
Sulfation of Phenolic Acids: Chemoenzymatic vs. Chemical Synthesis

 , , ,
, , ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
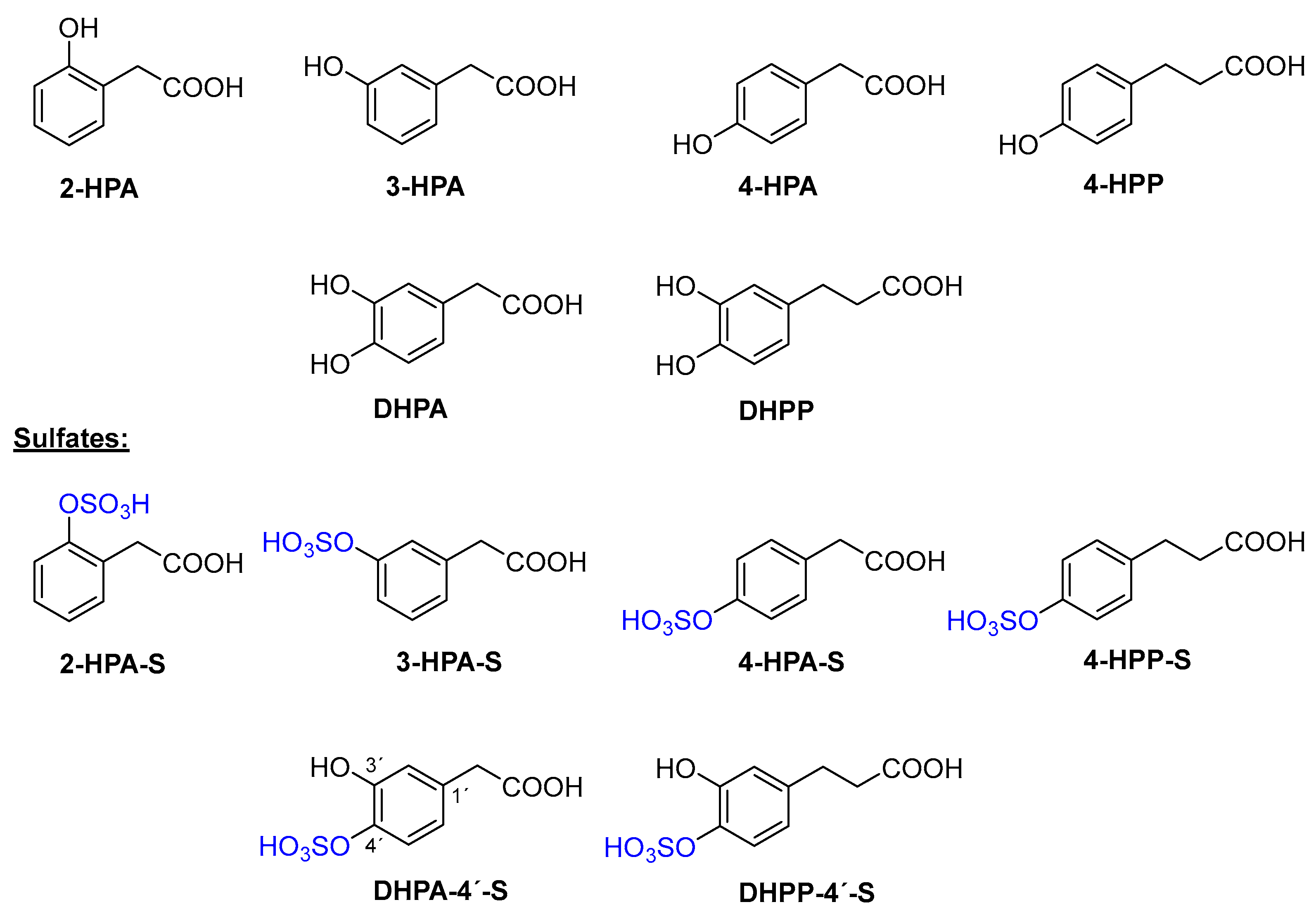
2.1. Chemical Sulfation of Monohydroxyphenolic Acids
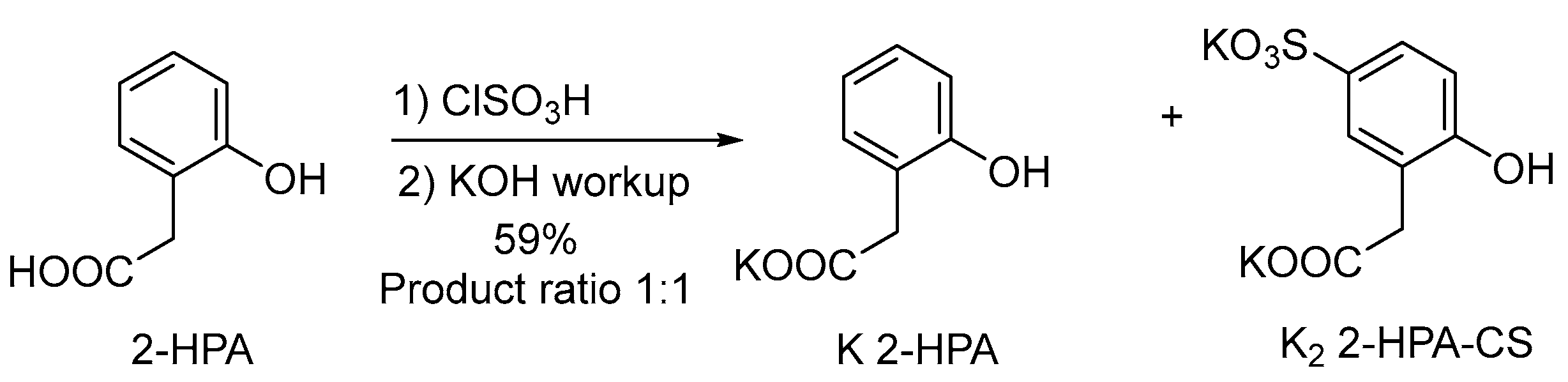
2.1.1. Synthesis of Potassium Salts
2.1.2. Synthesis of Sodium Salts
2.1.3. Other Attempted Chemical Methods
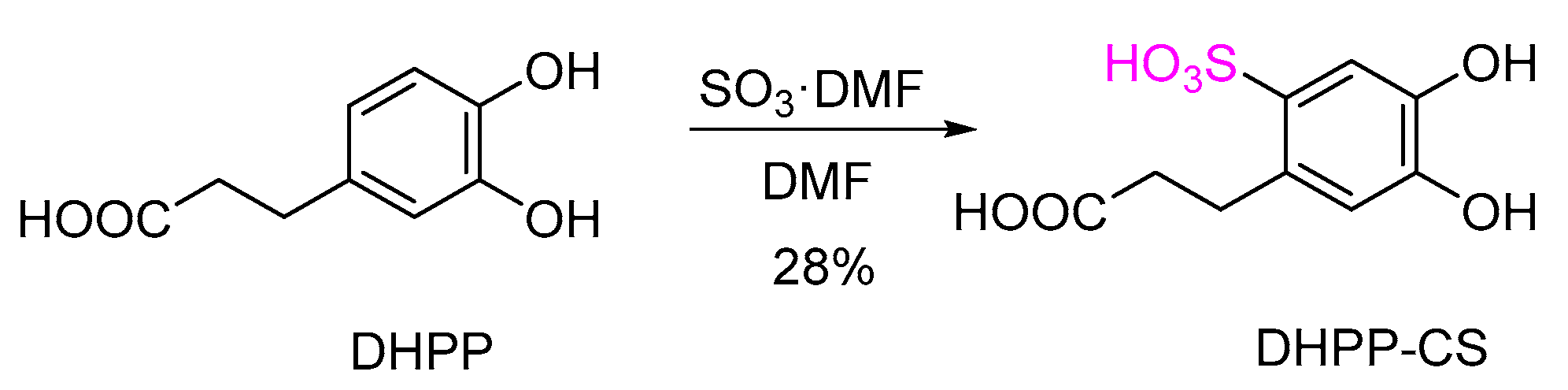
2.2. Chemical Sulfation of Dihydroxyphenolic Acids
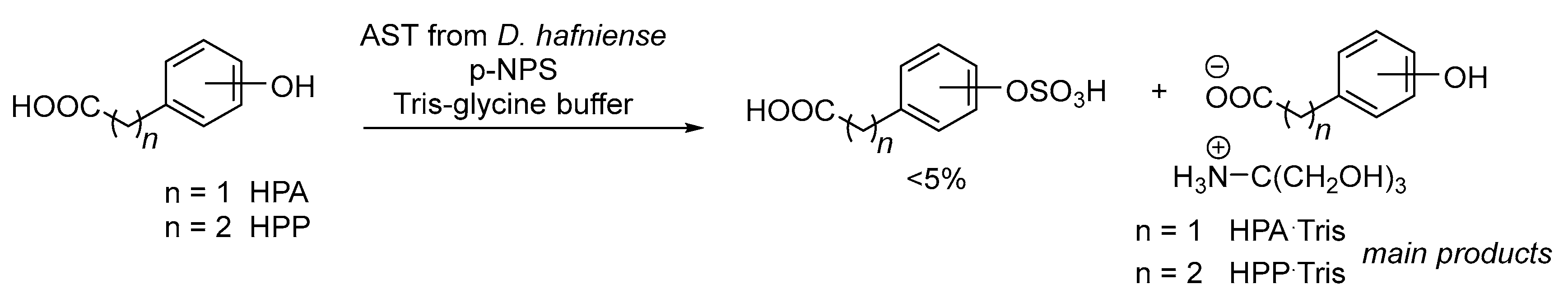
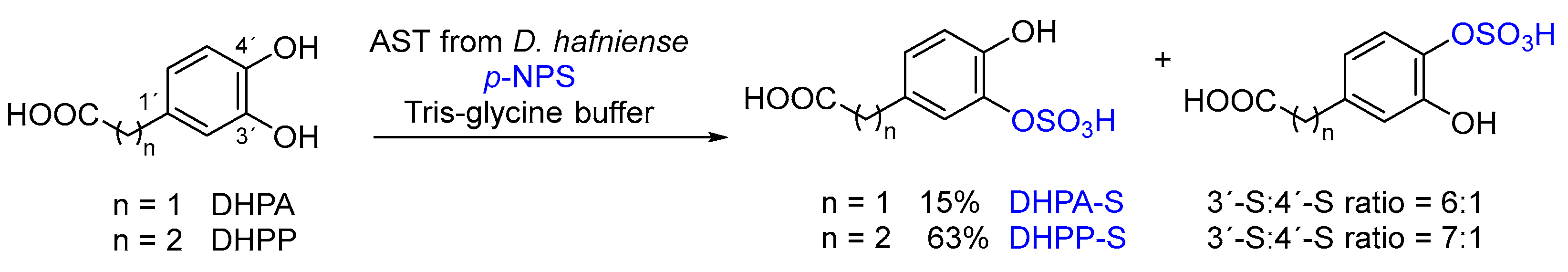
2.3. Enzymatic Sulfation of Phenolic Acids
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. HPLC
3.3. LRMS
3.4. HRMS
3.5. IR
3.6. NMR
3.7. Chemical Synthesis of Monohydroxyphenolic Acid Sulfates
3.7.1. General Procedure A for the Synthesis of Potassium Salts
3.7.2. General Procedure B for the Synthesis of Sodium Salts
3.7.3. Sulfation of 2-HPA with Chlorosulfonic Acid
3.8. Chemical Synthesis of Dihydroxyphenolic Acid Sulfates
3.8.1. General Procedure for the Synthesis of Dihydroxyphenolic Acid Sulfates
3.8.2. Preparation of Sodium Salts of Dihydroxyphenolic Acids
3.8.3. Preparation of Benzenesulfonic Acid DHPP-CS
3.9. Chemoenzymatic Sulfation
3.9.1. Preparation of Aryl Sulfotransferase from Desulfitobacterium Hafniense
3.9.2. Preparation and Purification of Sulfates
3.9.3. Chemical Synthesis of Tris Salts of Phenolic Acids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARE | Antioxidant responsive element |
| DCM | Dichloromethane |
| DEA | Diethylamine |
| DHPA | 3,4-Dihydroxyphenylacetic acid |
| DHPP | 3,4-Dihydroxyphenylpropionic acid |
| 2-HPA | 2-Hydroxyphenylacetic acid |
| 3-HPA | 3-Hydroxyphenylacetic acid |
| 4-HPA | 4-Hydroxyphenylacetic acid |
| 4-HPP | 3-(4-Hydroxyphenyl)propionic acid |
| p-NPS | p-Nitrophenyl sulfate |
| p-NP | p-Nitrophenol |
| Nrf2 | NF-E2-related factor 2 |
| Tris | Tris(hydroxymethyl)aminomethane |
References
