Cerebrovascular Inflammation and Aβ Deposition in an Alzheimer Mouse Model with Diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef]
- Grillo, C.A.; Woodruff, J.L.; Macht, V.A.; Reagan, L.P. Insulin Resistance and Hippocampal Dysfunction: Disentangling Peripheral and Brain Causes from Consequences. Exp. Neurol. 2019, 318, 71–77. [Google Scholar] [CrossRef]
- Grillo, C.A.; Piroli, G.G.; Kaigler, K.F.; Wilson, S.P.; Wilson, M.A.; Reagan, L.P. Downregulation of hypothalamic insulin receptor expression elicits depressive-like behaviors in rats. Behav. Brain Res. 2011, 222, 230–235. [Google Scholar] [CrossRef]
- Rozanska, O.; Uruska, A.; Zozulinska-Ziolkiewicz, D. Brain-Derived Neurotrophic Factor and Diabetes. Int. J. Mol. Sci. 2020, 21, 841. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-Q.; Townsend, M. Insulin Resistance and Amyloidogenesis as Common Molecular Foundation for Type 2 Diabetes and Alzheimer’s Disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2009, 1792, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Ittner, L.M.; Lim, Y.-A. Common Features between Diabetes Mellitus and Alzheimer’s Disease. Cell. Mol. Life Sci. CMLS 2009, 66, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.-A.; Rhein, V.; Baysang, G.; Meier, F.; Poljak, A.; Raftery, M.J.; Guilhaus, M.; Ittner, L.M.; Eckert, A.; Götz, J. Abeta and Human Amylin Share a Common Toxicity Pathway via Mitochondrial Dysfunction. Proteomics 2010, 10, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Kirkitadze, M.D.; Kowalska, A. Molecular Mechanisms Initiating Amyloid Beta-Fibril Formation in Alzheimer’s Disease. Acta Biochim. Pol. 2005, 52, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Checler, F.; Alves da Costa, C.; Dumanchin-Njock, C.; Lopez-Perez, E.; Marambaud, P.; Paitel, E.; Petit, A.; Vincent, B. Métabolisme du précurseur du peptide amyloïde et présénilines. médecine/sciences 2002, 18, 717–724. [Google Scholar] [CrossRef]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.-Y.; Klein, P.S. GSK-3alpha Regulates Production of Alzheimer’s Disease Amyloid-Beta Peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Ly, P.T.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-Mediated BACE1 Expression Reduces Alzheimer-Associated Phenotypes. J. Clin. Investig. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Li, X.; Song, D.; Leng, S.X. Link between Type 2 Diabetes and Alzheimer’s Disease: From Epidemiology to Mechanism and Treatment. Clin. Interv. Aging 2015, 10, 549–560. [Google Scholar] [CrossRef]
- Gao, C.; Hölscher, C.; Liu, Y.; Li, L. GSK3: A Key Target for the Development of Novel Treatments for Type 2 Diabetes Mellitus and Alzheimer Disease. Rev. Neurosci. 2011, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of β-Amyloid Precursor Protein Trafficking by Insulin Reduces Intraneuronal β-Amyloid and Requires Mitogen-Activated Protein Kinase Signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Trejo, J.L.; Spuch, C.; Bohl, D.; Heard, J.M.; Torres-Aleman, I. Blockade of the Insulin-like Growth Factor I Receptor in the Choroid Plexus Originates Alzheimer’s-like Neuropathology in Rodents: New Cues into the Human Disease? Neurobiol. Aging 2006, 27, 1618–1631. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Trejo, J.L.; Gerber, A.; Loetscher, H.; Torrado, J.; Metzger, F.; Torres-Aleman, I. Therapeutic Actions of Insulin-like Growth Factor I on APP/PS2 Mice with Severe Brain Amyloidosis. Neurobiol. Aging 2006, 27, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons Regulate Extracellular Levels of Amyloid β-Protein via Proteolysis by Insulin-Degrading Enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-Degrading Enzyme Regulates the Levels of Insulin, Amyloid β-Protein, and the β-Amyloid Precursor Protein Intracellular Domain in Vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-Degrading Enzyme as a Downstream Target of Insulin Receptor Signaling Cascade: Implications for Alzheimer’s Disease Intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef]
