
Human β-Defensin 3 Inhibits Porphyromonas Gingivalis Lipopolysaccharide-Induced Oxidative and Inflammatory Responses of Microglia by Suppression of Cathepsins B and L
Abstract
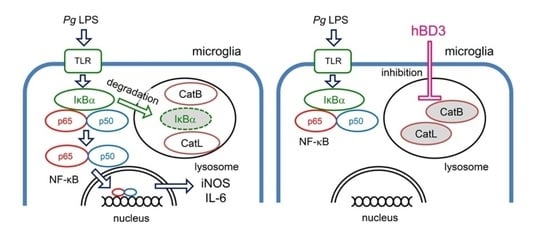
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effects of hBDs on Cell Viability of MG6
2.2. Effects of hBDs on Pg LPS-Induced Nitric Oxide (NO) Production and Inducible NO Synthase (iNOS) Expression by Microglia
2.3. Effects of hBDs on Pg LPS-Induced Interleukin-6 (IL-6) mRNA and Protein by Microglia
2.4. Effects of hBD3 on Pg LPS-Induced Nuclear Translocation of p65 and Proteolytic Degradation of IκBα
2.5. Inhibitory Effects of hBD3 on Activities of Human CatB and Cathepsin L (CatL)
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Assay for Cell Survival
4.4. Assay of NO Production
4.5. qRT-PCR
4.6. Cytokine Antibody Array
4.7. ELISA
4.8. Immunoblotting Analyses
4.9. Enzymatic Activity Assay of CatB and CatL
4.10. Fluorescence Imaging of Enzymatic Activities of CatB and CatL
4.11. Immunostaining
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nakanishi, H.; Nonaka, S.; Wu, Z. Microglial cathepsin B and Porphyromonas gingivalis gingipains as potential therapeutic targets for sporadic Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2020, 19, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, Z.; Nakanishi, Y.; Ni, J.; Hayashi, Y.; Takayama, F.; Zhou, Y.; Kadowaki, T.; Nakanishi, H. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci. Rep. 2017, 7, 11759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, S.; Nakanishi, H. Secreted gingipains from Porphyromonas gingivalis induce microglia migration through endosomal signaling by protease-activated receptor 2. Neurochem. Int. 2020, 140, 104840. [Google Scholar] [CrossRef]
- Nonaka, S.; Kadowaki, T.; Nakanishi, H. Secreted gingipains from Porphyromonas gingivalis increase permeability in human cerebral microvascular endothelial cells through intracellular degradation of tight junction proteins. Neurochem. Int. 2022, 154, 105282. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [Green Version]
- Poole, S.; Singhrao, S.K.; Kesavalu, L.; Curtis, M.A.; Crean, S. Determining the presence of periodontopathic virulence factors in short-term postmorem Alzheimer’s disease brain tissue. J. Alzheimers Dis. 2013, 36, 665–677. [Google Scholar] [CrossRef]
- Ide, M.; Harris, M.; Stevens, A.; Sussams, R.; Hopkins, V.; Culliford, D.; Fuller, J.; Ibbett, P.; Raybould, R.; Thomas, R.; et al. Periodontitis and cognitive decline in Alzheimer’s disease. PLoS ONE 2016, 11, e0151081. [Google Scholar] [CrossRef] [Green Version]
- Holmer, J.; Eriksdotter, M.; Schultzberg, M.; Pussinen, P.J.; Buhlin, K. Association between periodontitis and risk of Alzheimer’s disease, mild cognitive impairment and subjective cognitive decline: A case-control study. J. Clin. Periodontol. 2018, 45, 1287–1298. [Google Scholar] [CrossRef]
- Kanagasingam, S.; Chukkapalli, S.S.; Welbury, R.; Singhrao, S.K. Porphyromonas gingivalis is a strong factor for Alzheimer’s disease. J. Alzheimers Dis. Rep. 2020, 4, 501–511. [Google Scholar] [CrossRef]
- Olsen, I.; Singhrao, S.K. Interaction between genetic factors, Porphyromonas gingivalis and microglia to promote Alzheimer’s disease. J. Oral. Microbiol. 2020, 12, 1820834. [Google Scholar] [CrossRef]
