
Biological Age Predictors: The Status Quo and Future Trends

 , , , ,
, , , ,
Abstract
:1. Introduction
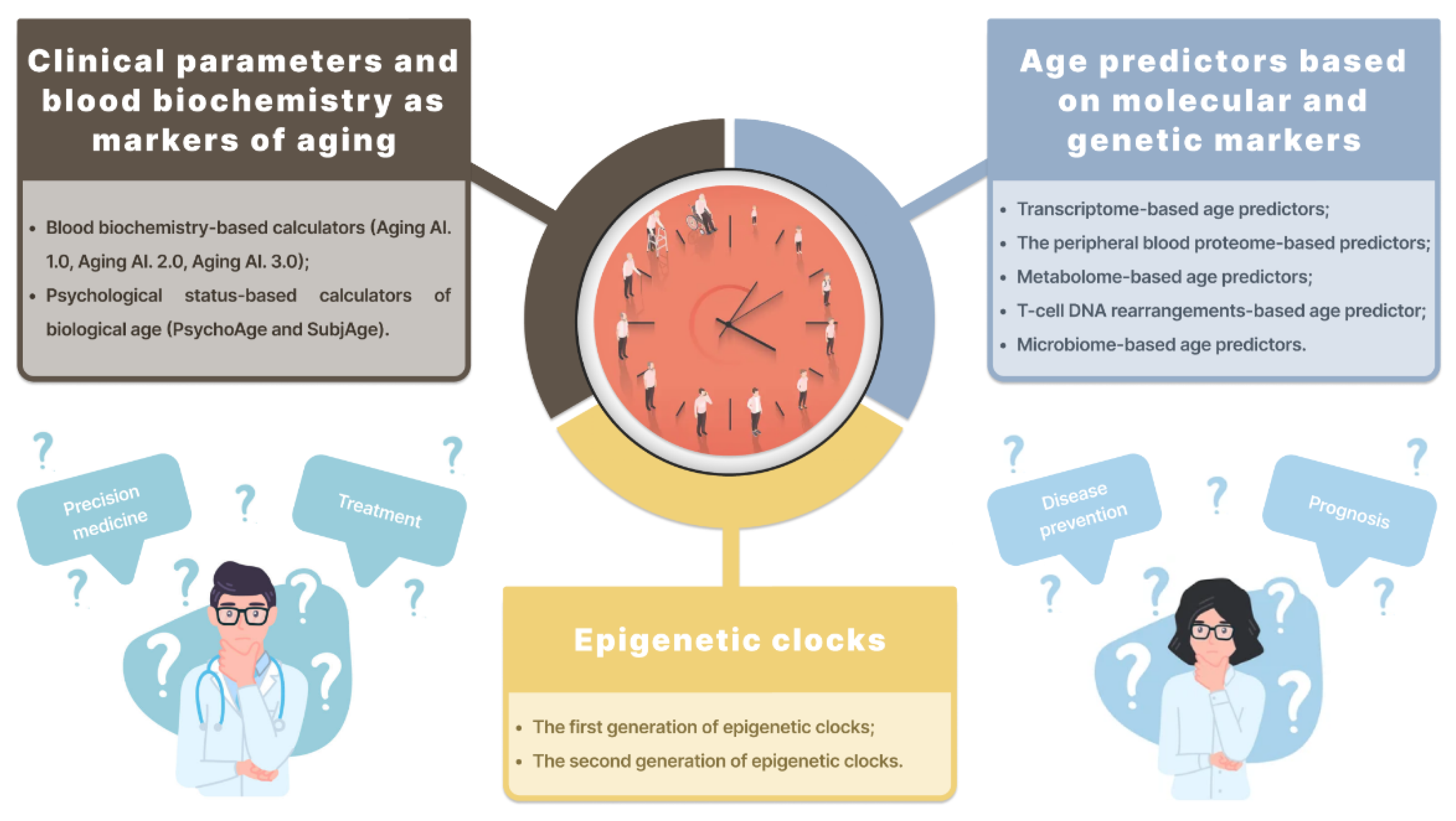
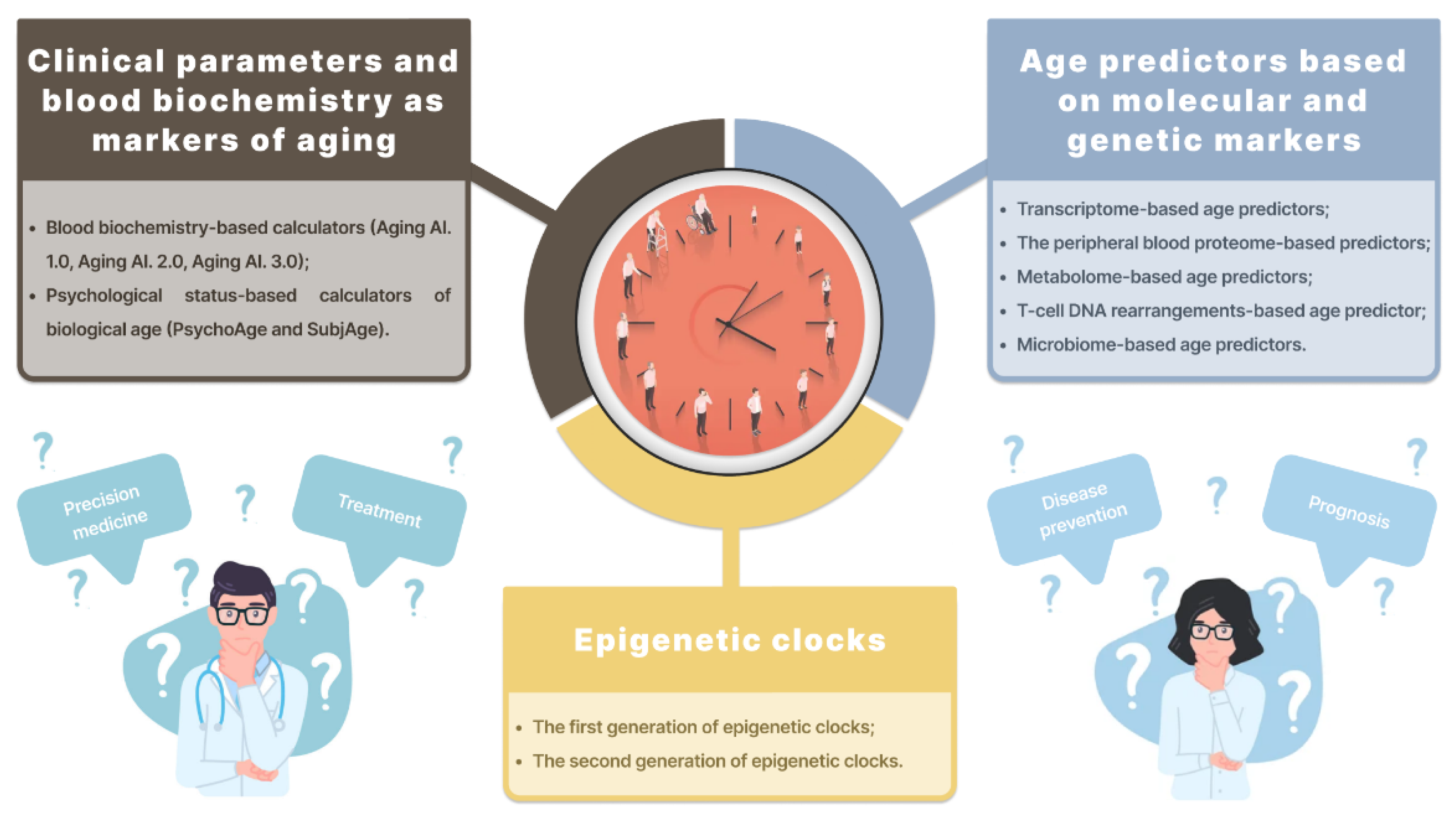
2. Biological Age Predictors
2.1. Clinical Parameters and Blood Biochemistry as Markers of Aging
Blood Biochemistry-Based Calculators

{kind=link}
| Study | Sample | Validation Strategy and Model Characteristics | Model Parameters | Comments |
|---|---|---|---|---|
| Putin E. et al., 2016 [10] | 62,419 samples from the Eastern European population (90% of Russia) |
| Best performance (DNN): 81.5% epsilon-accuracy r = 0.90, R2 = 0.80, MAE = 6.07 years within a 10-year frame. Overall performance (ensemble): 83.5% epsilon-accuracy r = 0.91, R2 = 0.82, MAE = 5.55 years. | 5 most important markers for chronological age estimation identified: albumin, glucose, alkaline phosphatase, urea, and erythrocytes An algorithm developed based on a single source of clinical data. |
| Mamoshina P. et al., 2018 [24] | 20,699 samples for the Canadian population, 65,760 samples for the South Korean population, and 55,920 samples for the Eastern European population | 55,751 samples from the NHANES with blood test values used to measure the predictive power of the models | The best-performing predictor trained on the Eastern European population-specific dataset demonstrated an MAE of 6.25, an R2 of 0.69 Best performing predictors: Canadian population: MAE = 6.36 years, R2 = 0.52 South Korean population: MAE = 5.59, R2 = 0.49 Eastern European population: MAE = 6.25, R2 = 0.69 | Population type identified as a major feature for age estimation in all 3 populations |
| Zhavoronkov A. et al., 2020 [27] | The final dataset contained 6071 participants (U.S. residents) |
| Best performance (DNN): MAE = 6.70 years and epsilon accuracy = 0.78 for PsychoAge; MAE = 7.32 years and epsilon accuracy = 0.74 for SubjAge Model validation (using other datasets): MAE = 7.18/7.73 years and epsilon accuracy = 0.73/0.70 for PsychoAge; MAE = 8.53/8.56 years and epsilon accuracy = 0.66/0.65 for SubjAge | Additionally, variables that remain highly important (top-25) across all age groups were defined. These variables form the psychological aging core. |
2.2. Age Predictors Based on Molecular and Genetic Markers
2.2.1. Transcriptome-Based Age Predictors
2.2.2. Predictors Based on the Peripheral Blood Proteome
2.2.3. Metabolome-Based Age Predictors
2.2.4. Age Predictors Based on T-Cell DNA Rearrangements
2.2.5. Microbiome-Based Age Predictors
| Reference | Methodology | Accuracy Reported | Sample Types | Number of Samples (Overall) | Age Range (Years) | Comments | Associations with Age-Related Conditions and Diseases |
|---|---|---|---|---|---|---|---|
| Fleischer J. et al. [30] | Linear regression10-fold cross-validation | MAE = 7.7 years MAE (median) = 4 years | Human dermal fibroblasts | 133 (people) 10 Hutchinson-Gilford progeria syndrome patients | 1–94 2–9 | Predicts progeria patients as 15–24 years older than age-matched controls; hence, provides an accurate estimation of biological age The only method that predicts accelerated aging in HGPS patients | No associations reported |
