4-O-Methylascochlorin-Mediated BNIP-3 Expression Controls the Balance of Apoptosis and Autophagy in Cervical Carcinoma Cells

 ,
,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
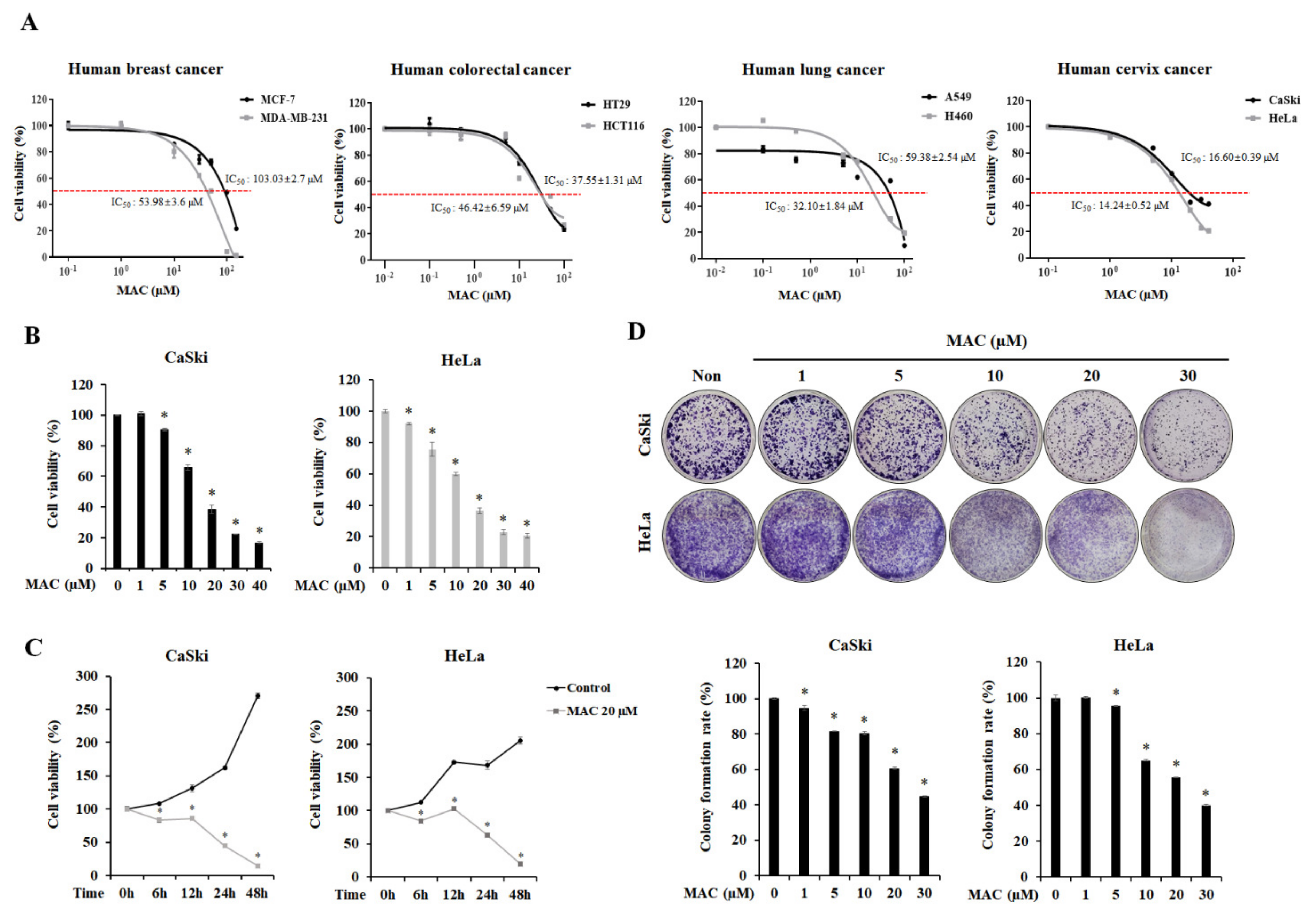
2.1. MAC Is Cytotoxic and Inhibits the Proliferation of Cervical Carcinoma Cells
2.2. MAC Induces Apoptosis of Cervical Carcinoma Cells
2.3. MAC Facilitates Autophagosome-Lysosome Fusion and Induces Autophagy in Cervical Carcinoma Cells
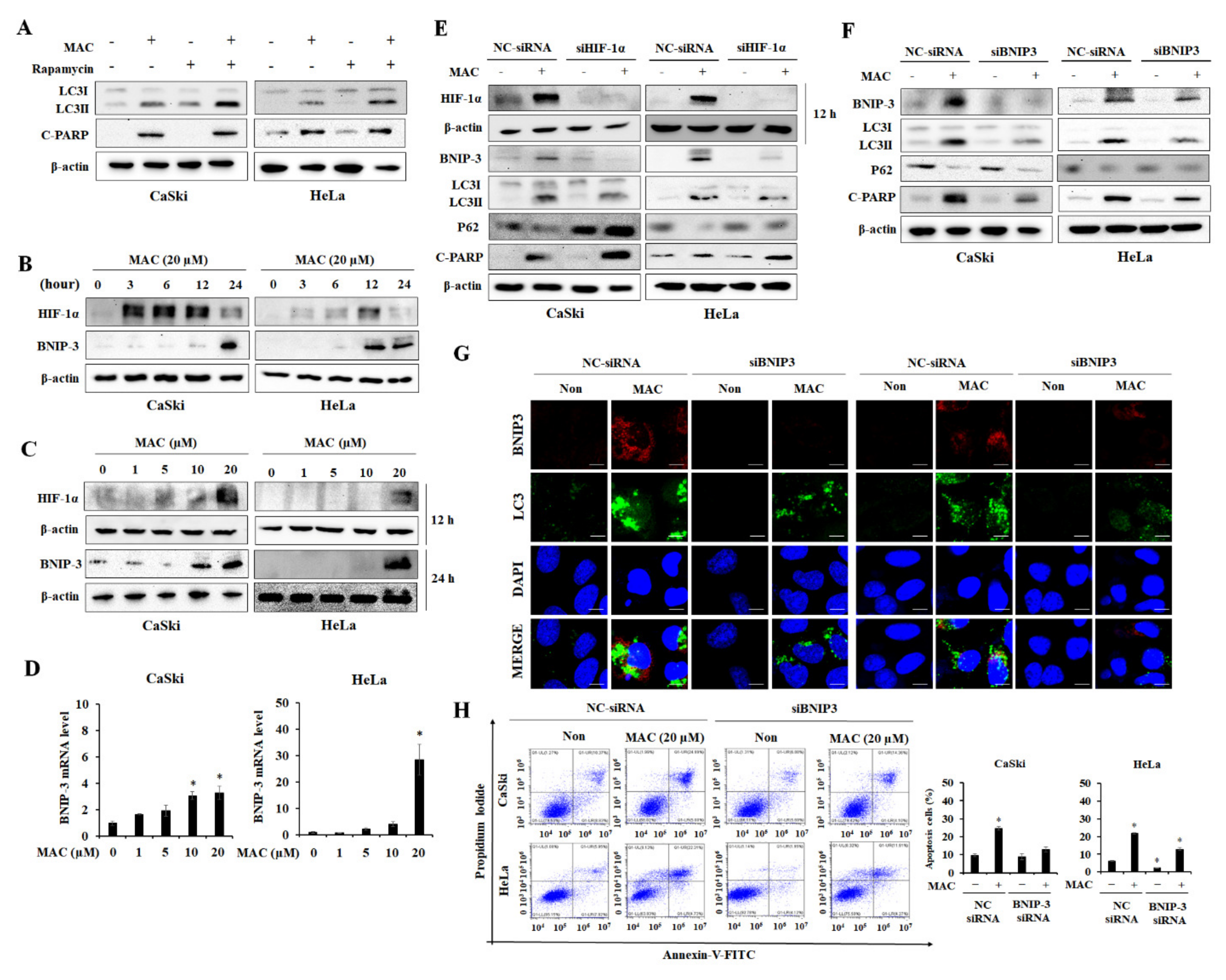
2.4. BNIP-3 Mediates MAC-Induced Autophagy and Apoptosis in Cervical Carcinoma Cells
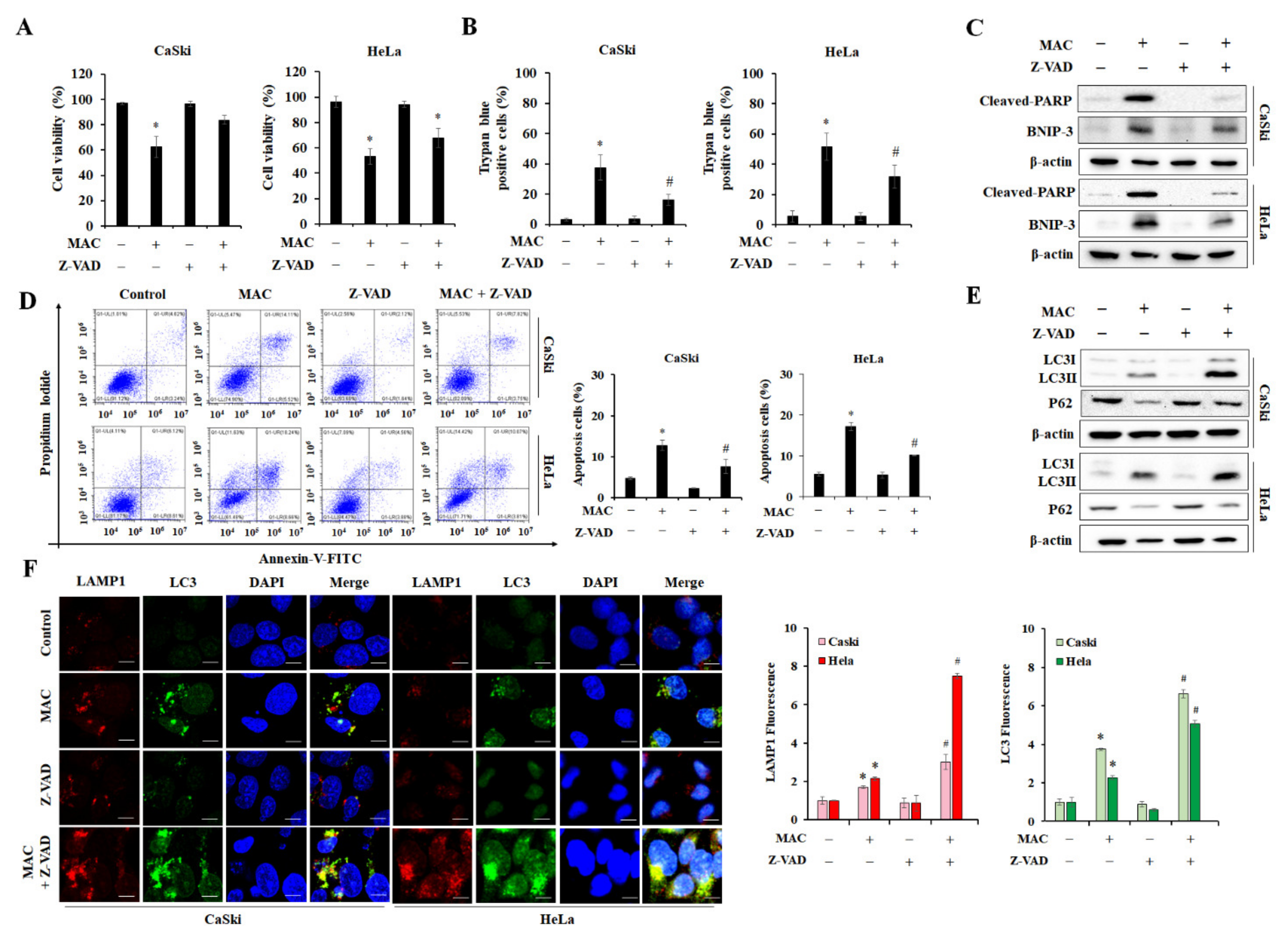
2.5. Inhibition of Apoptosis Enhances MAC-Induced Autophagy in Cervical Carcinoma Cells
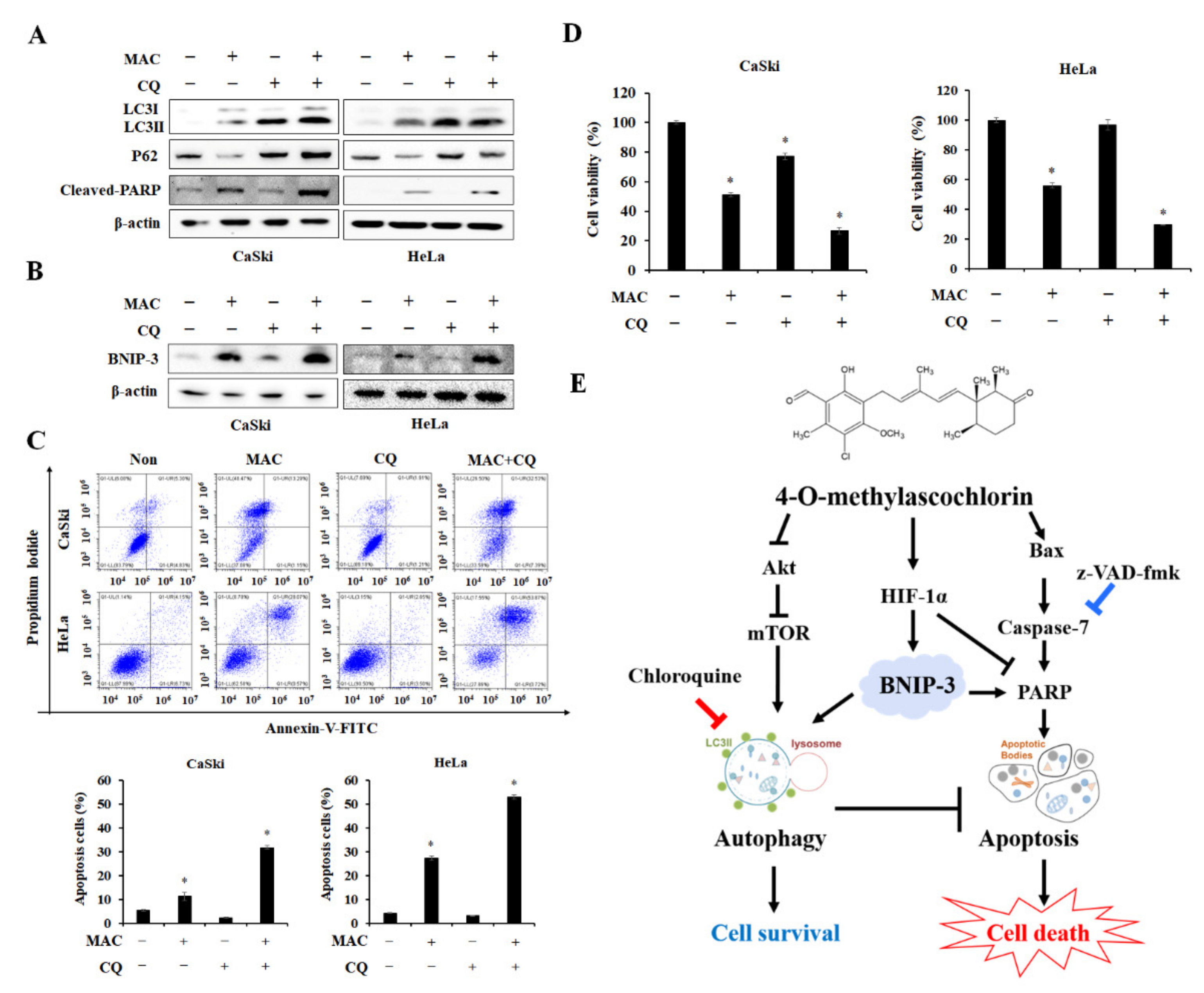
2.6. Inhibition of Autophagosome-Lysosome Fusion Enhances MAC-Induced Apoptosis in Cervical Carcinoma Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Materials
4.2. Cell Viability Assay
4.3. Colony Formation Assay
4.4. Cell Cycle Analysis
4.5. Trypan Blue Staining
4.6. Hoechst 33,342 and PI Double Staining
4.7. Flow Cytometry for Apoptosis
4.8. RT-PCR
4.9. Western Blot Analysis
4.10. Immunofluorescence
4.11. GFP-RFP-LC3 Plasmid Transfection and Autophagy Flux
4.12. siRNA Transfection
4.13. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MAC | 4-O-methylascochlorin |
| Akt | Serine-threonine kinase B |
| mTOR | mammalian target of rapamycin |
| p70S6K | 70-kDa ribosomal protein S6 Kinase |
| BNIP-3 | Bcl-2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| HIF-1α | Hypoxia-inducible factor-1 alpha |
| PARP | Poly (ADP-ribose) polymerase |
| CQ | Chloroquine |
| ATG | Autophagic-related gene |
| AMPK | AMP-activated protein kinase |
| IC50 | Half-maximal-inhibitory concentrations |
| LAMP1 | Lysosomal-associated membrane protein 1 |
