
Many Functions of Telomerase Components: Certainties, Doubts, and Inconsistencies



Abstract

1. Introduction
2. Telomerase: Function and Components
2.1. Telomerase Function
2.2. TERT Structure
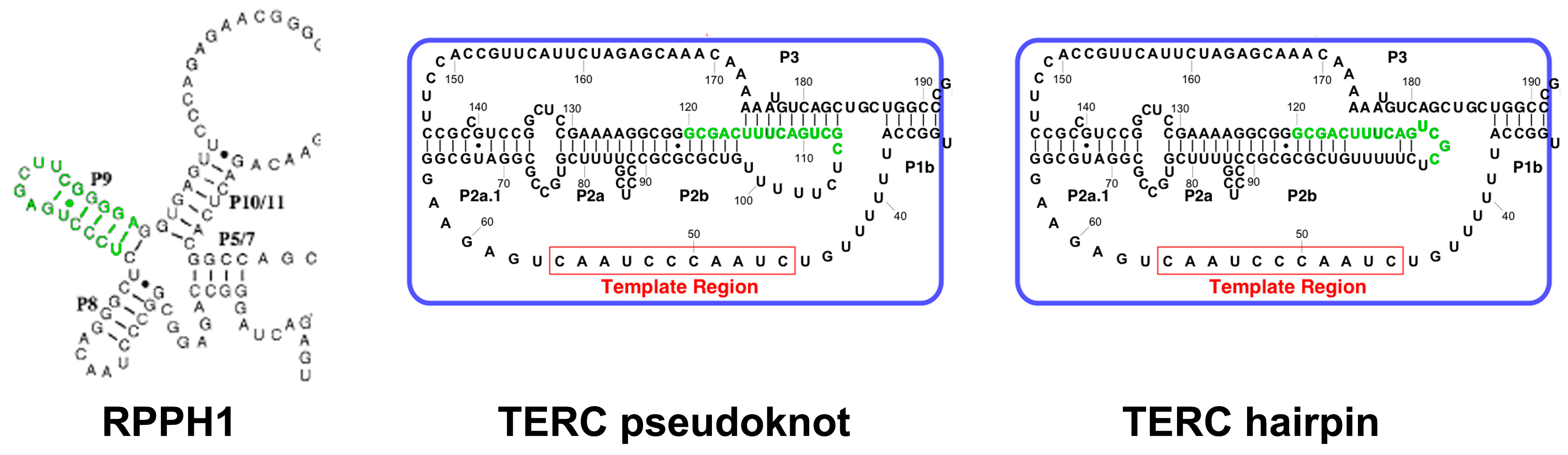
2.3. TERC Structure
2.4. Secondary and Accessory Proteins
2.5. Has Telomerase Any Non-Telomeric Function?
2.5.1. Addition of Telomeric Repeats at Double-Strand Break Sites?
2.5.2. NOP2-Dependent Recruitment of Telomerase to Cyclin D1 Promoter
3. TERT Non-Telomeric Functions
3.1. Gene Regulation
3.1.1. NF-κB Pathway
3.1.2. Wnt/β-Catenin Pathway
3.1.3. pRb and Cyclins
3.1.4. Ribosomal DNA
3.1.5. RNA Polymerase III Target Genes
3.1.6. Transcriptomic Studies
3.2. An RNA-Dependent RNA Polymerase?
3.3. TERT and Mitochondria
3.3.1. Shuttling of TERT between Nucleus, Cytoplasm and Mitochondria
3.3.2. TERT Association with mtDNA
3.3.3. Controversial Effects of TERT in Mitochondria
3.4. Cellular Effects
3.5. Alternative Isoforms of TERT and Their Functions
4. TERC Non-Telomeric Functions
4.1. Gene Regulation
4.2. New Actors in the Plot
4.2.1. RPL22
4.2.2. DNA-PK
4.3. TERC and Mitochondria
4.4. TERC-Derived Small RNA
4.5. Translation of TERC?
4.5.1. Prediction of TERC Protein-Coding Ability
4.5.2. Evidence of Absence or Absence of Evidence?
4.5.3. TERP
4.6. Cellular Effects
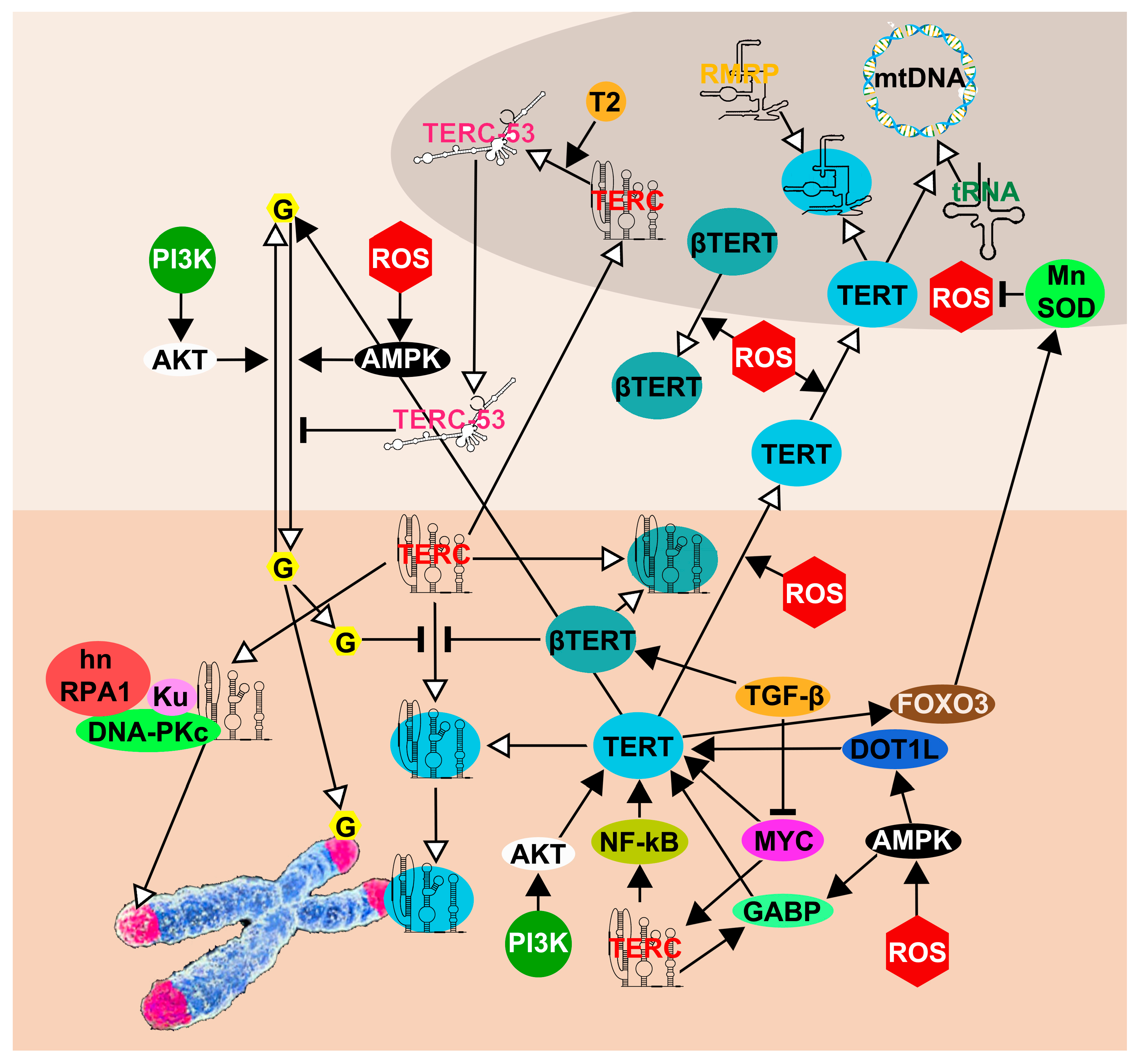
5. The Network of TERT and TERC Functions and Interactions
6. Open Questions for Future Research
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greider, C.W.; Blackburn, E.H. Identification of a Specific Telomere Terminal Transferase Activity in Tetrahymena Extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. Principle of Marginotomy in Template Synthesis of Polynucleotides. Dokl. Akad. Nauk SSSR 1971, 201, 1496–1499. [Google Scholar] [PubMed]
- Shay, J.W.; Wright, W.E. Telomerase: A Target for Cancer Therapeutics. Cancer Cell 2002, 2, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Prowse, K.R.; Ho, P.; Weissman, I.L. Telomerase Activity in Hematopoietic Cells Is Associated with Self-Renewal Potential. Immunity 1996, 5, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Härle-Bachor, C.; Boukamp, P. Telomerase Activity in the Regenerative Basal Layer of the Epidermis Inhuman Skin and in Immortal and Carcinoma-Derived Skin Keratinocytes. Proc. Natl. Acad. Sci. USA 1996, 93, 6476–6481. [Google Scholar] [CrossRef]
- Colitz, C.M.H.; Davidson, M.G.; McGahan, M.C. Telomerase Activity in Lens Epithelial Cells of Normal and Cataractous Lenses. Exp. Eye Res. 1999, 69, 641–649. [Google Scholar] [CrossRef]
- Rosen, J.; Jakobs, P.; Ale-Agha, N.; Altschmied, J.; Haendeler, J. Non-Canonical Functions of Telomerase Reverse Transcriptase - Impact on Redox Homeostasis. Redox Biol. 2020, 34. [Google Scholar] [CrossRef]
- Gould, S.J. Leonardo’s Mountain of Clams and the Diet of Worms: Essays on Natural History; Harmony Books: New York, NY, USA, 1998; ISBN 9780609601402. [Google Scholar]
- Liang, L.; Zhong, Z.; Rousseau, R. Scientists’ Referencing (Mis)Behavior Revealed by the Dissemination Network of Referencing Errors. Scientometrics 2014, 101, 1973–1986. [Google Scholar] [CrossRef]
- Udroiu, I.; Russo, V.; Persichini, T.; Colasanti, M.; Sgura, A. Telomeres and Telomerase in Basal Metazoa. Invertebr. Surviv. J. 2017, 14, 233–240. [Google Scholar] [CrossRef]
- Autexier, C.; Lue, N.F. The Structure and Function of Telomerase Reverse Transcriptase. Annu. Rev. Biochem. 2006, 75, 493–517. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A Telomeric Sequence in the RNA of Tetrahymena Telomerase Required for Telomere Repeat Synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Artandi, S.E. TCAB1: Driving Telomerase to Cajal Bodies. Cell Cycle 2009, 8, 1329–1331. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Collins, K. Control of Telomerase Action at Human Telomeres. Nat. Struct. Mol. Biol. 2015, 22, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kim, N.K.; Feigon, J. Architecture of Human Telomerase RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20325–20332. [Google Scholar] [CrossRef] [PubMed]
- Morin, G.B. The Human Telomere Terminal Transferase Enzyme Is a Ribonucleoprotein That Synthesizes TTAGGG Repeats. Cell 1989, 59, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomerase Is Processive. Mol. Cell. Biol. 1991, 11, 4572–4580. [Google Scholar] [CrossRef]
- Weise, J.M.; Günes, C. Differential Regulation of Human and Mouse Telomerase Reverse Transcriptase (TERT) Promoter Activity during Testis Development. Mol. Reprod. Dev. 2009, 76, 309–317. [Google Scholar] [CrossRef]
- Avilion, A.A.; Piatyszek, M.A.; Gupta, J.; Shay, J.W.; Bacchetti, S.; Greider, C.W. Human Telomerase RNA and Telomerase Activity in Immortal Cell Lines and Tumor Tissues. Cancer Res. 1996, 56. [Google Scholar]
- Castle, J.C.; Armour, C.D.; Löwer, M.; Haynor, D.; Biery, M.; Bouzek, H.; Chen, R.; Jackson, S.; Johnson, J.M.; Rohl, C.A.; et al. Digital Genome-Wide NcRNA Expression, Including SnoRNAs, across 11 Human Tissues Using PolyA-Neutral Amplification. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Hartmann, N.; Reichwald, K.; Lechel, A.; Graf, M.; Kirschner, J.; Dorn, A.; Terzibasi, E.; Wellner, J.; Platzer, M.; Rudolph, K.L.; et al. Telomeres Shorten While Tert Expression Increases during Ageing of the Short-Lived Fish Nothobranchius Furzeri. Mech. Ageing Dev. 2009, 130, 290–296. [Google Scholar] [CrossRef]
- Agarwal, S.; Loh, Y.H.; McLoughlin, E.M.; Huang, J.; Park, I.H.; Miller, J.D.; Huo, H.; Okuka, M.; Dos Reis, R.M.; Loewer, S.; et al. Telomere Elongation in Induced Pluripotent Stem Cells from Dyskeratosis Congenita Patients. Nature 2010, 464, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Weinrich, S.L.; Pruzan, R.; Ma, L.; Ouellette, M.; Tesmer, V.M.; Holt, S.E.; Bodnar, A.G.; Lichtsteiner, S.; Kim, N.W.; Trager, J.B.; et al. Reconstitution of Human Telomerase with the Template RNA Component HTR and the Catalytic Protein Subunit HTRT. Nat. Genet. 1997, 17, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.C.; Liu, J.K.; Collins, K. Template Definition by Tetrahymena Telomerase Reverse Transcriptase. EMBO J. 2000, 19, 4412–4422. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, T.J.; Huard, S.; Dupuis, S.; Autexier, C. Functional Multimerization of Human Telomerase Requires an RNA Interaction Domain in the N Terminus of the Catalytic Subunit. Mol. Cell. Biol. 2002, 22, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Sealey, D.C.F.; Zheng, L.; Taboski, M.A.S.; Cruickshank, J.; Ikura, M.; Harrington, L.A. The N-Terminus of HTERT Contains a DNA-Binding Domain and Is Required for Telomerase Activity and Cellular Immortalization. Nucleic Acids Res. 2010, 38, 2019–2035. [Google Scholar] [CrossRef]