| Van den Akker E. et al. [41] | Linear regression 5-Fold-cross-validation |
R2 = 0.65 MAE = 7.3 years | Blood metabolome | 25,000 | - | Only a biological sample required; no additional metadata needed Participants with current metabolic syndrome or diabetes mellitus type 2 were estimated older than healthy counterparts | Cardiometabolic health; increased risk of hospitalization due to heart failure, cognitive decline and cardiovascular and all-cause mortality; in nonagenarians, lower instrumental activities of daily living and increased risk of all-cause mortality during 10 years of follow-up |
| Ren X. et al. [32] | Elastic net | Multiple | Multiple | 9662 | - | Transcriptional age is significantly impacted by race The first model to perform RNA-Seq-based identification of differential gene expression for each individual tissue type | Significant correlation between the transcriptional age acceleration and mutation burden, mortality risk, and cancer stage in several types of cancer; Complementary information to DNA methylation age |
| Meyer D. et al. [33] | Temporal scaling and binarization | R2 = 0.92 MAE = 6.63 years MAD = 5.24 | Blood | 1020 | - | Universal applicability, no methylation analysis required; Improved accuracy for HGPS patients compared with Fleischer’s transcriptome-based model; no DNA methylation in C. elegans, hence the effect of the epigenetic clocks in gene expression is unclear. | No associations reported |
| Wang J. et al. [35] | Multivariate linear regression (MLR) Regression tree (best performing) Bagging regressionRandom forest regression (RFR, best performing) Support vector regression (SVR) | MAE = 8.767 years (S.rho = 0.6983) MAE = 9.126 years (S.rho = 0.660) | Blood | 100 | 19–73 | Significantly smaller prediction MAE values for males than females (MAE = 6.133 years for males and 10.923 years for females in the regression tree model) | No associations reported |
| Peters M. et al. [29] | Meta-analysis | Multiple | Blood | 14,983 (individuals) | - | Lower predictive accuracy compared to epigenetic clocks | Higher systolic and diastolic blood pressure, total cholesterol, HDL cholesterol, fasting glucose levels and body mass index (BMI) |
| Zubakov D. et al. [42] | Linear regression with sjTREC as a single predictor | R2 = 0.835, p = 8.16 × 10−215 Standard error of the estimate ± 8.9 years; | Blood | 195 (individuals) | 0–80 | Storage time analysis showed no statistically significant difference between the sjTREC quantifications in fresh and 1.5-year-old blood samples of the same individuals | No associations reported |
| Galkin F. et al. [43] | Elastic Net (EN) Random Forest (RF) Gradient Boosting (XGB) Deep Neural Networks (DNNs) | MAE = 5.91 years | Stool | 4000 | 18–90 | Accuracy comparable to the existing DNAm solutions (MAE < 5 years) The microbiome composition (such as Akkermansia muciniphila, a marker of obesity, glucose metabolism, and overall intestinal health) could be used in diagnosing gut metabolism disorders; further research needed due to inconsistent results | No associations reported |