- Cook, D.G.; Leverenz, J.B.; McMillan, P.J.; Kulstad, J.J.; Ericksen, S.; Roth, R.A.; Schellenberg, G.D.; Jin, L.-W.; Kovacina, K.S.; Craft, S. Reduced Hippocampal Insulin-Degrading Enzyme in Late-Onset Alzheimer’s Disease Is Associated with the Apolipoprotein E-Ε4 Allele. Am. J. Pathol. 2003, 162, 313–319. [Google Scholar] [CrossRef]
- Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; de la Monte, S.M. Altered Expression of Insulin-Degrading Enzyme and Regulator of Calcineurin in the Rat Intracerebral Streptozotocin Model and Human Apolipoprotein E-Ε4–Associated Alzheimer’s Disease. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2019, 11, 392–404. [Google Scholar] [CrossRef]
- Wei, Z.; Koya, J.; Reznik, S.E. Insulin Resistance Exacerbates Alzheimer Disease via Multiple Mechanisms. Front. Neurosci. 2021, 15, 687157. [Google Scholar] [CrossRef]
- Shiiki, T.; Ohtsuki, S.; Kurihara, A.; Naganuma, H.; Nishimura, K.; Tachikawa, M.; Hosoya, K.; Terasaki, T. Brain Insulin Impairs Amyloid-β(1-40) Clearance from the Brain. J. Neurosci. 2004, 24, 9632–9637. [Google Scholar] [CrossRef] [PubMed]
- Messier, C.; Teutenberg, K. The Role of Insulin, Insulin Growth Factor, and Insulin-Degrading Enzyme in Brain Aging and Alzheimer’s Disease. Neural Plast. 2005, 12, 311–328. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.-Q.; Ferreira, S.T.; Klein, W.L. Protection of Synapses against Alzheimer’s-Linked Toxins: Insulin Signaling Prevents the Pathogenic Binding of Aβ Oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal Insulin Therapy for Alzheimer Disease and Amnestic Mild Cognitive Impairment: A Pilot Clinical Trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef]
- Yoon, S.O.; Park, D.J.; Ryu, J.C.; Ozer, H.G.; Tep, C.; Shin, Y.J.; Lim, T.H.; Pastorino, L.; Kunwar, A.J.; Walton, J.C.; et al. JNK3 Perpetuates Metabolic Stress Induced by Abeta Peptides. Neuron 2012, 75, 824–837. [Google Scholar] [CrossRef]
- Clarke, J.R.; Lyra e Silva, N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazão, R.; et al. Alzheimer-Associated Aβ Oligomers Impact the Central Nervous System to Induce Peripheral Metabolic Deregulation. EMBO Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Kaminsky, Y.G.; Tikhonova, L.A.; Kosenko, E.A. Critical Analysis of Alzheimer’s Amyloid-Beta Toxicity to Mitochondria. Front. Biosci. Landmark Ed. 2015, 20, 173–197. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid Beta, Mitochondrial Dysfunction and Synaptic Damage: Implications for Cognitive Decline in Aging and Alzheimer’s Disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Chen, J.X.; Yan, S.D. Amyloid-β-Induced Mitochondrial Dysfunction. J. Alzheimers Dis. JAD 2007, 12, 177–184. [Google Scholar] [CrossRef]
- Kaneto, H.; Matsuoka, T.; Katakami, N.; Kawamori, D.; Miyatsuka, T.; Yoshiuchi, K.; Yasuda, T.; Sakamoto, K.; Yamasaki, Y.; Matsuhisa, M. Oxidative Stress and the JNK Pathway Are Involved in the Development of Type 1 and Type 2 Diabetes. Curr. Mol. Med. 2007, 7, 674–686. [Google Scholar] [CrossRef]
- Yung, J.H.M.; Giacca, A. Role of C-Jun N-Terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Helmerhorst, E.; Taddei, K.; Plewright, B.; van Bronswijk, W.; Martins, R. Alzheimer’s β-Amyloid Peptides Compete for Insulin Binding to the Insulin Receptor. J. Neurosci. 2002, 22, RC221. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Mehta, T.; Selkoe, D.J. Soluble Abeta Inhibits Specific Signal Transduction Cascades Common to the Insulin Receptor Pathway. J. Biol. Chem. 2007, 282, 33305–33312. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Klein, W.L. The Aβ Oligomer Hypothesis for Synapse Failure and Memory Loss in Alzheimer’s Diseas. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef]