| TFEB | Transcription factor EB |
| PI | Propidium iodide |
| DMEM | Dulbecco’s Modified Eagle Medium |
| RPMI | Roswell Park Memorial Institute |
| FBS | Fetal bovine serum |
| PBS | Phosphate-buffered saline |
| FITC | Fluorescein isothiocyanate |
| Trypsin EDTA | Trypsin disodium ethylenediaminetetraacetic acid |
References
- Saleem, A.; Tristram, A.; Fiander, A.; Hibbitts, S. Prophylactic HPV vaccination: A major breakthrough in the fight against cervical cancer? Minerva Med. 2009, 100, 503–523. [Google Scholar] [PubMed]
- Small, W., Jr.; Bacon, M.A.; Bajaj, A.; Chuang, L.T.; Fisher, B.J.; Harkenrider, M.M.; Jhingran, A.; Kitchener, H.C.; Mileshkin, L.R.; Viswanathan, A.N.; et al. Cervical cancer: A global health crisis. Cancer 2017, 123, 2404–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Zhou, H. Data-driven diagnosis of cervical cancer with support vector machine-based approaches. IEEE Access 2017, 5, 25189–25195. [Google Scholar] [CrossRef]
- Dereje, N.; Gebremariam, A.; Addissie, A.; Worku, A.; Assefa, M.; Abraha, A.; Tigeneh, W.; Kantelhardt, E.J.; Jemal, A. Factors associated with advanced stage at diagnosis of cervical cancer in Addis Ababa, Ethiopia: A population-based study. BMJ Open 2020, 10, e040645. [Google Scholar] [CrossRef]
- Zhu, H.; Luo, H.; Zhang, W.; Shen, Z.; Hu, X.; Zhu, X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des. Dev. Ther. 2016, 10, 1885–1895. [Google Scholar] [CrossRef] [Green Version]
- Zaman, M.S.; Chauhan, N.; Yallapu, M.M.; Gara, R.K.; Maher, D.M.; Kumari, S.; Sikander, M.; Khan, S.; Zafar, N.; Jaggi, M.; et al. Curcumin Nanoformulation for Cervical Cancer Treatment. Sci. Rep. 2016, 6, 20051. [Google Scholar] [CrossRef] [Green Version]