| Lahallier B. et al. [40] | SomaScan assay | Multiple | Plasma | 2925 | 18–95 | At peaks 2 and 3 (at the ages of 60 and 78), the proteins were associated with cardiovascular diseases, as well as Alzheimer’s disease and Down syndrome |
2.3. Epigenetic Clocks
| Epigenetic Clocks (Based Solely on Methylation Sites) | |||||||
| Title | Sample | Aged | Biomaterial | Methods | Regression Model | Results | Model Parameters |
| Bocklandt S. et al., 2011 [47] | 128 | 21–55 | Saliva | Microarray analysis: Illumina Human Methylation 27 microarrays Validation: Mass Array (Sequenom) and pyrosequencing | Multivariate regression and leave-one-out analysis | A total of 88 CpGs identified A linear model built based on 2 methylation sites in Edaradd и NPTX2 | MAE for males only, 5.3 y. MAE for females only, 6.2 y. MAE combined, 5.2 y. |
| Hannum J. et al., 2013 [48] | 656 | 19–101 | Whole blood | Illumina Human Methylation 450 BeadChip assay. | Penalized multivariate regression method (Elastic Net) combined with a bootstrap approach | A linear model built based on 71 CpGs and included gender and BMI | Training data: R = 96%, RMSE = 3.9 y. Validation data: R = 91%, RMSE = 4.9 y. |
| Horvath S., 2013 [49] | 7844 non-cancer 5826 cancer | 0–100 | Various human tissues and cell types | Illumina 27 K and Illumina 450 K platforms (for 21,369 CpGs present in both) | Penalized multivariate regression method (Elastic Net) | An aging clock formed by 353 CpGs automatically selected | Training data: R = 0.97, error = 2.9 y. Validation data: R = 0.96, error = 3.6 y. |
| Weidner C. et al., 2014 [50] | 575 | 0–78 | Whole blood | Human Methylation 27 BeadChip platform, Illumina Human Methylation 450 BeadChip assayBisulfite pyrosequencing for the 3 CpGs-based model | Multivariate linear regression | A predictive model developed by training on 102 CpGs and validated on 3 datasets and data from Hannum et al., covering 99 CpGs 3 CpGs selected by the multivariate linear model | Training data: MAD = 3.34 years, RMSE = 4.26 years, R2 = 0.98. Validation data: 3 datasets—MAD = 5.79, 5.52, and 4.02 years, respectively, Hannum et al. dataset—MAD = 4.12 years, RMSE = 5.34 years, R2 = 0.87. 3 CpG model: MAD = 5.4 years, RMSE = 7.2 years; validation, MAD = 4.5 years and RMSE = 5.6 years |
| Horvath S. et al., 2018 [51] | 2222 | 0–92 | Whole and cord blood, skin and buccal epithelium, fibroblasts | Infinium 450 K и EPIC array 850 K | ElasticNet regression | Epigenetic age estimator based on 391 CpGs. | Fibroblasts: R2 = 0.91, err = 2.6; Epithelium: R2 = 0.94, err = 6.3) Buccal cells: R2 = 0.88, err = 2); Keratinocytes: R2 = 0.99, err = 1); Skin: R2 = 0.99, err = 2.9 |