- Zhao, W.-Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid Beta Oligomers Induce Impairment of Neuronal Insulin Receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef]
- Pearson-Leary, J.; McNay, E.C. Intrahippocampal Administration of Amyloid-Β1–42 Oligomers Acutely Impairs Spatial Working Memory, Insulin Signaling, and Hippocampal Metabolism. J. Alzheimers Dis. JAD 2012, 30, 413–422. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Gomez-Isla, T.; LeRoith, D.; Torres-Aleman, I. Serum Insulin-like Growth Factor I Regulates Brain Amyloid-Beta Levels. Nat. Med. 2002, 8, 1390–1397. [Google Scholar] [CrossRef]
- Stein, T.D.; Anders, N.J.; DeCarli, C.; Chan, S.L.; Mattson, M.P.; Johnson, J.A. Neutralization of Transthyretin Reverses the Neuroprotective Effects of Secreted Amyloid Precursor Protein (APP) in APPSw Mice Resulting in Tau Phosphorylation and Loss of Hippocampal Neurons: Support for the Amyloid Hypothesis. J. Neurosci. 2004, 24, 7707–7717. [Google Scholar] [CrossRef]
- Ostrowski, P.P.; Barszczyk, A.; Forstenpointner, J.; Zheng, W.; Feng, Z.-P. Meta-Analysis of Serum Insulin-Like Growth Factor 1 in Alzheimer’s Disease. PLoS ONE 2016, 11, e0155733. [Google Scholar] [CrossRef]
- Jafferali, S.; Dumont, Y.; Sotty, F.; Robitaille, Y.; Quirion, R.; Kar, S. Insulin-like Growth Factor-I and Its Receptor in the Frontal Cortex, Hippocampus, and Cerebellum of Normal Human and Alzheimer Disease Brains. Synapse 2000, 38, 450–459. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimers Dis. JAD 2017, 59, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Q.; Chen, S.; Xu, C. Functions of Amyloid Precursor Protein in Metabolic Diseases. Metabolism. 2021, 115, 154454. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Qing, H.; Radenovic, A.; Kis, A.; Vileno, B.; Làszló, F.; Miller, L.; Martins, R.N.; Waeber, G.; Mooser, V.; et al. Beta Amyloid and Hyperphosphorylated Tau Deposits in the Pancreas in Type 2 Diabetes. Neurobiol. Aging 2010, 31, 1503–1515. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, A.F.; Ferreira, S.; Martins, I.C.; Menezes, R. Islet Amyloid Polypeptide: A Partner in Crime With Aβ in the Pathology of Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.J.; Willis, A.C.; Clark, A.; Turner, R.C.; Sim, R.B.; Reid, K.B. Purification and Characterization of a Peptide from Amyloid-Rich Pancreases of Type 2 Diabetic Patients. Proc. Natl. Acad. Sci. USA 1987, 84, 8628–8632. [Google Scholar] [CrossRef]
- Sanke, T.; Bell, G.I.; Sample, C.; Rubenstein, A.H.; Steiner, D.F. An Islet Amyloid Peptide Is Derived from an 89-Amino Acid Precursor by Proteolytic Processing. J. Biol. Chem. 1988, 263, 17243–17246. [Google Scholar] [CrossRef]
- Luca, S.; Yau, W.-M.; Leapman, R.; Tycko, R. Peptide Conformation and Supramolecular Organization in Amylin Fibrils: Constraints from Solid State NMR. Biochemistry 2007, 46, 13505–13522. [Google Scholar] [CrossRef]
- Kahn, S.E.; D’Alessio, D.A.; Schwartz, M.W.; Fujimoto, W.Y.; Ensinck, J.W.; Taborsky, G.J., Jr.; Porte, D., Jr. Evidence of Cosecretion of Islet Amyloid Polypeptide and Insulin by β-Cells. Diabetes 1990, 39, 634–638. [Google Scholar] [CrossRef]
- Fawver, J.N.; Ghiwot, Y.; Koola, C.; Carrera, W.; Rodriguez-Rivera, J.; Hernandez, C.; Dineley, K.T.; Kong, Y.; Li, J.; Jhamandas, J.; et al. Islet Amyloid Polypeptide (IAPP): A Second Amyloid in Alzheimer’s Disease. Curr. Alzheimer Res. 2014, 11, 928–940. [Google Scholar] [CrossRef]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.; DeCarli, C.; Despa, F. Amylin Deposition in the Brain: A Second Amyloid in Alzheimer’s Disease ? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef]