- Lai, C.K.; Mitchell, J.R.; Collins, K. RNA Binding Domain of Telomerase Reverse Transcriptase. Mol. Cell. Biol. 2001, 21, 990–1000. [Google Scholar] [CrossRef]
- Moriarty, T.J.; Marie-Egyptienne, D.T.; Autexier, C. Functional Organization of Repeat Addition Processivity and DNA Synthesis Determinants in the Human Telomerase Multimer. Mol. Cell. Biol. 2004, 24, 3720–3733. [Google Scholar] [CrossRef][Green Version]
- Mitchell, J.R.; Collins, K. Human Telomerase Activation Requires Two Independent Interactions between Telomerase RNA and Telomerase Reverse Transcriptase. Mol. Cell 2000, 6, 361–371. [Google Scholar] [CrossRef]
- Friedman, K.L.; Cech, T.R. Essential Functions of Amino-Terminal Domains in the Yeast Telomerase Catalytic Subunit Revealed by Selection for Viable Mutants. Genes Dev. 1999, 13, 2863–2874. [Google Scholar] [CrossRef]
- Willers, H.; McCarthy, E.E.; Wu, B.; Wunsch, H.; Tang, W.; Taghian, D.G.; Xia, F.; Powell, S.N. Dissociation of P53-Mediated Suppression of Homologous Recombination from G1/S Cell Cycle Checkpoint Control. Oncogene 2000, 19, 632–639. [Google Scholar] [CrossRef][Green Version]
- Beattie, T.L.; Zhou, W.; Robinson, M.O.; Harrington, L. Reconstitution of Human Telomerase Activity in Vitro. Curr. Biol. 1998, 8, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase Catalytic Subunit Homologs from Fission Yeast and Human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Bosoy, D.; Lue, N.F. Functional Analysis of Conserved Residues in the Putative “Finger” Domain of Telomerase Reverse Transcriptase. J. Biol. Chem. 2001, 276, 46305–46312. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Podlevsky, J.D.; Qi, X.; Bley, C.J.; Chen, J.J.L. A Novel Motif in Telomerase Reverse Transcriptase Regulates Telomere Repeat Addition Rate and Processivity. Nucleic Acids Res. 2010, 38, 1982–1996. [Google Scholar] [CrossRef]
- Gillis, A.J.; Schuller, A.P.; Skordalakes, E. Structure of the Tribolium Castaneum Telomerase Catalytic Subunit TERT. Nature 2008, 455, 633–637. [Google Scholar] [CrossRef]
- Seimiya, H.; Sawada, H.; Muramatsu, Y.; Shimizu, M.; Ohko, K.; Yamane, K.; Tsuruo, T. Involvement of 14-3-3 Proteins in Nuclear Localization of Telomerase. EMBO J. 2000, 19, 2652–2661. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen Peroxide Triggers Nuclear Export of Telomerase Reverse Transcriptase via Src Kinase Family-Dependent Phosphorylation of Tyrosine 707. Mol. Cell. Biol. 2003, 23, 4598–4610. [Google Scholar] [CrossRef]
- Ye, A.J.; Romero, D.P. Phylogenetic Relationships amongst Tetrahymenine Ciliates Inferred by a Comparison of Telomerase RNAs. Int. J. Syst. Evol. Microbiol. 2002, 52, 2297–2302. [Google Scholar] [CrossRef]
- Chakrabarti, K.; Pearson, M.; Grate, L.; Sterne-Weiler, T.; Deans, J.; Donohue, J.P.; Ares, M. Structural RNAs of Known and Unknown Function Identified in Malaria Parasites by Comparative Genomics and RNA Analysis. RNA 2007, 13, 1923–1939. [Google Scholar] [CrossRef]
- Song, J.; Logeswaran, D.; Castillo-González, C.; Li, Y.; Bose, S.; Aklilu, B.B.; Ma, Z.; Polkhovskiy, A.; Chen, J.J.L.; Shippen, D.E. The Conserved Structure of Plant Telomerase RNA Provides the Missing Link for an Evolutionary Pathway from Ciliates to Humans. Proc. Natl. Acad. Sci. USA 2019, 116, 24542–24550. [Google Scholar] [CrossRef]
- Qi, X.; Li, Y.; Honda, S.; Hoffmann, S.; Marz, M.; Mosig, A.; Podlevsky, J.D.; Stadler, P.F.; Selker, E.U.; Chen, J.J.L. The Common Ancestral Core of Vertebrate and Fungal Telomerase RNAs. Nucleic Acids Res. 2013, 41, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Logeswaran, D.; Li, Y.; Podlevsky, J.D.; Chen, J.J.L. Monophyletic Origin and Divergent Evolution of Animal Telomerase RNA. Mol. Biol. Evol. 2021, 38, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Podlevsky, J.D.; Chen, J.J.L. Evolutionary Perspectives of Telomerase RNA Structure and Function. RNA Biol. 2016, 13, 720–732. [Google Scholar] [CrossRef]
- Chen, J.L.; Blasco, M.A.; Greider, C.W. Secondary Structure of Vertebrate Telomerase RNA. Cell 2000, 100, 503–514. [Google Scholar] [CrossRef]
- Theimer, C.A.; Feigon, J. Structure and Function of Telomerase RNA. Curr. Opin. Struct. Biol. 2006, 16, 307–318. [Google Scholar] [CrossRef]
- Chen, J.L.; Greider, C.W. Template Boundary Definition in Mammalian Telomerase. Genes Dev. 2003, 17, 2747–2752. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.; Singh, S.; Lue, N.F. Functional Analysis of the C-Terminal Extension of Telomerase Reverse Transcriptase. A Putative “Thumb” Domain. J. Biol. Chem. 2002, 277, 36174–36180. [Google Scholar] [CrossRef] [PubMed]
- Förstemann, K.; Lingner, J. Telomerase Limits the Extent of Base Pairing between Template RNA and Telomeric DNA. EMBO Rep. 2005, 6, 361–366. [Google Scholar] [CrossRef]
- Brown, Y.; Abraham, M.; Pearl, S.; Kabaha, M.M.; Elboher, E.; Tzfati, Y. A Critical Three-Way Junction Is Conserved in Budding Yeast and Vertebrate Telomerase RNAs. Nucleic Acids Res. 2007, 35, 6280–6289. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Collins, K. Telomerase: An RNP Enzyme Synthesizes DNA. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–9. [Google Scholar] [CrossRef]
- Huang, J.; Brown, A.F.; Wu, J.; Xue, J.; Bley, C.J.; Rand, D.P.; Wu, L.; Zhang, R.; Chen, J.J.L.; Lei, M. Structural Basis for Protein-RNA Recognition in Telomerase. Nat. Struct. Mol. Biol. 2014, 21, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.R.; Cheng, J.; Collins, K. A Box H/ACA Small Nucleolar RNA-like Domain at the Human Telomerase RNA 3’ End. Mol. Cell. Biol. 1999, 19, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Vulliamy, T.J.; Marrone, A.; Knight, S.W.; Walne, A.; Mason, P.J.; Dokal, I. Mutations in Dyskeratosis Congenita: Their Impact on Telomere Length and the Diversity of Clinical Presentation. Blood 2006, 107, 2680–2685. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Unveiling Substrate RNA Binding to H/ACA RNPs: One Side Fits All. Curr. Opin. Struct. Biol. 2008, 18, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Reichow, S.L.; Hamma, T.; Ferré-D’Amaré, A.R.; Varani, G. The Structure and Function of Small Nucleolar Ribonucleoproteins. Nucleic Acids Res. 2007, 35, 1452–1464. [Google Scholar] [CrossRef] [PubMed]
- Cristofari, G.; Adolf, E.; Reichenbach, P.; Sikora, K.; Terns, R.M.; Terns, M.P.; Lingner, J. Human Telomerase RNA Accumulation in Cajal Bodies Facilitates Telomerase Recruitment to Telomeres and Telomere Elongation. Mol. Cell 2007, 27, 882–889. [Google Scholar] [CrossRef]
- Theimer, C.A.; Jády, B.E.; Chim, N.; Richard, P.; Breece, K.E.; Kiss, T.; Feigon, J. Structural and Functional Characterization of Human Telomerase RNA Processing and Cajal Body Localization Signals. Mol. Cell 2007, 27, 869–881. [Google Scholar] [CrossRef]