| Han et al., 2020 [59] | 973 40 38 | 1–101 | Whole blood | 450 K Illumina Bead Chip Pyrosequencing Droplet digital PCR Bisulfite barcoded amplicon sequencing | Linear correlation with the logarithm of age A multivariable linear regression model A multivariable model A multivariable linear regression model | 65 CpGs-based model 6 CpGs-based models 7 CpGs-based models 9 CpGs-based models | Training set R2 = 0.95; MAE = 3.0 years; Validation on 3674 samples: R2 = 0.82; MAE = 3.3 years. Validation on 40 samples: R2 = 0.86; several months later median error = 6.8 years. Validation on 40 samples: R2 = 0.89; median error = 2.9 years. The training set (R2 = 0.95; median error = 2.8 years); validation on 39 samples: R2 = 0.87; median error = 2.4 years. |
| Galkin et al. [60] | 6411 | ≈0–100 | Whole blood | Infinium Human Methylation 450 K and 27 K BeadChip platforms | Deep neural network | 1000 CpG-based model | MAE = 3.80 y. MedAE = 2.77 y. R2 = 0.93 |
| Combination Clocks (Epigenetic + Clinical Biomarkers) | |||||||
| Levine M. et al., 2018 [61] | 456 | 21–100 | Whole blood | Illumina 27 K and Illumina 450 K platforms, EPIC array 850 K (20,169 CpGs) | Elastic-net regression | 513 CpGs-based model | Strong associations between DNAm PhenoAge and all-cause mortality, mortality from aging-related diseases, CVD and coronary heart disease (CHD) mortality, cancer incidence and mortality, and Alzheimer’s disease. |
| Lu A. et al., 2019 [62] | 2356 | Whole blood | Illumina 450 K platforms, EPIC array 850 K | Elastic-net regression | 1030 CpGs-based model | Accurate prediction of time-to-death, time-to-cancer, time-to-CVD diseases, time-to-fatty liver, and time-to-menopause | |
3. Clinical Application of Biological Age Predictors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J. Cardiovasc. Dev. Dis. 2019, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Age and Cancer. Cancer Research UK. Available online: https://www.cancerresearchuk.org/about-cancer/causes-of-cancer/age-and-cancer (accessed on 30 August 2022).
- Osman, S.; Ziegler, C.; Gibson, R.; Mahmood, R.; Moraros, J. The Association between Risk Factors and Chronic Obstructive Pulmonary Disease in Canada: A Cross-sectional Study Using the 2014 Canadian Community Health Survey. Int. J. Prev. Med. 2017, 8, 86. [Google Scholar] [PubMed]
- Risk Factors: Who Gets Alzheimer’s Disease? Alzheimer’s Society. Available online: https://www.alzheimers.org.uk/about-dementia/types-dementia/who-gets-alzheimers-disease (accessed on 30 August 2022).
- Molani, S.; Hernandez, P.V.; Roper, R.T.; Duvvuri, V.R.; Baumgartner, A.M.; Goldman, J.D.; Ertekin-Taner, N.; Funk, C.C.; Price, N.D.; Rappaport, N.; et al. Risk factors for severe COVID-19 differ by age for hospitalized adults. Sci. Rep. 2022, 12, 6568. [Google Scholar] [CrossRef] [PubMed]
- Jee, H.; Jeon, B.H.; Kim, Y.H.; Kim, H.-K.; Choe, J.; Park, J.; Jin, Y. Development and application of biological age prediction models with physical fitness and physiological components in Korean adults. Gerontology 2012, 58, 344–353. [Google Scholar]