- Seymour, T.; Zhang, J. Porphyromonas gingivalis in the pathogenesis of Alzheimer’s disease and its therapeutic target. J. Explor. Res. Pharmacol. 2022, 7, 45–53. [Google Scholar] [CrossRef]
- Wu, Z.; Ni, J.; Liu, Y.; Teeling, J.L.; Takayama, F.; Collcutt, A.; Ibbett, P.; Nakanishi, H. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav. Immun. 2017, 65, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, C.; Zhang, X.; Chen, H.; Dong, J.; Lu, W.; Song, Z.; Zhou, W. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J. Neuroinflam. 2018, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, H.; Zhang, J.; Zhang, X.; Xia, X.; Qiu, C.; Liao, Y.; Chen, H.; Song, Z.; Zhou, W. Periodontitis induced by P. gingivalis-LPS is associated with neuroinflammation and learning and memory impairment in sprague-dawley rats. Front. Neurosci. 2020, 14, 658. [Google Scholar] [CrossRef] [PubMed]
- Colleran, A.; Ryan, A.; O’Gorman, A.; Mureau, C.; Liptrot, C.; Dockery, P.; Fearnhead, H.; Egan, L.J. Autophagosomal IκBα degradation plays a role in the long term control of tumor necrosis factor-α-induced nuclear factor-κB (NF-κB) activity. J. Biol. Chem. 2011, 286, 22886–22893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criollo, A.; Chereau, F.; Malik, S.A.; Niso-Santano, M.; Mariño, G.; Galluzzi, L.; Maiuri, M.C.; Baud, V.; Kroemer, G. Autophagy is required for the activation of NFκB. Cell Cycle 2012, 11, 194–199. [Google Scholar] [CrossRef]
- Wang, Y.R.; Qin, S.; Han, R.; Wu, J.C.; Liang, Z.Q.; Qin, Z.H.; Wang, Y. Cathepsin L play a role in quinolinic acid-induced NF-κB activation and excitotoxicity in rat striatal neurons. PLoS ONE 2013, 8, e75702. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Wu, Z.; Peterts, C.; Yamamoto, K.; Qing, H.; Nakanishi, H. The critical role of proteolytic relay through cathepsins B and E in the phenotypic change of microglia/ macrophage. J. Neurosci. 2015, 35, 12488–12501. [Google Scholar] [CrossRef]
- Nakanishi, H. Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural. Regen. Res. 2020, 15, 25–29. [Google Scholar] [CrossRef]
- Nakanishi, H. Cathepsin regulation on microglial function. Biochim. Biophys. Acta. Proteins Proteom. 2020, 1868, 140465. [Google Scholar] [CrossRef]
- Kamer, A.R.; Dasanayake, A.P.; Craig, R.G.; Glodzik-Sobanska, L.; Bry, M.; de Leon, M.J. Alzheimer’s disease and peripheral infections: The possible contribution from periodontal infections, model and hypothesis. J. Alzheimers Dis. 2008, 13, 437–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welling, M.M.; Nabuurs, R.J.; van der Weerd, L. Potential role of antimicrobial peptides in the early onset of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.N.; Zhao, J.; Lotoczky, G.; Grever, W.E.; Lyman, W.D. Induction of human β-defensin-2 expression in human astrocytes by lipopolysaccharide and cytokines. J. Neurochem. 2001, 77, 1027–1035. [Google Scholar] [CrossRef]
- Kazakos, E.I.; Kountouras, J.; Polyzos, S.A.; Deretzi, G. Novel aspects of defensins’ involvement in virus-induced autoimmunity in the central nervous system. Med. Hypotheses 2017, 102, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Williams, W.M.; Castellani, R.J.; Weinberg, A.; Perry, G.; Smith, M.K. Do β-defensins and other aminomicrobial peptides play a role in neuroimmune function and neurodegeneration? Sci. World J. 2012, 905785. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, K.; Okamura, N.; Arai, H.; Sekizawa, K.; Sasaki, H. Expression of human β-defensin-1 in the choroid plexus. Ann. Neurol. 1999, 45, 685. [Google Scholar] [CrossRef]