- Kiss, T.; Fayet-Lebaron, E.; Jády, B.E. Box H/ACA Small Ribonucleoproteins. Mol. Cell 2010, 37, 597–606. [Google Scholar] [CrossRef]
- Egan, E.D.; Collins, K. Specificity and Stoichiometry of Subunit Interactions in the Human Telomerase Holoenzyme Assembled in Vivo. Mol. Cell. Biol. 2010, 30, 2775–2786. [Google Scholar] [CrossRef]
- Lafontaine, D.L.J.; Bousquet-Antonelli, C.; Henry, Y.; Caizergues-Ferrer, M.; Tollervey, D. The Box H + ACA SnoRNAs Carry Cbf5p, the Putative RRNA Pseudouridine Synthase. Genes Dev. 1998, 12, 527–537. [Google Scholar] [CrossRef]
- Ganot, P.; Bortolin, M.L.; Kiss, T. Site-Specific Pseudouridine Formation in Preribosomal RNA Is Guided by Small Nucleolar RNAs. Cell 1997, 89, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.M.; Jády, B.E.; Darzacq, X.; Verheggen, C.; Bertrand, E.; Kiss, T. A Cajal Body-Specific Pseudouridylation Guide RNA Is Composed of Two Box H/ACA SnoRNA-like Domains. Nucleic Acids Res. 2002, 30, 4643–4649. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Darzacq, X.; Bertrand, E.; Jády, B.E.; Verheggen, C.; Kiss, T. A Common Sequence Motif Determines the Cajal Body-Specific Localization of Box H/ACA ScaRNAs. EMBO J. 2003, 22, 4283–4293. [Google Scholar] [CrossRef]
- Wang, C.; Meier, U.T. Architecture and Assembly of Mammalian H/ACA Small Nucleolar and Telomerase Ribonucleoproteins. EMBO J. 2004, 23, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Grozdanov, P.N.; Roy, S.; Kittur, N.; Meier, U.T. SHQ1 Is Required Prior to NAF1 for Assembly of H/ACA Small Nucleolar and Telomerase RNPs. RNA 2009, 15, 1188–1197. [Google Scholar] [CrossRef]
- Girard, J.P.; Lehtonen, H.; Caizergues-Ferrer, M.; Amalric, F.; Tollervey, D.; Lapeyre, B. GAR1 Is an Essential Small Nucleolar RNP Protein Required for Pre-RRNA Processing in Yeast. EMBO J. 1992, 11, 673. [Google Scholar] [CrossRef]
- Maiorano, D.; Brimage, L.J.E.; Leroy, D.; Kearsey, S.E. Functional Conservation and Cell Cycle Localization of the Nhp2 Core Component of H + ACA SnoRNPs in Fission and Budding Yeasts. Exp. Cell Res. 1999, 252, 165–174. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Abreu, E.B.; Meng, Z.; McCann, K.E.; Terns, R.M.; Veenstra, T.D.; Terns, M.P.; Artandi, S.E. A Human Telomerase Holoenzyme Protein Required for Cajal Body Localization and Telomere Synthesis. Science 2009, 323, 644–648. [Google Scholar] [CrossRef]
- Freund, A.; Zhong, F.L.; Venteicher, A.S.; Meng, Z.; Veenstra, T.D.; Frydman, J.; Artandi, S.E. Proteostatic Control of Telomerase Function through TRiC-Mediated Folding of TCAB1. Cell 2014, 159, 1389–1403. [Google Scholar] [CrossRef]
- Flint, J.; Craddock, C.F.; Villegas, A.; Bentley, D.P.; Williams, H.J.; Galanello, R.; Cao, A.; Wood, W.G.; Ayyub, H.; Higgs, D.R. Healing of Broken Human Chromosomes by the Addition of Telomeric Repeats. Am. J. Hum. Genet. 1994, 55, 505. [Google Scholar]
- Nergadze, S.G.; Santagostino, M.A.; Salzano, A.; Mondello, C.; Giulotto, E. Contribution of Telomerase RNA Retrotranscription to DNA Double-Strand Break Repair during Mammalian Genome Evolution. Genome Biol. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, J.; Cech, T.R. Finding the End: Recruitment of Telomerase to Telomeres. Nat. Rev. Mol. Cell Biol. 2013, 14, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Morgan, R.; Berger, C.S.; Sandberg, A.A. Presence of Telomeric Sequences on Deleted Chromosomes and Their Absence on Double Minutes in Cell Line HL-60. Cancer Genet. Cytogenet. 1993, 70, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, P.S.; Guan, X.Y.; Trent, J.M. Telomere Capture Stabilizes Chromosome Breakage. Nat. Genet. 1993 43 1993, 4, 252–255. [Google Scholar] [CrossRef]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234. [Google Scholar] [CrossRef]
- Q, F.; M, Y. New Telomere Formation Coupled with Site-Specific Chromosome Breakage in Tetrahymena Thermophila. Mol. Cell. Biol. 1996, 16, 1267–1274. [Google Scholar] [CrossRef]
- Matsumoto, T.; Fukui, K.; Niwa, O.; Sugawara, N.; Szostak, J.W.; Yanagida, M. Identification of Healed Terminal DNA Fragments in Linear Minichromosomes of Schizosaccharomyces Pombe. Mol. Cell. Biol. 1987, 7, 4424–4430. [Google Scholar] [CrossRef]
- Kramer, K.M.; Haber, J.E. New Telomeres in Yeast Are Initiated with a Highly Selected Subset of TG1-3 Repeats. Genes Dev. 1993, 7, 2345–2356. [Google Scholar] [CrossRef]
- Wang, S.S.; Zakian, V.A. Telomere-Telomere Recombination Provides an Express Pathway for Telomere Acquisition. Nature 1990, 345, 456–458. [Google Scholar] [CrossRef]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968. [Google Scholar] [CrossRef]
- Hong, J.; Lee, J.H.; Chung, I.K. Telomerase Activates Transcription of Cyclin D1 Gene through an Interaction with NOL1. J. Cell Sci. 2016, 129, 1566–1579. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N.; et al. TERT Promotes Epithelial Proliferation through Transcriptional Control of a Myc- and Wnt-Related Developmental Program. PLoS Genet. 2008, 4, 0124–0138. [Google Scholar] [CrossRef]
- Ding, D.; Xi, P.; Zhou, J.; Wang, M.; Cong, Y.S. Human Telomerase Reverse Transcriptase Regulates MMP Expression Independently of Telomerase Activity via NF-ΚB-Dependent Transcription. FASEB J. 2013, 27, 4375–4383. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Hubbard, A.K.; Giardina, C. NF-Kappa B Regulates Transcription of the Mouse Telomerase Catalytic Subunit. J. Biol. Chem. 2000, 275, 36671–36675. [Google Scholar] [CrossRef] [PubMed]
- Sinha-Datta, U.; Horikawa, I.; Michishita, E.; Datta, A.; Sigler-Nicot, J.C.; Brown, M.; Kazanji, M.; Barrett, J.C.; Nicot, C. Transcriptional Activation of HTERT through the NF-KappaB Pathway in HTLV-I-Transformed Cells. Blood 2004, 104, 2523–2531. [Google Scholar] [CrossRef]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase Directly Regulates NF-ΚB-Dependent Transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase Modulates Wnt Signalling by Association with Target Gene Chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Björkholm, M.; et al. Telomerase Reverse Transcriptase Promotes Epithelial-Mesenchymal Transition and Stem Cell-like Traits in Cancer Cells. Oncogene 2013, 32, 4203–4213. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without Cyclins and Cyclin-Dependent Kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef]