- Engelfriet, P.M.; Jansen, E.H.J.M.; Picavet, H.S.J.; Dollé, M.E.T. Biochemical markers of aging for longitudinal studies in humans. Epidemiol. Rev. 2013, 35, 132–151. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.-H.; Cameron-Smith, D.; Wessner, B.; Franzke, B. Biomarkers of Aging: From Function to Molecular Biology. Nutrients 2016, 8, 338. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell. 2017, 16, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Putin, E.; Mamoshina, P.; Aliper, A.; Korzinkin, M.; Moskalev, A.; Kolosov, A.; Ostrovskiy, A.; Cantor, C.; Vijg, J.; Zhavoronkov, A. Deep biomarkers of human aging: Application of deep neural networks to biomarker development. Aging 2016, 8, 1021–1033. [Google Scholar] [CrossRef] [Green Version]
- Niedernhofer, L.J.; Kirkland, J.L.; Ladiges, W. Molecular pathology endpoints useful for aging studies. Ageing Res. Rev. 2017, 35, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Dubowitz, N.; Xue, W.; Long, Q.; Ownby, J.G.; Olson, D.E.; Barb, D.; Rhee, M.K.; Mohan, A.V.; Watson-Williams, P.I.; Jackson, S.L.; et al. Aging is associated with increased HbA1c levels, independently of glucose levels and insulin resistance, and also with decreased HbA1c diagnostic specificity. Diabet. Med. 2014, 31, 927–935. [Google Scholar] [CrossRef]
- Chandni, R.; Paul, B.J.; Udayabhaskaran, V.; Ramamoorthy, K.P. A study of non-obese diabetes mellitus in adults in a tertiary care hospital in Kerala, India. Int. J. Diabetes Dev. Ctries 2013, 33, 83–85. [Google Scholar] [CrossRef]
- Weinstein, J.R.; Anderson, S. The aging kidney: Physiological changes. Adv. Chronic Kidney Dis. 2010, 17, 302–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maffei, V.J.; Kim, S.; Blanchard, E., 4th; Luo, M.; Jazwinski, S.M.; Taylor, C.M.; Welsh, D.A. Biological Aging and the Human Gut Microbiota. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1474–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonassi, S.; Coskun, E.; Ceppi, M.; Lando, C.; Bolognesi, C.; Burgaz, S.; Holland, N.; Kirsh-Volders, M.; Knasmueller, S.; Zeiger, E.; et al. The HUman MicroNucleus project on eXfoLiated buccal cells (HUMNXL): The role of life-style, host factors, occupational exposures, health status, and assay protocol. Mutat. Res. Mol. Mech. Mutagen. 2011, 728, 88–97. [Google Scholar] [CrossRef]
- Lulkiewicz, M.; Bajsert, J.; Kopczynski, P.; Barczak, W.; Rubis, B. Telomere length: How the length makes a difference. Mol. Biol. Rep. 2020, 47, 7181–7188. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Noren Hooten, N.; Abdelmohsen, K.; Gorospe, M.; Ejiogu, N.; Zonderman, A.B.; Evans, M.K. microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 2010, 5, e10724. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Brunet, A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012, 22, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Zhavoronkov, A.; Mamoshina, P.; Vanhaelen, Q.; Scheibye-Knudsen, M.; Moskalev, A.; Aliper, A. Artificial intelligence for aging and longevity research: Recent advances and perspectives. Ageing Res. Rev. 2019, 49, 49–66. [Google Scholar] [CrossRef]