- Finocchario-Kessler, S.; Wexler, C.; Maloba, M.; Mabachi, N.; Ndikum-Moffor, F.; Bukusi, E. Cervical cancer prevention and treatment research in Africa: A systematic review from a public health perspective. BMC Womens Health 2016, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Su, M.; Mei, Y.; Sinha, S. Role of the Crosstalk between Autophagy and Apoptosis in Cancer. J. Oncol. 2013, 2013, 102735. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Shimizu, S. Autophagic Cell Death and Cancer Chemotherapeutics. In Innovative Medicine: Basic Research and Development; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015; pp. 219–226. [Google Scholar]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef]
- Bindreither, D.; Ecker, S.; Gschirr, B.; Kofler, A.; Kofler, R.; Rainer, J. The synthetic glucocorticoids prednisolone and dexamethasone regulate the same genes in acute lymphoblastic leukemia cells. BMC Genom. 2014, 15, 662. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, J.T.; Wells, J.A. Direct activation of the apoptosis machinery as a mechanism to target cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 7533–7538. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, A.B. Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr. Cardiol. 2011, 32, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Burton, T.R.; Gibson, S.B. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009, 16, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.H.; Kang, J.H.; Hwang, S.L.; Cho, H.J.; Park, K.K.; Park, Y.Y.; Chung, I.K.; Chang, H.W.; Kim, C.H.; Min, K.S.; et al. 4-O-methylascochlorin, methylated derivative of ascochlorin, stabilizes HIF-1alpha via AMPK activation. Biochem. Biophys. Res. Commun. 2011, 406, 353–358. [Google Scholar] [CrossRef]