- Farwell, D.G.; Shera, K.A.; Koop, J.I.; Bonnet, G.A.; Matthews, C.P.; Reuther, G.W.; Coltrera, M.D.; McDougall, J.K.; Klingelhutz, A.J. Genetic and Epigenetic Changes in Human Epithelial Cells Immortalized by Telomerase. Am. J. Pathol. 2000, 156, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Wang, J.; Mao, Y.; Wan-Cheng Li, D.; Liu, M.; Reddy, V.N. Human Telomerase Accelerates Growth of Lens Epithelial Cells through Regulation of the Genes Mediating RB/E2F Pathway. Oncogene 2002, 21, 3784–3791. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Przyborski, S.; Cooke, M.J.; Zhang, X.; Stewart, R.; Anyfantis, G.; Atkinson, S.P.; Saretzki, G.; Armstrong, L.; Lako, M. A Key Role for Telomerase Reverse Transcriptase Unit in Modulating Human Embryonic Stem Cell Proliferation, Cell Cycle Dynamics, and in Vitro Differentiation. Stem Cells 2008, 26, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, O.G.; Assfalg, R.; Koch, S.; Schelling, A.; Meena, J.K.; Kraus, J.; Lechel, A.; Katz, S.F.; Benes, V.; Scharffetter-Kochanek, K.; et al. Telomerase Stimulates Ribosomal DNA Transcription under Hyperproliferative Conditions. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Khattar, E.; Kumar, P.; Liu, C.Y.; Can Akincilar, S.; Raju, A.; Lakshmanan, M.; Maury, J.J.P.; Qiang, Y.; Li, S.; Tan, E.Y.; et al. Telomerase Reverse Transcriptase Promotes Cancer Cell Proliferation by Augmenting TRNA Expression. J. Clin. Invest. 2016, 126, 4045–4060. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Q.; Ge, Y.; Zhao, Q.; Zheng, X.; Zhao, Y. HTERT Promotes Cell Adhesion and Migration Independent of Telomerase Activity. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Kumar, P.; Kumar, M.; Yadava, P.K. HTERT Promotes Tumor Progression by Enhancing TSPAN13 Expression in Osteosarcoma Cells. Mol. Carcinog. 2018, 57, 1038–1054. [Google Scholar] [CrossRef]
- Jaiswal, R.K.; Kumar, P.; Sharma, A.; Mishra, D.K.; Yadava, P.K. Proteomic Identification of Proteins Differentially Expressed Following Overexpression of HTERT (Human Telomerase Reverse Transcriptase) in Cancer Cells. PLoS One 2017, 12. [Google Scholar] [CrossRef]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-Dependent RNA Polymerase Formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [Google Scholar] [CrossRef]
- Mattijssen, S.; Hinson, E.R.; Onnekink, C.; Hermanns, P.; Zabel, B.; Cresswell, P.; Pruijn, G.J.M. Viperin MRNA Is a Novel Target for the Human RNase MRP/RNase P Endoribonuclease. Cell. Mol. Life Sci. 2011, 68, 2469–2480. [Google Scholar] [CrossRef]
- Lemieux, B.; Laterreur, N.; Perederina, A.; Noël, J.F.; Dubois, M.L.; Krasilnikov, A.S.; Wellinger, R.J. Active Yeast Telomerase Shares Subunits with Ribonucleoproteins RNase P and RNase MRP. Cell 2016, 165, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Yuno, M.; Okuzaki, D.; Ohki, R.; Fujii, H. Identification of Non-Coding RNAs Associated with Telomeres Using a Combination of EnChIP and RNA Sequencing. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial HTERT Exacerbates Free-Radical-Mediated MtDNA Damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; Van Houten, B. Mitochondrial Localization of Telomerase as a Determinant for Hydrogen Peroxide-Induced Mitochondrial DNA Damage and Apoptosis. Hum. Mol. Genet. 2006, 15, 1757–1768. [Google Scholar] [CrossRef]
- Saretzki, G. Extra-Telomeric Functions of Human Telomerase: Cancer, Mitochondria and Oxidative Stress. Curr. Pharm. Des. 2014, 20, 6386–6403. [Google Scholar] [CrossRef]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial Telomerase Reverse Transcriptase Binds to and Protects Mitochondrial DNA and Function from Damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef]
- Naamati, A.; Regev-Rudzki, N.; Galperin, S.; Lill, R.; Pines, O. Dual Targeting of Nfs1 and Discovery of Its Novel Processing Enzyme, Icp55. J. Biol. Chem. 2009, 284, 30200–30208. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Rahman, S.; Zeiher, A.M.; Dimmeler, S. Regulation of Telomerase Activity and Anti-Apoptotic Function by Protein-Protein Interaction and Phosphorylation. FEBS Lett. 2003, 536, 180–186. [Google Scholar] [CrossRef]
- Büchner, N.; Zschauer, T.C.; Lukosz, M.; Altschmied, J.; Haendeler, J. Downregulation of Mitochondrial Telomerase Reverse Transcriptase Induced by H2O2 Is Src Kinase Dependent. Exp. Gerontol. 2010, 45, 558–562. [Google Scholar] [CrossRef]
- Sharma, N.K.; Reyes, A.; Green, P.; Caron, M.J.; Bonini, M.G.; Gordon, D.M.; Holt, I.J.; Santos, J.H. Human Telomerase Acts as a HTR-Independent Reverse Transcriptase in Mitochondria. Nucleic Acids Res. 2012, 40, 712–725. [Google Scholar] [CrossRef]
- Chen, L.Y.; Zhang, Y.; Zhang, Q.; Li, H.; Luo, Z.; Fang, H.; Kim, S.H.; Qin, L.; Yotnda, P.; Xu, J.; et al. Mitochondrial Localization of Telomeric Protein TIN2 Links Telomere Regulation to Metabolic Control. Mol. Cell 2012, 47, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Kaguni, L.S. DNA Polymerase Gamma, the Mitochondrial Replicase. Annu. Rev. Biochem. 2004, 73, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, M.; Reis, R.J.S.; Ayyadevara, S.; Wang, X.; Ganne, A.; Khaidakov, M. Involvement of TRNAs in Replication of Human Mitochondrial DNA and Modifying Effects of Telomerase. Mech. Ageing Dev. 2017, 166, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; von Zglinicki, T.; Saretzki, G. Telomerase Does Not Counteract Telomere Shortening but Protects Mitochondrial Function under Oxidative Stress. J. Cell Sci. 2008, 121, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Tanaka, Y.; Kondo, Y.; Hitomi, M.; Barnett, G.H.; Ishizaka, Y.; Liu, J.; Haqqi, T.; Nishiyama, A.; Villeponteau, B.; et al. Antisense Telomerase Treatment: Induction of Two Distinct Pathways, Apoptosis and Differentiation. FASEB J. 1998, 12, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Folini, M.; Brambilla, C.; Villa, R.; Gandellini, P.; Vignati, S.; Paduano, F.; Daidone, M.G.; Zaffaroni, N. Antisense Oligonucleotide-Mediated Inhibition of HTERT, but Not HTERC, Induces Rapid Cell Growth Decline and Apoptosis in the Absence of Telomere Shortening in Human Prostate Cancer Cells. Eur. J. Cancer 2005, 41, 624–634. [Google Scholar] [CrossRef]
- Massard, C.; Zermati, Y.; Pauleau, A.L.; Larochette, N.; Métivier, D.; Sabatier, L.; Kroemer, G.; Soria, J.C. HTERT: A Novel Endogenous Inhibitor of the Mitochondrial Cell Death Pathway. Oncogene 2006, 25, 4505–4514. [Google Scholar] [CrossRef]
- Del Bufalo, D.; Rizzo, A.; Trisciuoglio, D.; Cardinali, G.; Torrisi, M.R.; Zangemeister-Wittke, U.; Zupi, G.; Biroccio, A. Involvement of HTERT in Apoptosis Induced by Interference with Bcl-2 Expression and Function. Cell Death Differ. 2005, 12, 1429–1438. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Pereira-Smith, O.M. Evidence That High Telomerase Activity May Induce a Senescent-like Growth Arrest in Human Fibroblasts. J. Biol. Chem. 2003, 278, 7692–7698. [Google Scholar] [CrossRef]
- Kovalenko, O.A.; Caron, M.J.; Ulema, P.; Medrano, C.; Thomas, A.P.; Kimura, M.; Bonini, M.G.; Herbig, U.; Santos, J.H. A Mutant Telomerase Defective in Nuclear-Cytoplasmic Shuttling Fails to Immortalize Cells and Is Associated with Mitochondrial Dysfunction. Aging Cell 2010, 9, 203–219. [Google Scholar] [CrossRef]