- Hong, M.; Lee, V.M.-Y. Insulin and Insulin-like Growth Factor-1 Regulate Tau Phosphorylation in Cultured Human Neurons *. J. Biol. Chem. 1997, 272, 19547–19553. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Marítin, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a Pivotal Kinase in Alzheimer Disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Frame, S.; Cohen, P. GSK3 Takes Centre Stage More than 20 Years after Its Discovery. Biochem. J. 2001, 359 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen Synthase Kinase-3 Signaling in Alzheimer’s Disease. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Ávila, J. GSK-3 and Tau: A Key Duet in Alzheimer’s Disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H. Glycogen Synthase Kinase 3: An Emerging Therapeutic Target. Trends Mol. Med. 2002, 8, 126–132. [Google Scholar] [CrossRef]
- Moon, R.T.; Kohn, A.D.; De Ferrari, G.V.; Kaykas, A. WNT and Beta-Catenin Signalling: Diseases and Therapies. Nat. Rev. Genet. 2004, 5, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple Isoforms of Human Microtubule-Associated Protein Tau: Sequences and Localization in Neurofibrillary Tangles of Alzheimer’s Disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Himmler, A.; Drechsel, D.; Kirschner, M.W.; Martin, D.W. Tau Consists of a Set of Proteins with Repeated C-Terminal Microtubule-Binding Domains and Variable N-Terminal Domains. Mol. Cell. Biol. 1989, 9, 1381–1388. [Google Scholar]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and Sequencing of the CDNA Encoding an Isoform of Microtubule-Associated Protein Tau Containing Four Tandem Repeats: Differential Expression of Tau Protein MRNAs in Human Brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef]
- Panda, D.; Samuel, J.C.; Massie, M.; Feinstein, S.C.; Wilson, L. Differential Regulation of Microtubule Dynamics by Three- and Four-Repeat Tau: Implications for the Onset of Neurodegenerative Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 9548–9553. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, G.; Cole, R.D. Phosphorylation Affects the Ability of Tau Protein to Promote Microtubule Assembly. J. Biol. Chem. 1984, 259, 5301–5305. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.-M.; Thies, E.; Trinczek, B.; Biernat, J.; Mandelkow, E. MARK/PAR1 Kinase Is a Regulator of Microtubule-Dependent Transport in Axons. J. Cell Biol. 2004, 167, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.; Gautam, D.; Surjo, D.; Ueki, K.; Baudler, S.; Schubert, D.; Kondo, T.; Alber, J.; Galldiks, N.; Küstermann, E.; et al. Role for Neuronal Insulin Resistance in Neurodegenerative Diseases. Proc. Natl. Acad. Sci. USA 2004, 101, 3100–3105. [Google Scholar] [CrossRef]
- Hernández, F.; Gómez de Barreda, E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A Possible Link between Beta Amyloid Peptide and Tau Protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Ishiguro, K.; Uchida, T.; Takashima, A.; Lemere, C.A.; Imahori, K. Preferential Labeling of Alzheimer Neurofibrillary Tangles with Antisera for Tau Protein Kinase (TPK) I/Glycogen Synthase Kinase-3 Beta and Cyclin-Dependent Kinase 5, a Component of TPK II. Acta Neuropathol. 1996, 92, 232–241. [Google Scholar] [CrossRef]
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Distribution of Active Glycogen Synthase Kinase 3beta (GSK-3beta) in Brains Staged for Alzheimer Disease Neurofibrillary Changes. J. Neuropathol. Exp. Neurol. 1999, 58, 1010–1019. [Google Scholar] [CrossRef]
- Arnaud, L.; Robakis, N.K.; Figueiredo-Pereira, M.E. It May Take Inflammation, Phosphorylation and Ubiquitination to “tangle” in Alzheimer’s Disease. Neurodegener. Dis. 2006, 3, 313–319. [Google Scholar] [CrossRef]
- Li, L.; Jiang, Y.; Wang, J.-Z.; Liu, R.; Wang, X. Tau Ubiquitination in Alzheimer’s Disease. Front. Neurol. 2022, 12, 786353. [Google Scholar] [CrossRef]