- Williams, W.M.; Torres, S.; Siedlak, S.L.; Castellani, R.J.; Perry, G.; Smith, M.A.; Zhu, X. Antimicrobial peptide β-defensin-1 expression is upregulated in Alzheimer’s brain. J. Neuroinflam. 2013, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szekeres, M.; Ivitz, E.; Datki, Z.; Kálmán, J.; Pákáski, M.; Várhelyi, Z.P.; Klivényi, P.; Zadori, D.; Somogyvári, F.; Szolnoki, Z.; et al. Relevance of defensin β-2 and α defensins (HNP1–3) in Alzheimer’s disease. Psychiatry Res. 2016, 239, 342–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, F.; Webb, S.; Li, H.-N.; Patel, H.B.; Perretti, M.; Jackson, I.J.; Gray, M.; Davidson, D.J.; Dorin, J.R. Human β-defensin 3 has immunosuppressive activity in vivo and in vitro. Eur. J. Immunol. 2010, 40, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Lyu, J.; Li, H.; Lei, L.; Bian, T.; Li, L.; Yan, F. Human β-defensin 3 inhibits periodontitis development by suppressing inflammatory responses in macrophages. Mol. Immunol. 2017, 91, 65–74. [Google Scholar] [CrossRef]
- Hirano, T. Interleukin 6 and its receptor: Ten years later. Int. Rev. Immunol. 1998, 16, 249–284. [Google Scholar] [CrossRef]
- Chang, S.F.; Lin, S.S.; Yang, H.C.; Chou, Y.Y.; Gao, J.I.; Lu, S.C. LPS-induced G-CSF expression in macrophages is mediated by ERK2, but not ERK1. PLoS ONE 2015, 10, 0129685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-κB transcription factor. Mol. Cell Biol. 1990, 10, 2327–2334. [Google Scholar] [CrossRef]
- Xie, Q.W.; Kashiwabara, Y.; Nathan, C. Role of transcription factor NF-κB/ Rel in induction of nitric oxide synthase. J. Biol. Chem. 1994, 269, 4705–4708. [Google Scholar] [CrossRef]
- Semple, F.; MacPherson, H.; Webb, S.; Cox, S.L.; Mallin, L.J.; Tyrrell, C.; Grimes, G.R.; Semple, C.A.; Nix, M.A.; Millhauser, G.L.; et al. Human β-defensin 3 affects the activity of pro-inflammatory pathways associated with MyD88 and TRIF. Eur. J. Immunol. 2011, 41, 3291–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Suh, J.S.; Kim, J.M.; Kim, J.H.; Park, H.J.; Park, Y.J.; Chung, C.P. Identification of a cell-penetrating peptide domain from human β-defensin 3 and characterization of its anti-inflammatory activity. Int. J. Nanomed. 2015, 10, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Nativel, B.; Couret, D.; Giraud, P.; Meilhac, O.; Lefebvre d’Hellencourt, C.; Viranaïcken, W.; Da Silva, C.R. Porphyromonas gingivalis lipopolysaccharides act exclusively through TLR4 with a resilience between mouse and human. Sci. Rep. 2017, 7, 15789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qui, C.; Yuan, Z.; He, Z.; Chen, H.; Liao, Y.; Li, S.; Zhou, W.; Song, Z. Lipopolysaccharide preparation derived from Porphyromonas gingivalis induces a weaker immuno-inflammatory response in BV-2 microglial cells than Escherichia coli by differentially activating TLR2/4-mediated NF-κB/STAT3 signaling pathways. Front. Cell. Infect. Microbiol. 2021, 11, 606986. [Google Scholar] [CrossRef]