- Hong, S.; Park, K.K.; Magae, J.; Ando, K.; Lee, T.S.; Kwon, T.K.; Kwak, J.Y.; Kim, C.H.; Chang, Y.C. Ascochlorin inhibits matrix metalloproteinase-9 expression by suppressing activator protein-1-mediated gene expression through the ERK1/2 signaling pathway: Inhibitory effects of ascochlorin on the invasion of renal carcinoma cells. J. Biol. Chem. 2005, 280, 25202–25209. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.M.; Jeong, Y.J.; Cho, H.J.; Magae, J.; Bae, Y.S.; Chang, Y.C. Suppression of c-Myc induces apoptosis via an AMPK/mTOR-dependent pathway by 4-O-methyl-ascochlorin in leukemia cells. Apoptosis 2016, 21, 657–668. [Google Scholar] [CrossRef]
- Tsuruga, M.; Nakajima, H.; Ozawa, S.; Togashi, M.; Chang, Y.C.; Ando, K.; Magae, J. Characterization of 4-O-methyl-ascochlorin-induced apoptosis in comparison with typical apoptotic inducers in human leukemia cell lines. Apoptosis 2004, 9, 429–435. [Google Scholar] [CrossRef]
- Seok, J.Y.; Jeong, Y.J.; Hwang, S.K.; Kim, C.H.; Magae, J.; Chang, Y.C. Upregulation of AMPK by 4-O-methylascochlorin promotes autophagy via the HIF-1alpha expression. J. Cell. Mol. Med. 2018, 22, 6345–6356. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Aspects Med. 2006, 27, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Pei, F.; Wang, H.S.; Chen, Z.; Zhang, L. Autophagy regulates odontoblast differentiation by suppressing NF-kappaB activation in an inflammatory environment. Cell Death Dis. 2016, 7, e2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Gao, S.; Wang, T.; Yan, J.; Xu, G.; Li, Y.; Niu, H.; Huang, R.; Wu, S. A novel contribution of spvB to pathogenesis of Salmonella Typhimurium by inhibiting autophagy in host cells. Oncotarget 2016, 7, 8295–8309. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Zhong, W.; Hao, L.; Sun, X.; Zhou, Z. Activation of mTORC1 by Free Fatty Acids Suppresses LAMP2 and Autophagy Function via ER Stress in Alcohol-Related Liver Disease. Cells 2021, 10, 2730. [Google Scholar] [CrossRef]