- Scalbert, A.; Williamson, G. Dietary intake and bioavailability of polyphenols. J. Nutr. 2000, 130, 2073S–2085S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, A.; O’Reilly, E.J.; Kay, C.; Sampson, L.; Franz, M.; Forman, J.P.; Curhan, G.; Rimm, E.B. Habitual intake of flavonoid subclasses and incident hypertension in adults. Am. J. Clin. Nutr. 2011, 93, 338–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.; Kroon, P.A.; Rimm, E.B.; Cohn, J.S.; Harvey, I.; Le Cornu, K.A.; Ryder, J.J.; Hall, W.L.; Cassidy, A. Flavonoids, flavonoid-rich foods, and cardiovascular risk: A meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2008, 88, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mink, P.J.; Scrafford, C.G.; Barraj, L.M.; Harnack, L.; Hong, C.P.; Nettleton, J.A.; Jacobs, D.R., Jr. Flavonoid intake and cardiovascular disease mortality: A prospective study in postmenopausal women. Am. J. Clin. Nutr. 2007, 85, 895–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullough, M.L.; Peterson, J.J.; Patel, R.; Jacques, P.F.; Shah, R.; Dwyer, J.T. Flavonoid intake and cardiovascular disease mortality in a prospective cohort of US adults. Am. J. Clin. Nutr. 2012, 95, 454–464. [Google Scholar] [CrossRef] [Green Version]
- Milenkovic, D.; Jude, B.; Morand, C. miRNA as molecular target of polyphenols underlying their biological effects. Free Radic. Biol. Med. 2013, 64, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Mladěnka, P.; Zatloukalová, L.; Filipský, T.; Hrdina, R. Cardiovascular effects of flavonoids are not caused only by direct antioxidant activity. Free Radic. Biol. Med. 2010, 49, 963–975. [Google Scholar] [CrossRef]
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014, 66, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Ursini, F. Para-hormesis: An innovative mechanism for the health protection brought by antioxidants in wine. Nutr. Aging 2014, 2, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef]
- Rothwell, J.A.; Urpi-Sarda, M.; Boto-Ordonez, M.; Llorach, R.; Farran-Codina, A.; Barupal, D.K.; Neveu, V.; Manach, C.; Andres-Lacueva, C.; Scalbert, A. Systematic analysis of the polyphenol metabolome using the Phenol-Explorer database. Mol. Nutr. Food Res. 2016, 60, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, A.F.; Borge, G.I.A.; Piskula, M.; Tudose, A.; Tudoreanu, L.; Valentová, K.; Williamson, G.; Santos, C.N. Bioavailability of quercetin in humans with a focus on interindividual variation. Compr. Rev. Food Sci. Food Saf. 2018, 17, 