- Pingel, L.C.; Kohlgraf, K.G.; Hansen, C.J.; Eastman, C.G.; Dietrich, D.E.; Burnell, K.K.; Srikantha, R.N.; Xiao, X.; Bélanger, M.; Progulske-Fox, A.; et al. Human β-defensin 3 binds to hemagglutinin B (rHagB), a non-fimbrial adhesin from Porphyromonas gingivalis, and attenuates a pro-inflammatory cytokine response. Immunol. Cell Biol. 2008, 86, 643–649. [Google Scholar] [CrossRef]
- Taggart, C.C.; Greene, C.M.; Smith, S.G.; Levine, R.L.; McCray, P.B., Jr.; O’Neill, S.; McElvaney, N.G. Inactivation of human β-defensins 2 and 3 by elastolytic cathepsins. J. Immunol. 2003, 171, 931–937. [Google Scholar] [CrossRef]
- Nie, R.; Wu, Z.; Ni, J.; Zeng, F.; Yu, W.; Zhang, Y.; Kadowaki, T.; Kashiwazaki, H.; Teeling, J.L.; Zhou, Y. Porphyromonas gingivalis infection induces amyloid-β accumulation in monocytes/macrophages. J. Alzheimers Dis. 2019, 72, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Bogyo, M.; Verhelst, S.; Bellingard-Dubouchaud, V.; Toba, S.; Greenbaum, D. Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chem. Biol. 2000, 7, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Montaser, M.; Lalmanach, G.; Mach, L. CA-074, but not its methyl ester CA-074Me, is a selective inhibitor of cathepsin B within living cells. Biol. Chem. 2002, 383, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; To, M.; Saruta, J.; Sato, C.; Yamamoto, Y.; Kondo, Y.; Shimizu, T.; Kamata, Y.; Tsukinoki, K. Salivary lactoferrin is transferred into the brain via the sublingual route. Biosci. Biotech. Biochem. 2017, 81, 1300–1304. [Google Scholar] [CrossRef] [Green Version]
- Contini, C.; Olianas, A.; Serrao, S.; Deriu, C.; Iavarone, F.; Boroumand, M.; Bizzarro, A.; Lauria, A.; Faa, G.; Castagnola, M.; et al. Top-down proteomics of human saliva highlights anti-inflammatory, antioxidant, and antimicrobial defense responses in Alzheimer disease. Front. Neurosci. 2021, 15, 743596. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inoue, E.; Minatozaki, S.; Katsuta, Y.; Nonaka, S.; Nakanishi, H. Human β-Defensin 3 Inhibits Porphyromonas Gingivalis Lipopolysaccharide-Induced Oxidative and Inflammatory Responses of Microglia by Suppression of Cathepsins B and L. Int. J. Mol. Sci. 2022, 23, 15099. https://doi.org/10.3390/ijms232315099
Inoue E, Minatozaki S, Katsuta Y, Nonaka S, Nakanishi H. Human β-Defensin 3 Inhibits Porphyromonas Gingivalis Lipopolysaccharide-Induced Oxidative and Inflammatory Responses of Microglia by Suppression of Cathepsins B and L. International Journal of Molecular Sciences. 2022; 23(23):15099. https://doi.org/10.3390/ijms232315099
Chicago/Turabian StyleInoue, Erika, Shiyo Minatozaki, Yui Katsuta, Saori Nonaka, and Hiroshi Nakanishi. 2022. "Human β-Defensin 3 Inhibits Porphyromonas Gingivalis Lipopolysaccharide-Induced Oxidative and Inflammatory Responses of Microglia by Suppression of Cathepsins B and L" International Journal of Molecular Sciences 23, no. 23: 15099. https://doi.org/10.3390/ijms232315099
APA StyleInoue, E., Minatozaki, S., Katsuta, Y., Nonaka, S., & Nakanishi, H. (2022). Human β-Defensin 3 Inhibits Porphyromonas Gingivalis Lipopolysaccharide-Induced Oxidative and Inflammatory Responses of Microglia by Suppression of Cathepsins B and L. International Journal of Molecular Sciences, 23(23), 15099. https://doi.org/10.3390/ijms232315099