- Su, F.; Shi, M.; Zhang, J.; Zheng, Q.; Zhang, D.; Zhang, W.; Wang, H.; Li, X. Simvastatin Protects Heart from Pressure Overload Injury by Inhibiting Excessive Autophagy. Int. J. Med. Sci. 2018, 15, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Berry, E.A.; Huang, L.S.; Lee, D.W.; Daldal, F.; Nagai, K.; Minagawa, N. Ascochlorin is a novel, specific inhibitor of the mitochondrial cytochrome bc1 complex. Biochim. Biophys. Acta 2010, 1797, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Park, J.Y.; Abekura, F.; Choi, H.; Kim, H.D.; Magae, J.; Chang, Y.C.; Lee, Y.C.; Kim, C.H. 4-O-methylascochlorin attenuates inflammatory responses induced by lipopolysaccharide in RAW 264.7 macrophages. Int. Immunopharmacol. 2021, 90, 107184. [Google Scholar] [CrossRef]
- Hwang, S.-K.; Jeong, Y.-J.; Shin, J.-M.; Magae, J.; Kim, C.-H.; Chang, Y.-C. MAC inhibits c-Myc and induces autophagy by downregulation of CIP2A in leukemia cells. Mol. Cell. Toxicol. 2018, 14, 417–424. [Google Scholar] [CrossRef]
- Evan, G.I.; Brown, L.; Whyte, M.; Harrington, E. Apoptosis and the cell cycle. Curr. Opin. Cell Biol. 1995, 7, 825–834. [Google Scholar] [CrossRef]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1509–1518. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell. Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Sha, Y.; Rao, L.; Settembre, C.; Ballabio, A.; Eissa, N.T. STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 2017, 36, 2544–2552. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 1206, 67–83. [Google Scholar] [PubMed]