714–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimpao, R.C.; Ventura, M.R.; Ferreira, R.B.; Williamson, G.; Santos, C.N. Phenolic sulfates as new and highly abundant metabolites in human plasma after ingestion of a mixed berry fruit purée. J. Nutr. 2015, 113, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Al-Horani, R.A.; Desai, U.R. Chemical sulfation of small molecules—Advances and challenges. Tetrahedron 2010, 66, 2907–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, A.F.; Santos, C.N.; Ventura, M.R. Synthesis of new sulfated and glucuronated metabolites of dietary phenolic compounds identified in human biological samples. J. Agric. Food Chem. 2017, 65, 6460–6466. [Google Scholar] [CrossRef] [PubMed]
- Gomes, V.P.; Torres, C.; Rodriguez-Borges, J.E.; Paiva-Martins, F. A convenient synthesis of hydroxytyrosol monosulfate metabolites. J. Agric. Food Chem. 2015, 63, 9565–9571. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Zetina, S.M.; Gonzalez-Manzano, S.; Perez-Alonso, J.J.; Gonzalez-Paramas, A.M.; Santos-Buelga, C. Preparation and characterization of protocatechuic acid sulfates. Molecules 2019, 24, 307. [Google Scholar] [CrossRef] [Green Version]
- Duenas, M.; Gonzalez-Manzano, S.; Surco-Laos, F.; Gonzalez-Paramas, A.; Santos-Buelga, C. Characterization of sulfated quercetin and epicatechin metabolites. J. Agric. Food Chem. 2012, 60, 3592–3598. [Google Scholar] [CrossRef]
- Gill, D.M.; Male, L.; Jones, A.M. Sulfation made simple: A strategy for synthesising sulfated molecules. Chem. Commun. 2019, 55, 4319–4322. [Google Scholar] [CrossRef] [Green Version]
- Raghuraman, A.; Riaz, M.; Hindle, M.; Desai, U.R. Rapid and efficient microwave-assisted synthesis of highly sulfated organic scaffolds. Tetrahedron Lett. 2007, 48, 6754–6758. [Google Scholar] [CrossRef]
- Needs, P.W.; Kroon, P.A. Convenient syntheses of metabolically important quercetin glucuronides and sulfates. Tetrahedron 2006, 62, 6862–6868. [Google Scholar] [CrossRef]
- Todd, J.S.; Zimmerman, R.C.; Crews, P.; Alberte, R.S. The antifouling activity of natural and synthetic phenol acid sulphate esters. Phytochemistry 1993, 34, 401–404. [Google Scholar] [CrossRef]