- Kovalenko, O.A.; Kaplunov, J.; Herbig, U.; detoledo, S.; Azzam, E.I.; Santos, J.H. Expression of (NES-)HTERT in Cancer Cells Delays Cell Cycle Progression and Increases Sensitivity to Genotoxic Stress. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Hande, M.P.; Pervaiz, S. HTERT Overexpression Alleviates Intracellular ROS Production, Improves Mitochondrial Function, and Inhibits ROS-Mediated Apoptosis in Cancer Cells. Cancer Res. 2011, 71, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.; Schmid, B.; Akintola, O.; Saretzki, G. Telomerase Does Not Improve DNA Repair in Mitochondria upon Stress but Increases MnSOD Protein under Serum-Free Conditions. Int. J. Mol. Sci. 2019, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, L.; Dai, G.; Xia, K.; Liu, G.; Song, Q.; Tao, C.; Gao, T.; Guo, W. Telomerase Reverse Transcriptase Promotes Chemoresistance by Suppressing Cisplatin-Dependent Apoptosis in Osteosarcoma Cells. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Hahn, W.C.; Schultze, J.L.; Nadler, L.M. The Telomerase Catalytic Subunit Is a Widely Expressed Tumor-Associated Antigen Recognized by Cytotoxic T Lymphocytes. Immunity 1999, 10, 673–679. [Google Scholar] [CrossRef]
- Hrdličková, R.; Nehyba, J.; Bose, H.R. Alternatively Spliced Telomerase Reverse Transcriptase Variants Lacking Telomerase Activity Stimulate Cell Proliferation. Mol. Cell. Biol. 2012, 32, 4283–4296. [Google Scholar] [CrossRef]
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The Major Reverse Transcriptase-Incompetent Splice Variant of the Human Telomerase Protein Inhibits Telomerase Activity but Protects from Apoptosis. Cancer Res. 2013, 73, 2817–2828. [Google Scholar] [CrossRef]
- Wick, M.; Zubov, D.; Hagen, G. Genomic Organization and Promoter Characterization of the Gene Encoding the Human Telomerase Reverse Transcriptase (HTERT). Gene 1999, 232, 97–106. [Google Scholar] [CrossRef]
- Withers, J.B.; Ashvetiya, T.; Beemon, K.L. Exclusion of Exon 2 Is a Common MRNA Splice Variant of Primate Telomerase Reverse Transcriptases. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Zhu, S.; Rousseau, P.; Lauzon, C.; Gandin, V.; Topisirovic, I.; Autexier, C. Inactive C-Terminal Telomerase Reverse Transcriptase Insertion Splicing Variants Are Dominant-Negative Inhibitors of Telomerase. Biochimie 2014, 101, 93–103. [Google Scholar] [CrossRef]
- Yi, X.; White, D.M.; Aisner, D.L.; Baur, J.A.; Wright, W.E.; Shay, J.W. An Alternate Splicing Variant of the Human Telomerase Catalytic Subunit Inhibits Telomerase Activity. Neoplasia 2000, 2, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, H.; Ohyashiki, K.; Ohyashiki, J.H.; Nagao, K.; Kanamaru, T.; Hirata, H.; Hibi, N.; Tsukada, Y. Expression Profile of a Gamma-Deletion Variant of the Human Telomerase Reverse Transcriptase Gene. Neoplasia 2003, 5, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.A.; Hu, J.-F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Telomerase Activity in Human Development Is Regulated by Human Telomerase Reverse Transcriptase (HTERT) Transcription and by Alternate Splicing of HTERT Transcripts. Cancer Res. 1998, 58. [Google Scholar]
- Zhdanov, D.D.; Vasina, D.A.; Grachev, V.A.; Orlova, E.V.; Orlova, V.S.; Pokrovskaya, M.V.; Alexandrova, S.S.; Sokolov, N.N. Alternative Splicing of Telomerase Catalytic Subunit HTERT Generated by Apoptotic Endonuclease EndoG Induces Human CD4 + T Cell Death. Eur. J. Cell Biol. 2017, 96, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, D.D.; Pokrovsky, V.S.; Orlova, E.V.; Orlova, V.S.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Sokolov, N.N. Intracellular Localization of Apoptotic Endonuclease EndoG and Splice-Variants of Telomerase Catalytic Subunit HTERT. Biochem. (Mosc.) 2017, 82, 894–905. [Google Scholar] [CrossRef]
- Vařecha, M.; Potěšilová, M.; Matula, P.; Kozubek, M. Endonuclease G Interacts with Histone H2B and DNA Topoisomerase II Alpha during Apoptosis. Mol. Cell. Biochem. 2012, 363, 301–307. [Google Scholar] [CrossRef]
- Lee, J.S.; Seo, T.W.; Yi, J.H.; Shin, K.S.; Yoo, S.J. CHIP Has a Protective Role against Oxidative Stress-Induced Cell Death through Specific Regulation of Endonuclease G. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Lee, J.H.; Khadka, P.; Baek, S.H.; Chung, I.K. CHIP Promotes Human Telomerase Reverse Transcriptase Degradation and Negatively Regulates Telomerase Activity. J. Biol. Chem. 2010, 285, 42033–42045. [Google Scholar] [CrossRef]
- Zhdanov, D.D.; Novachly, N.S.; Pokrovskaya, M.V.; Aleksandrova, S.S.; Kabardokov, T.A.; Sokolov, N.N. Identification of Genes Whose MRNAs Are Subjected to Alternative Splicing by Endonuclease EndoG Action in Human and Murine CD4+ T Lymphocytes. Biomed. Chem. Res. Methods 2020, 3, e00128. [Google Scholar] [CrossRef]
- Ludlow, A.T.; Wong, M.S.; Robin, J.D.; Batten, K.; Yuan, L.; Lai, T.P.; Dahlson, N.; Zhang, L.; Mender, I.; Tedone, E.; et al. NOVA1 Regulates HTERT Splicing and Cell Growth in Non-Small Cell Lung Cancer. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Li, S.; Crothers, J.; Haqq, C.M.; Blackburn, E.H. Cellular and Gene Expression Responses Involved in the Rapid Growth Inhibition of Human Cancer Cells by RNA Interference-Mediated Depletion of Telomerase RNA. J. Biol. Chem. 2005, 280, 23709–23717. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.K.; Varshney, A.; Sharma, A.; Das, B.C.; Yadava, P.K. Expression of Targeted Ribozyme against Telomerase RNA Causes Altered Expression of Several Other Genes in Tumor Cells. Tumour Biol. 2014, 35, 5539–5550. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, S.; Nosrati, M.; Li, S.; Fong, S.; Torabian, S.; Rangel, J.; Moore, D.H.; Federman, S.; LaPosa, R.R.; Baehner, F.L.; et al. Genes and Pathways Downstream of Telomerase in Melanoma Metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 11306–11311. [Google Scholar] [CrossRef]
- Liu, H.; Yang, Y.; Ge, Y.; Liu, J.; Zhao, Y. TERC Promotes Cellular Inflammatory Response Independent of Telomerase. Nucleic Acids Res. 2019, 47, 8084–8095. [Google Scholar] [CrossRef] [PubMed]
- Balakumaran, A.; Mishra, P.J.; Pawelczyk, E.; Yoshizawa, S.; Sworder, B.J.; Cherman, N.; Kuznetsov, S.A.; Bianco, P.; Giri, N.; Savage, S.A.; et al. Bone Marrow Skeletal Stem/Progenitor Cell Defects in Dyskeratosis Congenita and Telomere Biology Disorders. Blood 2015, 125, 793–802. [Google Scholar] [CrossRef]
- Gazzaniga, F.S.; Blackburn, E.H. An Antiapoptotic Role for Telomerase RNA in Human Immune Cells Independent of Telomere Integrity or Telomerase Enzymatic Activity. Blood 2014, 124, 3675–3684. [Google Scholar] [CrossRef]
- Sung, L.Y.; Chang, W.F.; Zhang, Q.; Liu, C.C.; Liou, J.Y.; Chang, C.C.; Ou-Yang, H.; Guo, R.; Fu, H.; Cheng, W.T.K.; et al. Telomere Elongation and Naive Pluripotent Stem Cells Achieved from Telomerase Haplo-Insufficient Cells by Somatic Cell Nuclear Transfer. Cell Rep. 2014, 9, 1603–1609. [Google Scholar] [CrossRef]