- Kondo, T.; Takeda, K.; Muko, R.; Ito, A.; Chang, Y.C.; Magae, J.; Kataoka, T. 4-O-Methylascochlorin inhibits the prolyl hydroxylation of hypoxia-inducible factor-1alpha, which is attenuated by ascorbate. J. Antibiot. 2019, 72, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, D.; Hu, H.; Zhang, P.; Xie, R.; Cui, W. HIF-1alpha/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2019, 120, 109464. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Hwang, S.K.; Yu, M.H.; Cho, Y.; Song, K.H.; Magae, J.; Jeon, E.J.; Chang, Y.C. 4-O-methylascochlorin-stimulated HIF-1alpha expression induces the epithelial mesenchymal transition and cell survival in breast cancer cells. Toxicol. In Vitro 2022, 81, 105342. [Google Scholar] [CrossRef]
- Chen, B.-C.; Weng, Y.-J.; Shibu, M.A.; Han, C.-K.; Chen, Y.-S.; Shen, C.-Y.; Lin, Y.-M.; Viswanadha, V.P.; Liang, H.-Y.; Huang, C.-Y. Estrogen and/or estrogen receptor α inhibits BNIP3-induced apoptosis and autophagy in H9c2 cardiomyoblast cells. Int. J. Mol. Sci. 2018, 19, 1298. [Google Scholar] [CrossRef] [Green Version]
- Dlugosz, P.J.; Billen, L.P.; Annis, M.G.; Zhu, W.; Zhang, Z.; Lin, J.; Leber, B.; Andrews, D.W. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 2006, 25, 2287–2296. [Google Scholar] [CrossRef] [Green Version]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Yang, X.; Zhao, Q.; Ying, F.; Cai, E.; Sun, S.; He, X. BNIP3 contributes to cisplatin-induced apoptosis in ovarian cancer cells. FEBS Open Bio 2020, 10, 1463–1473. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007, 78, 217–245. [Google Scholar]
- Masud Alam, M.; Kariya, R.; Kawaguchi, A.; Matsuda, K.; Kudo, E.; Okada, S. Inhibition of autophagy by chloroquine induces apoptosis in primary effusion lymphoma in vitro and in vivo through induction of endoplasmic reticulum stress. Apoptosis 2016, 21, 1191–1201. [Google Scholar] [CrossRef]