- Soidinsalo, O.; Wähälä, K. Synthesis of daidzein 7-O-β-d-glucuronide-4′-O-sulfate. Steroids 2007, 72, 851–854. [Google Scholar] [CrossRef]
- Liu, Y.; Lien, I.F.; Ruttgaizer, S.; Dove, P.; Taylor, S.D. Synthesis and protection of aryl sulfates using the 2,2,2-trichloroethyl moiety. Org. Lett. 2004, 6, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raheem, K.S.; Botting, N.P.; Slawin, A.M.Z.; Kay, C.D.; O’Hagan, D. Flavonoid metabolism: The synthesis of phenolic glucuronides and sulfates as candidate metabolites for bioactivity studies of dietary flavonoids. Tetrahedron 2012, 68, 4194–4201. [Google Scholar] [CrossRef]
- Taylor, S.D.; Desoky, A. Rapid and efficient chemoselective and multiple sulfations of phenols using sulfuryl imidazolium salts. Tetrahedron Lett. 2011, 52, 3353–3357. [Google Scholar] [CrossRef]
- Liu, C.; Yang, C.; Hwang, S.; Ferraro, S.L.; Flynn, J.P.; Niu, J. A General approach to O-sulfation by a sulfur(vi) fluoride exchange reaction. Angew. Chem. Int. Ed. 2020, 59, 18435–18441. [Google Scholar] [CrossRef]
- Purchartová, K.; Valentová, K.; Pelantová, H.; Marhol, P.; Cvačka, J.; Havlíček, L.; Křenková, A.; Vavříková, E.; Biedermann, D.; Chambers, C.S.; et al. Prokaryotic and eukaryotic aryl sulfotransferases: Sulfation of quercetin and its derivatives. ChemCatChem 2015, 7, 3152–3162. [Google Scholar] [CrossRef]
- Valentová, K.; Káňová, K.; Di Meo, F.; Pelantová, H.; Chambers, C.S.; Rydlová, L.; Petrásková, L.; Křenková, A.; Cvačka, J.; Trouillas, P.; et al. Chemoenzymatic preparation and biophysical properties of sulfated quercetin metabolites. Int. J. Mol. Sci. 2017, 18, 2231. [Google Scholar] [CrossRef]
- Brodsky, K.; Káňová, K.; Konvalinková, D.; Slámová, K.; Pelantová, H.; Valentová, K.; Bojarová, P.; Křen, V.; Petrásková, L. Bacterial aryl sulfotransferases in selective and sustainable sulfation of biologically active compounds using novel sulfate donors. ChemSusChem 2022, 15, e202201253. [Google Scholar] [CrossRef]