- Kang, Y.G.; Suh, E.; Lee, J.-W.; Kim, D.W.; Cho, K.H.; Bae, C.-Y. Biological age as a health index for mortality and major age-related disease incidence in Koreans: National Health Insurance Service-Health screening 11-year follow-up study. Clin. Interv. Aging 2018, 13, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.W.; Yaqub, A.; Ma, Y.; Koudstaal, W.; Hofman, A.; Ikram, M.A.; Ghanbari, M.; Goudsmit, J. Biological age in healthy elderly predicts aging-related diseases including dementia. Sci. Rep. 2021, 11, 15929. [Google Scholar] [CrossRef]
- Mamoshina, P.; Kochetov, K.; Putin, E.; Cortese, F.; Aliper, A.; Lee, W.-S.; Ahn, S.-M.; Uhn, L.; Skjodt, N.; Kovalchuk, O.; et al. Population Specific Biomarkers of Human Aging: A Big Data Study Using South Korean, Canadian, and Eastern European Patient Populations. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Insilico Medicine. Aging.AI. Available online: http://aging.ai/ (accessed on 25 November 2022).
- Cohen, A.A.; Morissette-Thomas, V.; Ferrucci, L.; Fried, L.P. Deep biomarkers of aging are population-dependent. Aging 2016, 8, 2253–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhavoronkov, A.; Kochetov, K.; Diamandis, P.; Mitina, M. PsychoAge and SubjAge: Development of deep markers of psychological and subjective age using artificial intelligence. Aging 2020, 12, 23548–23577. [Google Scholar] [CrossRef] [PubMed]
- Alkass, K.; Buchholz, B.A.; Ohtani, S.; Yamamoto, T.; Druid, H.; Spalding, K.L. Age estimation in forensic sciences: Application of combined aspartic acid racemization and radiocarbon analysis. Mol. Cell. Proteomics. 2010, 9, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K.; et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef] [Green Version]
- Fleischer, J.G.; Schulte, R.; Tsai, H.H.; Tyagi, S.; Ibarra, A.; Shokhirev, M.N.; Huang, L.; Hetzer, M.W.; Navlakha, S. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 2018, 19, 221. [Google Scholar] [CrossRef]
- Glass, D.; Viñuela, A.; Davies, M.N.; Ramasamy, A.; Parts, L.; Knowles, D.; A Brown, A.; Hedman, K.; Small, K.S.; Buil, A.; et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013, 14, R75. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Kuan, P.F. RNAAgeCalc: A multi-tissue transcriptional age calculator. PLoS ONE 2020, 15, e0237006. [Google Scholar] [CrossRef]
- Meyer, D.H.; Schumacher, B. BiT age: A transcriptome based aging clock near the theoretical limit of accuracy. Aging Cell. 2021, 20, e13320. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell. 2016, 15, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, C.; Wei, Y.; Zhao, Y.; Wang, C.; Lu, C.; Feng, J.; Li, S.; Cong, B. Circular RNA as a Potential Biomarker for Forensic Age Prediction. Front. Genet. 2022, 13, 825443. [Google Scholar] [CrossRef] [PubMed]
- Constantin, L. Circular RNAs and Neuronal Development. Adv. Exp. Med. Biol. 2018, 1087, 205–213. [Google Scholar] [PubMed]