- Quan, Y.; Lei, H.; Wahafu, W.; Liu, Y.; Ping, H.; Zhang, X. Inhibition of autophagy enhances the anticancer effect of enzalutamide on bladder cancer. Biomed. Pharmacother. 2019, 120, 109490. [Google Scholar] [CrossRef]
- Abbady, A.Q.; Twair, A.; Ali, B.; Murad, H. Characterization of Annexin V Fusion with the Superfolder GFP in Liposomes Binding and Apoptosis Detection. Front. Physiol. 2017, 8, 317. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, Y.; Jeong, Y.-J.; Song, K.-H.; Chung, I.-K.; Magae, J.; Kwon, T.K.; Choi, Y.-H.; Kwak, J.-Y.; Chang, Y.-C. 4-O-Methylascochlorin-Mediated BNIP-3 Expression Controls the Balance of Apoptosis and Autophagy in Cervical Carcinoma Cells. Int. J. Mol. Sci. 2022, 23, 15138. https://doi.org/10.3390/ijms232315138
Cho Y, Jeong Y-J, Song K-H, Chung I-K, Magae J, Kwon TK, Choi Y-H, Kwak J-Y, Chang Y-C. 4-O-Methylascochlorin-Mediated BNIP-3 Expression Controls the Balance of Apoptosis and Autophagy in Cervical Carcinoma Cells. International Journal of Molecular Sciences. 2022; 23(23):15138. https://doi.org/10.3390/ijms232315138
Chicago/Turabian StyleCho, Yuna, Yun-Jeong Jeong, Kwon-Ho Song, Il-Kyung Chung, Junji Magae, Taeg Kyu Kwon, Yung-Hyun Choi, Jong-Young Kwak, and Young-Chae Chang. 2022. "4-O-Methylascochlorin-Mediated BNIP-3 Expression Controls the Balance of Apoptosis and Autophagy in Cervical Carcinoma Cells" International Journal of Molecular Sciences 23, no. 23: 15138. https://doi.org/10.3390/ijms232315138
APA StyleCho, Y., Jeong, Y.-J., Song, K.-H., Chung, I.-K., Magae, J., Kwon, T. K., Choi, Y.-H., Kwak, J.-Y., & Chang, Y.-C. (2022). 4-O-Methylascochlorin-Mediated BNIP-3 Expression Controls the Balance of Apoptosis and Autophagy in Cervical Carcinoma Cells. International Journal of Molecular Sciences, 23(23), 15138. https://doi.org/10.3390/ijms232315138