- Puangmalai, N.; Sengupta, U.; Bhatt, N.; Gaikwad, S.; Montalbano, M.; Bhuyan, A.; Garcia, S.; McAllen, S.; Sonawane, M.; Jerez, C.; et al. Lysine 63-Linked Ubiquitination of Tau Oligomers Contributes to the Pathogenesis of Alzheimer’s Disease. J. Biol. Chem. 2022, 298, 101766. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S. The Ubiquitin-Proteasome System in Alzheimer’s Disease. J. Cell. Mol. Med. 2008, 12, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Magdesian, M.H.; Carvalho, M.M.V.F.; Mendes, F.A.; Saraiva, L.M.; Juliano, M.A.; Juliano, L.; Garcia-Abreu, J.; Ferreira, S.T. Amyloid-Beta Binds to the Extracellular Cysteine-Rich Domain of Frizzled and Inhibits Wnt/Beta-Catenin Signaling. J. Biol. Chem. 2008, 283, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- Maj, M.; Hoermann, G.; Rasul, S.; Base, W.; Wagner, L.; Attems, J. The Microtubule-Associated Protein Tau and Its Relevance for Pancreatic Beta Cells. J. Diabetes Res. 2015, 2016, e1964634. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Figeac, F.; Uzan, B.; Faro, M.; Chelali, N.; Portha, B.; Movassat, J. Neonatal Growth and Regeneration of Beta-Cells Are Regulated by the Wnt/Beta-Catenin Signaling in Normal and Diabetic Rats. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E245–E256. [Google Scholar] [CrossRef] [PubMed]
- Figeac, F.; Ilias, A.; Bailbe, D.; Portha, B.; Movassat, J. Local In Vivo GSK3β Knockdown Promotes Pancreatic β Cell and Acinar Cell Regeneration in 90% Pancreatectomized Rat. Mol. Ther. 2012, 20, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tanabe, K.; Bernal-Mizrachi, E.; Permutt, M.A. Mice with Beta Cell Overexpression of Glycogen Synthase Kinase-3beta Have Reduced Beta Cell Mass and Proliferation. Diabetologia 2008, 51, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Pitasi, C.L.; Liu, J.; Gausserès, B.; Pommier, G.; Delangre, E.; Armanet, M.; Cattan, P.; Mégarbane, B.; Hanak, A.-S.; Maouche, K.; et al. Implication of Glycogen Synthase Kinase 3 in Diabetes-Associated Islet Inflammation. J. Endocrinol. 2020, 244, 133–148. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Krebs, E.G. Phosphorylation of Insulin Receptor Substrate 1 by Glycogen Synthase Kinase 3 Impairs Insulin Action. Proc. Natl. Acad. Sci. USA 1997, 94, 9660–9664. [Google Scholar] [CrossRef]
- Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.; Carter, L.; Henry, R.R. Potential Role of Glycogen Synthase Kinase-3 in Skeletal Muscle Insulin Resistance of Type 2 Diabetes. Diabetes 2000, 49, 263–271. [Google Scholar] [CrossRef]
- Becker, W.; Sippl, W. Activation, Regulation, and Inhibition of DYRK1A. FEBS J. 2011, 278, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Walte, A.; Rüben, K.; Birner-Gruenberger, R.; Preisinger, C.; Bamberg-Lemper, S.; Hilz, N.; Bracher, F.; Becker, W. Mechanism of Dual Specificity Kinase Activity of DYRK1A. FEBS J. 2013, 280, 4495–4511. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Man-Un Ung, P.; Wang, P.; Wang, H.; Li, H.; Andrews, M.K.; Stewart, A.F.; Schlessinger, A.; DeVita, R.J. Novel Selective Thiadiazine DYRK1A Inhibitor Lead Scaffold with Human Pancreatic β-Cell Proliferation Activity. Eur. J. Med. Chem. 2018, 157, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Duchon, A.; Herault, Y. DYRK1A, a Dosage-Sensitive Gene Involved in Neurodevelopmental Disorders, Is a Target for Drug Development in Down Syndrome. Front. Behav. Neurosci. 2016, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-J.; Sternberg, L.R.; Kasten-Sportès, C.; Keuren, M.L.V.; Chung, S.-H.; Slack, A.C.; Miller, D.E.; Glover, T.W.; Chiang, P.-W.; Lou, L.; et al. Isolation of Human and Murine Homologues of TheDrosophilaMinibrain Gene: Human Homologue Maps to 21q22.2 in the Down Syndrome “Critical Region”. Genomics 1996, 38, 331–339. [Google Scholar] [CrossRef]