- Kedde, M.; Le Sage, C.; Duursma, A.; Zlotorynski, E.; Van Leeuwen, B.; Nijkamp, W.; Beijersbergen, R.; Agami, R. Telomerase-Independent Regulation of ATR by Human Telomerase RNA. J. Biol. Chem. 2006, 281, 40503–40514. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic Maps of Long Noncoding RNA Occupancy Reveal Principles of RNA-Chromatin Interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef]
- Ivanyi-Nagy, R.; Ahmed, S.M.; Peter, S.; Ramani, P.D.; Ong, P.F.; Dreesen, O.; Dröge, P. The RNA Interactome of Human Telomerase RNA Reveals a Coding-Independent Role for a Histone MRNA in Telomere Homeostasis. Elife 2018, 7. [Google Scholar] [CrossRef]
- Trapp, S.; Parcells, M.S.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Kumar, P.M.; Nair, V.K.; Osterrieder, N. A Virus-Encoded Telomerase RNA Promotes Malignant T Cell Lymphomagenesis. J. Exp. Med. 2006, 203, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Kaufer, B.B.; Arndt, S.; Trapp, S.; Osterrieder, N.; Jarosinski, K.W. Herpesvirus Telomerase RNA (VTR) with a Mutated Template Sequence Abrogates Herpesvirus-Induced Lymphomagenesis. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Kheimar, A.; Trimpert, J.; Groenke, N.; Kaufer, B.B. Overexpression of Cellular Telomerase RNA Enhances Virus-Induced Cancer Formation. Oncogene 2019, 38, 1778–1786. [Google Scholar] [CrossRef]
- Kaufer, B.B.; Trapp, S.; Jarosinski, K.W.; Osterrieder, N. Herpesvirus Telomerase RNA(VTR)-Dependent Lymphoma Formation Does Not Require Interaction of VTR with Telomerase Reverse Transcriptase (TERT). PLoS Pathog. 2010, 6, 87–88. [Google Scholar] [CrossRef]
- Le, S.; Sternglanz, R.; Greider, C.W. Identification of Two RNA-Binding Proteins Associated with Human Telomerase RNA. Mol. Biol. Cell 2000, 11, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Kheimar, A.; Kaufer, B.B. Epstein-Barr Virus-Encoded RNAs (EBERs) Complement the Loss of Herpesvirus Telomerase RNA (VTR) in Virus-Induced Tumor Formation. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Tycowski, K.T.; Guo, Y.E.; Lee, N.; Moss, W.N.; Vallery, T.K.; Xie, M.; Steitz, J.A. Viral Noncoding RNAs: More Surprises. Genes Dev. 2015, 29, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Lees-Miller, S.P.; Meek, K. Repair of DNA Double Strand Breaks by Non-Homologous End Joining. Biochimie 2003, 85, 1161–1173. [Google Scholar] [CrossRef]
- Stellwagen, A.E.; Haimberger, Z.W.; Veatch, J.R.; Gottschling, D.E. Ku Interacts with Telomerase RNA to Promote Telomere Addition at Native and Broken Chromosome Ends. Genes Dev. 2003, 17, 2384–2395. [Google Scholar] [CrossRef]
- Ting, N.S.Y.; Yu, Y.; Pohorelic, B.; Lees-Miller, S.P.; Beattie, T.L. Human Ku70/80 Interacts Directly with HTR, the RNA Component of Human Telomerase. Nucleic Acids Res. 2005, 33, 2090–2098. [Google Scholar] [CrossRef]
- Pfingsten, J.S.; Goodrich, K.J.; Taabazuing, C.; Ouenzar, F.; Chartrand, P.; Cech, T.R. Mutually Exclusive Binding of Telomerase RNA and DNA by Ku Alters Telomerase Recruitment Model. Cell 2012, 148, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Anisenko, A.N.; Knyazhanskaya, E.S.; Zatsepin, T.S.; Gottikh, M.B. Human Ku70 Protein Binds Hairpin RNA and Double Stranded DNA through Two Different Sites. Biochimie 2017, 132, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Ford, L.P.; Lenertz, L.; Wright, W.E.; Shay, J.W. Human Ku70/80 Associates Physically with Telomerase through Interaction with HTERT. J. Biol. Chem. 2002, 277, 47242–47247. [Google Scholar] [CrossRef]
- Tomlinson, R.L.; Abreu, E.B.; Ziegler, T.; Ly, H.; Counter, C.M.; Terns, R.M.; Terns, M.P. Telomerase Reverse Transcriptase Is Required for the Localization of Telomerase RNA to Cajal Bodies and Telomeres in Human Cancer Cells. Mol. Biol. Cell 2008, 19, 3793–3800. [Google Scholar] [CrossRef] [PubMed]
- Fiset, S.; Chabot, B. HnRNP A1 May Interact Simultaneously with Telomeric DNA and the Human Telomerase RNA in Vitro. Nucleic Acids Res. 2001, 29, 2268–2275. [Google Scholar] [CrossRef]
- Ting, N.S.Y.; Pohorelic, B.; Yu, Y.; Lees-Miller, S.P.; Beattie, T.L. The Human Telomerase RNA Component, HTR, Activates the DNA-Dependent Protein Kinase to Phosphorylate Heterogeneous Nuclear Ribonucleoprotein A1. Nucleic Acids Res. 2009, 37, 6105–6115. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Lin, Y.F.; Xu, K.; Lee, K.J.; Wang, D.; Chen, B.P.C. DNA-PKcs Phosphorylates HnRNP-A1 to Facilitate the RPA-to-POT1 Switch and Telomere Capping after Replication. Nucleic Acids Res. 2015, 43, 5971–5983. [Google Scholar] [CrossRef]
- Raghunandan, M.; Geelen, D.; Majerova, E.; Decottignies, A. NHP2 Downregulation Counteracts HTR-Mediated Activation of the DNA Damage Response at ALT Telomeres. EMBO J. 2021, 40. [Google Scholar] [CrossRef]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative Lengthening of Telomeres Renders Cancer Cells Hypersensitive to ATR Inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Vulliamy, T.; Beswick, R.; Kirwan, M.; Marrone, A.; Digweed, M.; Walne, A.; Dokal, I. Mutations in the Telomerase Component NHP2 Cause the Premature Ageing Syndrome Dyskeratosis Congenita. Proc. Natl. Acad. Sci. USA 2008, 105, 8073–8078. [Google Scholar] [CrossRef]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous Occurrence of Telomeric DNA Damage Response in the Absence of Chromosome Fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Amato, R.; Valenzuela, M.; Berardinelli, F.; Salvati, E.; Maresca, C.; Leone, S.; Antoccia, A.; Sgura, A. G-Quadruplex Stabilization Fuels the ALT Pathway in ALT-Positive Osteosarcoma Cells. Genes 2020, 11, 304. [Google Scholar] [CrossRef]
- Chang, D.D.; Clayton, D.A. Mouse RNAase MRP RNA Is Encoded by a Nuclear Gene and Contains a Decamer Sequence Complementary to a Conserved Region of Mitochondrial RNA Substrate. Cell 1989, 56, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Alfonzo, J.D.; Söll, D. Mitochondrial TRNA Import--the Challenge to Understand Has Just Begun. Biol. Chem. 2009, 390, 717–722. [Google Scholar] [CrossRef]
- Wang, G.; Chen, H.W.; Oktay, Y.; Zhang, J.; Allen, E.L.; Smith, G.M.; Fan, K.C.; Hong, J.S.; French, S.W.; McCaffery, J.M.; et al. PNPASE Regulates RNA Import into Mitochondria. Cell 2010, 142, 456–467. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The Human Mitochondrial Transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA Directly Enhances Mitochondrial Translation during Muscle Differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef]