- Zhang, Z.; Yang, T.; Xiao, J. Circular RNAs: Promising Biomarkers for Human Diseases. EBioMedicine 2018, 34, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Hanan, M.; Simchovitz, A.; Yayon, N.; Vaknine, S.; Cohen-Fultheim, R.; Karmon, M.; Madrer, N.; Rohrlich, T.M.; Maman, M.; Bennett, E.R.; et al. A Parkinson’s disease CircRNAs Resource reveals a link between circSLC8A1 and oxidative stress. EMBO Mol. Med. 2020, 12, e11942. [Google Scholar] [CrossRef]
- Haque, S.; Ames, R.M.; Moore, K.; Pilling, L.C.; Peters, L.L.; Bandinelli, S.; Ferrucci, L.; Harries, L.W. circRNAs expressed in human peripheral blood are associated with human aging phenotypes, cellular senescence and mouse lifespan. Geroscience 2020, 42, 183–199. [Google Scholar] [CrossRef] [Green Version]
- Lehallier, B.; Gate, D.; Schaum, N.; Nanasi, T.; Lee, S.E.; Yousef, H.; Losada, P.M.; Berdnik, D.; Keller, A.; Verghese, J.; et al. Undulating changes in human plasma proteome profiles across the lifespan are linked to disease. Alzheimers Dement. 2020, 16, e043868. [Google Scholar] [CrossRef]
- van den Akker, E.B.; Trompet, S.; Barkey Wolf, J.J.H.; Beekman, M.; Suchiman, H.E.D.; Deelen, J.; Asselbergs, F.W.; Boersma, E.; Cats, D.; Elders, P.M.; et al. Metabolic Age Based on the BBMRI-NL 1H-NMR Metabolomics Repository as Biomarker of Age-related Disease. Circ. Genom. Precis. Med. 2020, 13, 541–547. [Google Scholar] [CrossRef]
- Zubakov, D.; Liu, F.; van Zelm, M.C.; Vermeulen, J.; Oostra, B.A.; van Duijn, C.M.; Driessen, G.; Van Dongen, J.; Kayser, M.; Langerak, A. Estimating human age from T-cell DNA rearrangements. Curr. Biol. 2010, 20, R970–R971. [Google Scholar] [CrossRef] [Green Version]
- Galkin, F.; Mamoshina, P.; Aliper, A.; Putin, E.; Moskalev, V.; Gladyshev, V.N.; Zhavoronkov, A. Human Gut Microbiome Aging Clock Based on Taxonomic Profiling and Deep Learning. iScience 2020, 23, 101199. [Google Scholar] [CrossRef]
- Huang, S.; Haiminen, N.; Carrieri, A.-P.; Hu, R.; Jiang, L.; Parida, L.; Russell, B.; Allaband, C.; Zarrinpar, A.; Vázquez-Baeza, Y.; et al. Human Skin, Oral, and Gut Microbiomes Predict Chronological Age. mSystems 2020, 5, e00630-19. [Google Scholar] [CrossRef] [PubMed]
- Velegzhaninov, I.; Mezenceva, V.; Shostal, O.; Baranova, A.; Moskalev, A. Age dynamics of DNA damage and CpG methylation in the peripheral blood leukocytes of mice. Mutat. Res. Mol. Mech. Mutagen. 2015, 775, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Demanelis, K.; Jasmine, F.; Chen, L.S.; Chernoff, M.; Tong, L.; Delgado, D.; Zhang, C.; Shinkle, J.; Sabarinathan, M.; Lin, H.; et al. Determinants of telomere length across human tissues. Science 2020, 369, eaaz6876. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sánchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.-H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Lin, Q.; Weidner, C.I.; Costa, I.G.; Marioni, R.E. DNA Methylation Levels at Individual Age-Associated CpG Sites Can Be Indicative for Life Expectancy. In Aging. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/pmc4789590/ (accessed on 30 August 2022).