- Tejedor, F.J.; Hämmerle, B. MNB/DYRK1A as a Multiple Regulator of Neuronal Development. FEBS J. 2011, 278, 223–235. [Google Scholar] [CrossRef]
- Branca, C.; Shaw, D.M.; Belfiore, R.; Gokhale, V.; Shaw, A.Y.; Foley, C.; Smith, B.; Hulme, C.; Dunckley, T.; Meechoovet, B.; et al. Dyrk1 Inhibition Improves Alzheimer’s Disease-like Pathology. Aging Cell 2017, 16, 1146–1154. [Google Scholar] [CrossRef]
- Dowjat, W.K.; Adayev, T.; Kuchna, I.; Nowicki, K.; Palminiello, S.; Hwang, Y.W.; Wegiel, J. Trisomy-Driven Overexpression of DYRK1A Kinase in the Brain of Subjects with Down Syndrome. Neurosci. Lett. 2007, 413, 77–81. [Google Scholar] [CrossRef]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.-S.; Taverne, T.; Girard, A.; et al. A Novel DYRK1A (Dual Specificity Tyrosine Phosphorylation-Regulated Kinase 1A) Inhibitor for the Treatment of Alzheimer’s Disease: Effect on Tau and Amyloid Pathologies in Vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef]
- Wegiel, J.; Gong, C.-X.; Hwang, Y.-W. The Role of DYRK1A in Neurodegenerative Diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.-W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.-X. Overexpression of Dyrk1A Contributes to Neurofibrillary Degeneration in Down Syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.-R.; Cho, H.-J.; Lee, H.-W.; Jeong, H.K.; Radnaabazar, C.; Kim, Y.-S.; Kim, M.-J.; Son, M.-Y.; Seo, H.; Chung, S.-H.; et al. Dual-Specificity Tyrosine(Y)-Phosphorylation Regulated Kinase 1A-Mediated Phosphorylation of Amyloid Precursor Protein: Evidence for a Functional Link between Down Syndrome and Alzheimer’s Disease. J. Neurochem. 2008, 104, 1333–1344. [Google Scholar] [CrossRef]
- Park, J.; Song, W.-J.; Chung, K.C. Function and Regulation of Dyrk1A: Towards Understanding Down Syndrome. Cell. Mol. Life Sci. 2009, 66, 3235–3240. [Google Scholar] [CrossRef] [PubMed]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A Gene, Encoded in Chromosome 21 Down Syndrome Critical Region, Bridges between Beta-Amyloid Production and Tau Phosphorylation in Alzheimer Disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Woods, Y.L.; Cohen, P.; Becker, W.; Jakes, R.; Goedert, M.; Wang, X.; Proud, C.G. The Kinase DYRK Phosphorylates Protein-Synthesis Initiation Factor EIF2Bepsilon at Ser539 and the Microtubule-Associated Protein Tau at Thr212: Potential Role for DYRK as a Glycogen Synthase Kinase 3-Priming Kinase. Biochem. J. 2001, 355 Pt 3, 609–615. [Google Scholar] [CrossRef]
- Wegiel, J.; Dowjat, K.; Kaczmarski, W.; Kuchna, I.; Nowicki, K.; Frackowiak, J.; Mazur Kolecka, B.; Wegiel, J.; Silverman, W.P.; Reisberg, B.; et al. The Role of Overexpressed DYRK1A Protein in the Early Onset of Neurofibrillary Degeneration in Down Syndrome. Acta Neuropathol. 2008, 116, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhang, T.; Zhou, C.; Chohan, M.O.; Gu, X.; Wegiel, J.; Zhou, J.; Hwang, Y.-W.; Iqbal, K.; Grundke-Iqbal, I.; et al. Increased Dosage of Dyrk1A Alters Alternative Splicing Factor (ASF)-Regulated Alternative Splicing of Tau in Down Syndrome. J. Biol. Chem. 2008, 283, 28660–28669. [Google Scholar] [CrossRef]
- Yin, X.; Jin, N.; Shi, J.; Zhang, Y.; Wu, Y.; Gong, C.-X.; Iqbal, K.; Liu, F. Dyrk1A Overexpression Leads to Increase of 3R-Tau Expression and Cognitive Deficits in Ts65Dn Down Syndrome Mice. Sci. Rep. 2017, 7, 619. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Park, S.Y.; Jung, M.-S.; Yoon, S.-H.; Kwen, M.-Y.; Lee, S.-Y.; Choi, S.-H.; Radnaabazar, C.; Kim, M.-K.; Kim, H.; et al. Dyrk1A-Mediated Phosphorylation of Presenilin 1: A Functional Link between Down Syndrome and Alzheimer’s Disease. J. Neurochem. 2010, 115, 574–584. [Google Scholar] [CrossRef]