- Wang, G.; Shimada, E.; Koehler, C.M.; Teitell, M.A. PNPASE and RNA Trafficking into Mitochondria. Biochim. Biophys. Acta 2012, 1819, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, P.; Zheng, Q.; Gao, G.; Yuan, J.; Wang, P.; Huang, J.; Xie, L.; Lu, X.; Tong, T.; et al. Mitochondrial Trafficking and Processing of Telomerase RNA TERC. Cell Rep. 2018, 24, 2589–2595. [Google Scholar] [CrossRef]
- Denesyuk, N.A.; Thirumalai, D. Crowding Promotes the Switch from Hairpin to Pseudoknot Conformation in Human Telomerase RNA. J. Am. Chem. Soc. 2011, 133, 11858–11861. [Google Scholar] [CrossRef]
- Liu, P.; Huang, J.; Zheng, Q.; Xie, L.; Lu, X.; Jin, J.; Wang, G. Mammalian Mitochondrial RNAs Are Degraded in the Mitochondrial Intermembrane Space by RNASET2. Protein Cell 2017, 8, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Luhtala, N.; Parker, R. T2 Family Ribonucleases: Ancient Enzymes with Diverse Roles. Trends Biochem. Sci. 2010, 35, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Hillwig, M.S.; Contento, A.L.; Meyer, A.; Ebany, D.; Bassham, D.C.; MacIntosha, G.C. RNS2, a Conserved Member of the RNase T2 Family, Is Necessary for Ribosomal RNA Decay in Plants. Proc. Natl. Acad. Sci. USA 2011, 108, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Liu, P.; Gao, G.; Yuan, J.; Wang, P.; Huang, J.; Xie, L.; Lu, X.; Di, F.; Tong, T.; et al. Mitochondrion-Processed TERC Regulates Senescence without Affecting Telomerase Activities. Protein Cell 2019, 10, 631–648. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, C.; Pinto, A.R.; Li, H.; Li, N.; Wang, L.; Simpson, R.; Liu, J.P. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) Induces Cancer Cell Senescence by Interacting with Telomerase RNA Component. Proc. Natl. Acad. Sci. USA 2012, 109, 13308–13313. [Google Scholar] [CrossRef]
- Sawa, A.; Khan, A.A.; Hester, L.D.; Snyder, S.H. Glyceraldehyde-3-Phosphate Dehydrogenase: Nuclear Translocation Participates in Neuronal and Nonneuronal Cell Death. Proc. Natl. Acad. Sci. USA 1997, 94, 11669–11674. [Google Scholar] [CrossRef]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-Nitrosylated GAPDH Initiates Apoptotic Cell Death by Nuclear Translocation Following Siah1 Binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef]
- Sen, N.; Hara, M.R.; Kornberg, M.D.; Cascio, M.B.; Bae, B.I.; Shahani, N.; Thomas, B.; Dawson, T.M.; Dawson, V.L.; Snyder, S.H.; et al. Nitric Oxide-Induced Nuclear GAPDH Activates P300/CBP and Mediates Apoptosis. Nat. Cell Biol. 2008, 10, 866–873. [Google Scholar] [CrossRef]
- Chuang, D.M.; Ishitani, R.; Roses, A.D.; Burke, J.R.; Vance, J.M.; Strittmatter, W.J. A Role for GAPDH in Apoptosis and Neurodegeneration. Nat. Med. 1996, 2, 609–610. [Google Scholar] [CrossRef]
- Nagy, E.; Henics, T.; Eckert, M.; Miseta, A.; Lightowlers, R.N.; Kellermayer, M. Identification of the NAD(+)-Binding Fold of Glyceraldehyde-3-Phosphate Dehydrogenase as a Novel RNA-Binding Domain. Biochem. Biophys. Res. Commun. 2000, 275, 253–260. [Google Scholar] [CrossRef]
- Azam, S.; Jouvet, N.; Jilani, A.; Vongsamphanh, R.; Yang, X.; Yang, S.; Ramotar, D. Human Glyceraldehyde-3-Phosphate Dehydrogenase Plays a Direct Role in Reactivating Oxidized Forms of the DNA Repair Enzyme APE1. J. Biol. Chem. 2008, 283, 30632–30641. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.L.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Fish, L.; Zhang, S.; Yu, J.X.; Culbertson, B.; Zhou, A.Y.; Goga, A.; Goodarzi, H. Cancer Cells Exploit an Orphan RNA to Drive Metastatic Progression. Nat. Med. 2018, 24, 1743–1751. [Google Scholar] [CrossRef]
- Laudadio, I.; Orso, F.; Azzalin, G.; Calabrò, C.; Berardinelli, F.; Coluzzi, E.; Gioiosa, S.; Taverna, D.; Sgura, A.; Carissimi, C.; et al. AGO2 Promotes Telomerase Activity and Interaction between the Telomerase Components TERT and TERC. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Laudadio, I.; Carissimi, C.; Fulci, V. How RNAi Machinery Enters the World of Telomerase. Cell Cycle 2019, 18, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Rubtsova, M.; Naraykina, Y.; Vasilkova, D.; Meerson, M.; Zvereva, M.; Prassolov, V.; Lazarev, V.; Manuvera, V.; Kovalchuk, S.; Anikanov, N.; et al. Protein Encoded in Human Telomerase RNA Is Involved in Cell Protective Pathways. Nucleic Acids Res. 2018, 46, 8966–8977. [Google Scholar] [CrossRef]
- Hartford, C.C.R.; Lal, A. When Long Noncoding Becomes Protein Coding. Mol. Cell. Biol. 2020, 40. [Google Scholar] [CrossRef]
- Tseng, C.K.; Wang, H.F.; Burns, A.M.M.; Schroeder, M.R.R.; Gaspari, M.; Baumann, P. Human Telomerase RNA Processing and Quality Control. Cell Rep. 2015, 13, 2232–2243. [Google Scholar] [CrossRef]
- Mudge, J.M.; Jungreis, I.; Hunt, T.; Gonzalez, J.M.; Wright, J.C.; Kay, M.; Davidson, C.; Fitzgerald, S.; Seal, R.; Tweedie, S.; et al. Discovery of High-Confidence Human Protein-Coding Genes and Exons by Whole-Genome PhyloCSF Helps Elucidate 118 GWAS Loci. Genome Res. 2019, 29, 2073–2087. [Google Scholar] [CrossRef]
- van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.L.; et al. The Translational Landscape of the Human Heart. Cell 2019, 178, 242–260.e29. [Google Scholar] [CrossRef]
- Ruiz-Orera, J.; Albà, M.M. Conserved Regions in Long Non-Coding RNAs Contain Abundant Translation and Protein-RNA Interaction Signatures. NAR genomics Bioinforma. 2019, 1. [Google Scholar] [CrossRef]
- Nguyen, D.; Grenier St-Sauveur, V.; Bergeron, D.; Dupuis-Sandoval, F.; Scott, M.S.S.; Bachand, F. A Polyadenylation-Dependent 3’ End Maturation Pathway Is Required for the Synthesis of the Human Telomerase RNA. Cell Rep. 2015, 13, 2244–2257. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Stern-Ginossar, N.; Harris, M.S.; Talhouarne, G.J.S.; Jackson, S.E.; Wills, M.R.; Weissman, J.S. Ribosome Profiling Reveals Pervasive Translation Outside of Annotated Protein-Coding Genes. Cell Rep. 2014, 8, 1365–1379. [Google Scholar] [CrossRef] [PubMed]
- Brenner, K.A. A Noncanonical Function of the Telomerase RNA Component in Human Embryonic Stem Cells. Arts Sci. Electron. Theses Diss. 2019. [Google Scholar] [CrossRef]
- Feng, J.; Funk, W.D.; Wang, S.S.; Weinrich, S.L.; Avilion, A.A.; Chiu, C.P.; Adams, R.R.; Chang, E.; Allsopp, R.C.; Yu, J.; et al. The RNA Component of Human Telomerase. Science 1995, 269, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Hoare, S.F.; Bryce, L.A.; Bea, G.; Wisman, A.; Burns, S.; Going, J.J.; Van Der Zee, A.G.J.; Keith, W.N. Lack of Telomerase RNA Gene HTERC Expression in Alternative Lengthening of Telomeres Cells Is Associated with Methylation of the HTERC Promoter 1. CANCER Res. 2001, 61, 27–32. [Google Scholar]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative Lengthening of Telomeres in Mammalian Cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef]