- Valentová, K.; Purchartová, K.; Rydlová, L.; Roubalová, L.; Biedermann, D.; Petrásková, L.; Křenková, A.; Pelantová, H.; Holečková-Moravcová, V.; Tesařová, E.; et al. Sulfated metabolites of flavonolignans and 2,3-dehydroflavonolignans: Preparation and properties. Int. J. Mol. Sci. 2018, 19, 2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Káňová, K.; Petrásková, L.; Pelantová, H.; Rybková, Z.; Malachová, K.; Cvačka, J.; Křen, V.; Valentová, K. Sulfated metabolites of luteolin, myricetin, and ampelopsin: Chemoenzymatic preparation and biophysical properties. J. Agric. Food Chem. 2020, 68, 11197–11206. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Ganzera, M.; Karsten, U.; Skhirtladze, A.; Stuppner, H. Phytochemical and analytical characterization of novel sulfated coumarins in the marine green macroalga Dasycladus vermicularis (Scopoli) Krasser. Molecules 2018, 23, 2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshehri, J.A.; Gill, D.M.; Jones, A.M. A Sulfuryl group transfer strategy to selectively prepare sulfated steroids and isotopically labelled derivatives. Front. Mol. Biosci. 2021, 8, 776900. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.M.; Povinelli, A.P.R.; Zazeri, G.; Shamir, S.A.; Mahmoud, A.M.; Wilkinson, F.L.; Alexander, M.Y.; Cornelio, M.L.; Jones, A.M. The modulatory role of sulfated and non-sulfated small molecule heparan sulfate-glycomimetics in endothelial dysfunction: Absolute structural clarification, molecular docking and simulated dynamics, SAR analyses and ADMET studies. RSC Med. Chem. 2021, 12, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, J.A.; Benedetti, A.M.; Jones, A.M. A novel exchange method to access sulfated molecules. Sci. Rep. 2020, 10, 16559. [Google Scholar] [CrossRef] [PubMed]
- Cerfontain, H.; Koeberg-Telder, A. Sulfonation and sulfation on reaction of 1,4-dihydroxybenzene and its methyl ethers in concentrated sulfuric acid. Recl. Trav. Chim. Pays-Bas 2010, 107, 583–591. [Google Scholar] [CrossRef]
- Fumeaux, R.; Menozzi-Smarrito, C.; Stalmach, A.; Munari, C.; Kraehenbuehl, K.; Steiling, H.; Crozier, A.; Williamson, G.; Barron, D. First synthesis, characterization, and evidence for the presence of hydroxycinnamic acid sulfate and glucuronide conjugates in human biological fluids as a result of coffee consumption. Org. Biomol. Chem. 2010, 8, 5199–5211. [Google Scholar] [CrossRef]