- Lee, M.-S.; Kao, S.-C.; Lemere, C.A.; Xia, W.; Tseng, H.-C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.-H. APP Processing Is Regulated by Cytoplasmic Phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Hamdane, M.; Gompel, M.; Bégard, S.; Drobecq, H.; Ghestem, A.; Grosjean, M.-E.; Kostanjevecki, V.; Grognet, P.; Vanmechelen, E.; et al. Phosphorylation of Amyloid Precursor Carboxy-Terminal Fragments Enhances Their Processing by a Gamma-Secretase-Dependent Mechanism. Neurobiol. Dis. 2005, 20, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.-Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B Induces Human β-Cell Proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef] [PubMed]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dančík, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A Stimulates Human β-Cell Proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef]
- Belgardt, B.-F.; Lammert, E. DYRK1A: A Promising Drug Target for Islet Transplant–Based Diabetes Therapies. Diabetes 2016, 65, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Alvarez-Perez, J.-C.; Felsenfeld, D.P.; Liu, H.; Sivendran, S.; Bender, A.; Kumar, A.; Sanchez, R.; Scott, D.K.; Garcia-Ocaña, A.; et al. Induction of Human Pancreatic Beta Cell Replication by Inhibitors of Dual Specificity Tyrosine Regulated Kinase. Nat. Med. 2015, 21, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Kong, X.; Cui, Y.; Wei, Y.; Zhang, J.; Wei, W. Conversion from MCI to AD in Patients with the APOE Ε4 Genotype: Prediction by Plasma HCY and Serum BDNF. Neurosci. Lett. 2016, 626, 19–24. [Google Scholar] [CrossRef]
- Fujinami, A.; Ohta, K.; Obayashi, H.; Fukui, M.; Hasegawa, G.; Nakamura, N.; Kozai, H.; Imai, S.; Ohta, M. Serum Brain-Derived Neurotrophic Factor in Patients with Type 2 Diabetes Mellitus: Relationship to Glucose Metabolism and Biomarkers of Insulin Resistance. Clin. Biochem. 2008, 41, 812–817. [Google Scholar] [CrossRef]
- Zhen, Y.F.; Zhang, J.; Liu, X.Y.; Fang, H.; Tian, L.B.; Zhou, D.H.; Kosten, T.R.; Zhang, X.Y. Low BDNF Is Associated with Cognitive Deficits in Patients with Type 2 Diabetes. Psychopharmacol. 2013, 227, 93–100. [Google Scholar] [CrossRef] [PubMed]
- ter Haar, E.; Coll, J.T.; Austen, D.A.; Hsiao, H.-M.; Swenson, L.; Jain, J. Structure of GSK3β Reveals a Primed Phosphorylation Mechanism. Nat. Struct. Biol. 2001, 8, 593–596. [Google Scholar] [CrossRef]
- Cline, G.W.; Johnson, K.; Regittnig, W.; Perret, P.; Tozzo, E.; Xiao, L.; Damico, C.; Shulman, G.I. Effects of a Novel Glycogen Synthase Kinase-3 Inhibitor on Insulin-Stimulated Glucose Metabolism in Zucker Diabetic Fatty (Fa/Fa) Rats. Diabetes 2002, 51, 2903–2910. [Google Scholar] [CrossRef]
- Dokken, B.B.; Henriksen, E.J. Chronic Selective Glycogen Synthase Kinase-3 Inhibition Enhances Glucose Disposal and Muscle Insulin Action in Prediabetic Obese Zucker Rats. Am. J. Physiol.-Endocrinol. Metab. 2006, 291, E207–E213. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of Glycogen Synthase Kinase-3 by Lithium Correlates with Reduced Tauopathy and Degeneration in Vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; Diniz, B.S.; Radanovic, M.; Santos, F.S.; Talib, L.L.; Gattaz, W.F. Disease-Modifying Properties of Long-Term Lithium Treatment for Amnestic Mild Cognitive Impairment: Randomised Controlled Trial. Br. J. Psychiatry 2011, 198, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of Glycogen Synthase Kinase-3 Ameliorates β-Amyloid Pathology and Restores Lysosomal Acidification and Mammalian Target of Rapamycin Activity in the Alzheimer Disease Mouse Model: IN VIVO AND IN VITRO STUDIES *. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Griebel, G.; Stemmelin, J.; Lopez-Grancha, M.; Boulay, D.; Boquet, G.; Slowinski, F.; Pichat, P.; Beeské, S.; Tanaka, S.; Mori, A.; et al. The Selective GSK3 Inhibitor, SAR502250, Displays Neuroprotective Activity and Attenuates Behavioral Impairments in Models of Neuropsychiatric Symptoms of Alzheimer’s Disease in Rodents. Sci. Rep. 2019, 9, 18045. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Zhang, Q.; Zhang, C.; Wu, C.; Yang, K.; Song, Z.; Wang, Q.; Li, C.; Zhou, Y.; Chen, J.; et al. A Natural DYRK1A Inhibitor as a Potential Stimulator for β-Cell Proliferation in Diabetes. Clin. Transl. Med. 2021, 11, e494. [Google Scholar] [CrossRef]