- Herbig, U.; Jobling, W.A.; Chen, B.P.C.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, P53, and P21(CIP1), but Not P16(INK4a). Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Voss, M.; Kaiser, S.; Kapp, U.; Waller, C.F.; Martens, U.M. Lack of Telomerase Activity in Human Mesenchymal Stem Cells. Leukemia 2003, 17, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Baena-Del Valle, J.A.; Zheng, Q.; Esopi, D.M.; Rubenstein, M.; Hubbard, G.K.; Moncaliano, M.C.; Hruszkewycz, A.; Vaghasia, A.; Yegnasubramanian, S.; Wheelan, S.J.; et al. MYC Drives Overexpression of Telomerase RNA (HTR/TERC) in Prostate Cancer. J. Pathol. 2018, 244, 11–24. [Google Scholar] [CrossRef]
- Kang, S.S.; Kwon, T.; Kwon, D.Y.; Do, S. Il Akt Protein Kinase Enhances Human Telomerase Activity through Phosphorylation of Telomerase Reverse Transcriptase Subunit. J. Biol. Chem. 1999, 274, 13085–13090. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ge, Y.; Lin, K.; Liu, Q.; Zhou, H.; Hu, Q.; Zhao, Y.; He, W.; Ju, Z. Telomerase RNA TERC and the PI3K-AKT Pathway Form a Positive Feedback Loop to Regulate Cell Proliferation Independent of Telomerase Activity. Nucleic Acids Res. 2022, 50, 3764–3776. [Google Scholar] [CrossRef] [PubMed]
- Cerezo, A.; Kalthoff, H.; Schuermann, M.; Schäfer, B.; Boukamp, P. Dual Regulation of Telomerase Activity through C-Myc-Dependent Inhibition and Alternative Splicing of HTERT. J. Cell Sci. 2002, 115, 1305–1312. [Google Scholar] [CrossRef]
- Jo, D.; Park, R.; Kim, H.; Jang, M.; Lee, E.J.; Jang, I.S.; Park, J. AMP-Activated Protein Kinase Regulates the Expression of Human Telomerase Reverse Transcriptase. PLoS One 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Karnewar, S.; Neeli, P.K.; Panuganti, D.; Kotagiri, S.; Mallappa, S.; Jain, N.; Jerald, M.K.; Kotamraju, S. Metformin Regulates Mitochondrial Biogenesis and Senescence through AMPK Mediated H3K79 Methylation: Relevance in Age-Associated Vascular Dysfunction. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 1115–1128. [Google Scholar] [CrossRef]
- McKinney, A.; Amen, A.; Stevers, N.; Costello, J. CSIG-24. Gabp Links Ampk Signaling to Tert Regulation in a Tert Promoter Mutation Dependent Manner. Neuro. Oncol. 2019, 21, vi49. [Google Scholar] [CrossRef]
- Kwon, H.J.; Rhim, J.H.; Jang, I.S.; Kim, G.E.; Park, S.C.; Yeo, E.J. Activation of AMP-Activated Protein Kinase Stimulates the Nuclear Localization of Glyceraldehyde 3-Phosphate Dehydrogenase in Human Diploid Fibroblasts. Exp. Mol. Med. 2010, 42, 254–269. [Google Scholar] [CrossRef]
- Sundararaj, K.P.; Wood, R.E.; Ponnusamy, S.; Salas, A.M.; Szulc, Z.; Bielawska, A.; Obeid, L.M.; Hannun, Y.A.; Ogretmen, B. Rapid Shortening of Telomere Length in Response to Ceramide Involves the Inhibition of Telomere Binding Activity of Nuclear Glyceraldehyde-3-Phosphate Dehydrogenase. J. Biol. Chem. 2004, 279, 6152–6162. [Google Scholar] [CrossRef]
- Demarse, N.A.; Ponnusamy, S.; Spicer, E.K.; Apohan, E.; Baatz, J.E.; Ogretmen, B.; Davies, C. Direct Binding of Glyceraldehyde 3-Phosphate Dehydrogenase to Telomeric DNA Protects Telomeres against Chemotherapy-Induced Rapid Degradation. J. Mol. Biol. 2009, 394, 789–803. [Google Scholar] [CrossRef]
- Pariona-Llanos, R.; Pavani, R.S.; Reis, M.; Noël, V.; Silber, A.M.; Armelin, H.A.; Cano, M.I.N.; Elias, M.C. Glyceraldehyde 3-Phosphate Dehydrogenase-Telomere Association Correlates with Redox Status in Trypanosoma Cruzi. PLoS One 2015, 10. [Google Scholar] [CrossRef]
- Abdel-Haleem, A.M.; Lewis, N.E.; Jamshidi, N.; Mineta, K.; Gao, X.; Gojobori, T. The Emerging Facets of Non-Cancerous Warburg Effect. Front. Endocrinol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.M.; Kwon, S.; Pak, Y.K.; Seol, H.W.; Choi, Y.M.; Park, D.J.; Park, K.S.; Lee, H.K. Dynamic Changes in Mitochondrial Biogenesis and Antioxidant Enzymes during the Spontaneous Differentiation of Human Embryonic Stem Cells. Biochem. Biophys. Res. Commun. 2006, 348, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Folmes, C.D.L.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic Oxidative Bioenergetics Transitions into Pluripotency-Dependent Glycolysis to Facilitate Nuclear Reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Rodic, S.; Vincent, M.D. Reactive Oxygen Species (ROS) Are a Key Determinant of Cancer’s Metabolic Phenotype. Int. J. cancer 2018, 142, 440–448. [Google Scholar] [CrossRef]
- Ren, Y.; Shen, H.M. Critical Role of AMPK in Redox Regulation under Glucose Starvation. Redox Biol. 2019, 25. [Google Scholar] [CrossRef]
- Fujita, T.; Asano, Y.; Ohtsuka, J.; Takada, Y.; Saito, K.; Ohki, R.; Fujii, H. Identification of Telomere-Associated Molecules by Engineered DNA-Binding Molecule-Mediated Chromatin Immunoprecipitation (EnChIP). Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Déjardin, J.; Kingston, R.E. Purification of Proteins Associated with Specific Genomic Loci. Cell 2009, 136, 175–186. [Google Scholar] [CrossRef]
- Goldfarb, K.C. Noncoding MRP RNA Function Investigated by Genetic Manipulation and Biochemical Analysis. Ph.D. Thesis, University of Colorado at Boulder, Boulder, CO, USA, 2016. [Google Scholar]
- Wu, Y.L.; Dudognon, C.; Nguyen, E.; Hillion, J.; Pendino, F.; Tarkanyi, I.; Aradi, J.; Lanotte, M.; Tong, J.H.; Chen, G.Q.; et al. Immunodetection of Human Telomerase Reverse-Transcriptase (HTERT) Re-Appraised: Nucleolin and Telomerase Cross Paths. J. Cell Sci. 2006, 119, 2797–2806. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | TERT Expression | TERC Silencing | TERC Overexpression | TERC-53 Overexpression |
|---|---|---|---|---|
| Human Fibroblasts | No | No effects [205,206] | ||
| Human Fibroblasts | No | Faster senescence [185] | ||
| Human BMSCs | No [210] | Senescence [146] | ||
| Human CD4 T cells | Yes [147] | Apoptosis [147] | Reduction of DM-induced apoptosis [147] | |
| Human ESCs | Yes [205] | Apoptosis [205] | Reduction of DOX-induced apoptosis [205] | |
| HeLa and HCT116 cancer cells | Yes | Apoptosis [142,143] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udroiu, I.; Marinaccio, J.; Sgura, A. Many Functions of Telomerase Components: Certainties, Doubts, and Inconsistencies. Int. J. Mol. Sci. 2022, 23, 15189. https://doi.org/10.3390/ijms232315189
Udroiu I, Marinaccio J, Sgura A. Many Functions of Telomerase Components: Certainties, Doubts, and Inconsistencies. International Journal of Molecular Sciences. 2022; 23(23):15189. https://doi.org/10.3390/ijms232315189
Chicago/Turabian StyleUdroiu, Ion, Jessica Marinaccio, and Antonella Sgura. 2022. "Many Functions of Telomerase Components: Certainties, Doubts, and Inconsistencies" International Journal of Molecular Sciences 23, no. 23: 15189. https://doi.org/10.3390/ijms232315189
APA StyleUdroiu, I., Marinaccio, J., & Sgura, A. (2022). Many Functions of Telomerase Components: Certainties, Doubts, and Inconsistencies. International Journal of Molecular Sciences, 23(23), 15189. https://doi.org/10.3390/ijms232315189