- Uutela, P.; Reinilä, R.; Harju, K.; Piepponen, P.; Ketola, R.A.; Kostiainen, R. Analysis of intact glucuronides and sulfates of serotonin, dopamine, and their Phase I metabolites in rat brain microdialysates by liquid chromatography−tandem mass spectrometry. Anal. Chem. 2009, 81, 8417–8425. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starting Acid | Product | Product Abbreviation | Yield [%] | Purity [%] | Side Products |
|---|---|---|---|---|---|
| 2-HPA | No reaction | - | - | - | - |
| 3-HPA | 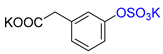 | K2 3-HPA-S | 44 | 88 | 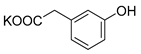 |
| 4-HPA | 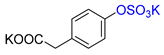 | K2 4-HPA-S | 24 | 94 | 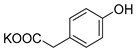 |
| 4-HPP |  | K2 4-HPP-S | 38 | 87 |  |
| Starting Acid | Product | Product Abbreviation | Yield [%] | Purity [%] |
|---|---|---|---|---|
| 2-HPA | No reaction | - | - | - |
| 3-HPA | 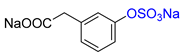 | Na2 3-HPA-S | 23 | >99 |
| 4-HPA | 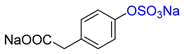 | Na2 4-HPA-S | 53 | >99 |
| 4-HPP | 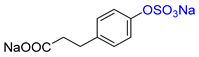 | Na2 4-HPP-S | 16 | >99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolaříková, V.; Brodsky, K.; Petrásková, L.; Pelantová, H.; Cvačka, J.; Havlíček, L.; Křen, V.; Valentová, K. Sulfation of Phenolic Acids: Chemoenzymatic vs. Chemical Synthesis. Int. J. Mol. Sci. 2022, 23, 15171. https://doi.org/10.3390/ijms232315171
Kolaříková V, Brodsky K, Petrásková L, Pelantová H, Cvačka J, Havlíček L, Křen V, Valentová K. Sulfation of Phenolic Acids: Chemoenzymatic vs. Chemical Synthesis. International Journal of Molecular Sciences. 2022; 23(23):15171. https://doi.org/10.3390/ijms232315171
Chicago/Turabian StyleKolaříková, Viola, Katerina Brodsky, Lucie Petrásková, Helena Pelantová, Josef Cvačka, Libor Havlíček, Vladimír Křen, and Kateřina Valentová. 2022. "Sulfation of Phenolic Acids: Chemoenzymatic vs. Chemical Synthesis" International Journal of Molecular Sciences 23, no. 23: 15171. https://doi.org/10.3390/ijms232315171
APA StyleKolaříková, V., Brodsky, K., Petrásková, L., Pelantová, H., Cvačka, J., Havlíček, L., Křen, V., & Valentová, K. (2022). Sulfation of Phenolic Acids: Chemoenzymatic vs. Chemical Synthesis. International Journal of Molecular Sciences, 23(23), 15171. https://doi.org/10.3390/ijms232315171