- Souchet, B.; Audrain, M.; Billard, J.M.; Dairou, J.; Fol, R.; Orefice, N.S.; Tada, S.; Gu, Y.; Dufayet-Chaffaud, G.; Limanton, E.; et al. Inhibition of DYRK1A Proteolysis Modifies Its Kinase Specificity and Rescues Alzheimer Phenotype in APP/PS1 Mice. Acta Neuropathol. Commun. 2019, 7, 46. [Google Scholar] [CrossRef]
- Zhu, B.; Parsons, T.; Stensen, W.; Mjøen Svendsen, J.S.; Fugelli, A.; Hodge, J.J.L. DYRK1a Inhibitor Mediated Rescue of Drosophila Models of Alzheimer’s Disease-Down Syndrome Phenotypes. Front. Pharmacol. 2022, 13, 881385. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Molecular Target | Disease | Experimental Model | Main Findings | References |
|---|---|---|---|---|
| GSK3β Inhibition | Diabetes | Zucker diabetic fatty (fa/fa) rats |
| [257] |
| Zucker prediabetic fatty (fa/fa) rats |
| [258] | ||
| Neonatal streptozotocin-induced diabetes in rats |
| [222] | ||
| 90%-pancreatectomized Wistar rats |
| [223] | ||
| Diabetic Goto–Kakizaki rats |
| [225] | ||
| AD | JNPL3 transgenic mice overexpressing mutant human tau |
| [259] | |
| Patients with amnestic mild cognitive impairment |
| [260] | ||
| 5XFAD mouse model of AD |
| [261] | ||
| P301L human tau transgenic mice |
| [262] | ||
| Rat embryonic hippocampal neurons |
| |||
| Swiss mice injected with Aβ25–35 Aged APP (SW)/tau (VLW) mice |
|
| Molecular Therapeutic Target | Disease | Experimental Model | Main Findings | References |
|---|---|---|---|---|
| DYRK1A Inhibition | Diabetes | R7T1 mouse β cells and rat and human islets |
| [249] |
| Diabetic mice transplanted with human islets |
| |||
| Rat and human β cells |
| [252] | ||
| Partial pancreatectomy mouse model |
| |||
| Diabetic NODSCID mice transplanted with marginal mass of human islets |
| |||
| Human β cells; NGS mice transplanted with human islets |
| [250] | ||
| INS-1 cells |
| [263] | ||
| db/db mice |
| |||
| AD | HEK293 cells, SH-SY5Y neuroblastoma cells, and rat primary cortical neurons |
| [236] | |
| Neuronal cells |
| |||
| HEK293 cells overexpressing APP |
| |||
| 3xTg-AD mice |
| [234] | ||
| Aged APP/PS1 mice |
| [264] | ||
| AD–DS drosophila models overexpressing human tau, human Aβ, or minibrain |
| [265] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamzé, R.; Delangre, E.; Tolu, S.; Moreau, M.; Janel, N.; Bailbé, D.; Movassat, J. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Shared Molecular Mechanisms and Potential Common Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 15287. https://doi.org/10.3390/ijms232315287
Hamzé R, Delangre E, Tolu S, Moreau M, Janel N, Bailbé D, Movassat J. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Shared Molecular Mechanisms and Potential Common Therapeutic Targets. International Journal of Molecular Sciences. 2022; 23(23):15287. https://doi.org/10.3390/ijms232315287
Chicago/Turabian StyleHamzé, Rim, Etienne Delangre, Stefania Tolu, Manon Moreau, Nathalie Janel, Danielle Bailbé, and Jamileh Movassat. 2022. "Type 2 Diabetes Mellitus and Alzheimer’s Disease: Shared Molecular Mechanisms and Potential Common Therapeutic Targets" International Journal of Molecular Sciences 23, no. 23: 15287. https://doi.org/10.3390/ijms232315287
APA StyleHamzé, R., Delangre, E., Tolu, S., Moreau, M., Janel, N., Bailbé, D., & Movassat, J. (2022). Type 2 Diabetes Mellitus and Alzheimer’s Disease: Shared Molecular Mechanisms and Potential Common Therapeutic Targets. International Journal of Molecular Sciences, 23(23), 15287. https://doi.org/10.3390/ijms232315287