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.-C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schönfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.-C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Ritchie, S.J.; Muniz-Terrera, G.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, M.E.; Hosgood, H.D.; Chen, B.; Absher, D.; Assimes, T.; Horvath, S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging 2015, 7, 690–700. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging 2015, 7, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Franzen, J.; Stiehl, T.; Gobs, M.; Kuo, C.-C.; Nikolić, M.; Hapala, J.; Koop, B.E.; Strathmann, K.; Ritz-Timme, S.; et al. New targeted approaches for epigenetic age predictions. BMC Biol. 2020, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Galkin, F.; Mamoshina, P.; Kochetov, K.; Sidorenko, D.; Zhavoronkov, A. DeepMAge: A Methylation Aging Clock Developed with Deep Learning. Aging Dis. 2021, 12, 1252–1262. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11, 171. [Google Scholar] [CrossRef]
- Galkin, F.; Parish, A.; Bischof, E.; Zhang, J.; Mamoshina, P.; Zhavoronkov, A. Increased Pace of Aging in COVID-Related Mortality. Life 2021, 11, 730. [Google Scholar] [CrossRef]
- Corley, M.J.; Pang, A.P.S.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, 110, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Ying, K.; Zhai, R.; Pyrkov, T.V.; Shindyapina, A.V.; Mariotti, M.; Fedichev, P.O.; Shen, X.; Gladyshev, V.N. Genetic and phenotypic analysis of the causal relationship between aging and COVID-19. Commun. Med. 2021, 1, 35. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-L.; Pilling, L.C.; Atkins, J.C.; Masoli, J.; Delgado, J.; Tignanelli, C.; Kuchel, G.A.; Melzer, D.; Beckman, K.B.; Levine, M.E. COVID-19 severity is predicted by earlier evidence of accelerated aging. medRxiv 2020. [CrossRef]
- Pang, A.P.S.; Higgins-Chen, A.T.; Comite, F.; Raica, I.; Arboleda, C.; Went, H.; Mendez, T.; Schotsaert, M.; Dwaraka, V.; Smith, R.; et al. Longitudinal Study of DNA Methylation and Epigenetic Clocks Prior to and Following Test-Confirmed COVID-19 and mRNA Vaccination. Front. Genet. 2022, 13, 819749. [Google Scholar] [CrossRef] [PubMed]
- Higgins-Chen, A.T.; Thrush, K.L.; Wang, Y.; Minteer, C.J.; Kuo, P.-L.; Wang, M.; Niimi, P.; Sturm, G.; Lin, J.; Moore, A.Z.; et al. A computational solution for bolstering reliability of epigenetic clocks: Implications for clinical trials and longitudinal tracking. Nat Aging 2022, 2, 644–661. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, W.; Wang, T.; Ran, D.; Davalos, V.; Planas-Serra, L.; Pujol, A.; Esteller, M.; Wang, X.; Yu, H. Accelerated biological aging in COVID-19 patients. Nat. Commun. 2022, 13, 2135. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erema, V.V.; Yakovchik, A.Y.; Kashtanova, D.A.; Bochkaeva, Z.V.; Ivanov, M.V.; Sosin, D.V.; Matkava, L.R.; Yudin, V.S.; Makarov, V.V.; Keskinov, A.A.; et al. Biological Age Predictors: The Status Quo and Future Trends. Int. J. Mol. Sci. 2022, 23, 15103. https://doi.org/10.3390/ijms232315103
Erema VV, Yakovchik AY, Kashtanova DA, Bochkaeva ZV, Ivanov MV, Sosin DV, Matkava LR, Yudin VS, Makarov VV, Keskinov AA, et al. Biological Age Predictors: The Status Quo and Future Trends. International Journal of Molecular Sciences. 2022; 23(23):15103. https://doi.org/10.3390/ijms232315103
Chicago/Turabian StyleErema, Veronika V., Anna Y. Yakovchik, Daria A. Kashtanova, Zanda V. Bochkaeva, Mikhail V. Ivanov, Dmitry V. Sosin, Lorena R. Matkava, Vladimir S. Yudin, Valentin V. Makarov, Anton A. Keskinov, and et al. 2022. "Biological Age Predictors: The Status Quo and Future Trends" International Journal of Molecular Sciences 23, no. 23: 15103. https://doi.org/10.3390/ijms232315103
APA StyleErema, V. V., Yakovchik, A. Y., Kashtanova, D. A., Bochkaeva, Z. V., Ivanov, M. V., Sosin, D. V., Matkava, L. R., Yudin, V. S., Makarov, V. V., Keskinov, A. A., Kraevoy, S. A., & Yudin, S. M. (2022). Biological Age Predictors: The Status Quo and Future Trends. International Journal of Molecular Sciences, 23(23), 15103. https://doi.org/10.3390/ijms232315103