
UCP2 as a Cancer Target through Energy Metabolism and Oxidative Stress Control

Abstract
1. Introduction
2. Role of UCP2 in the Physiological and Pathophysiological Context
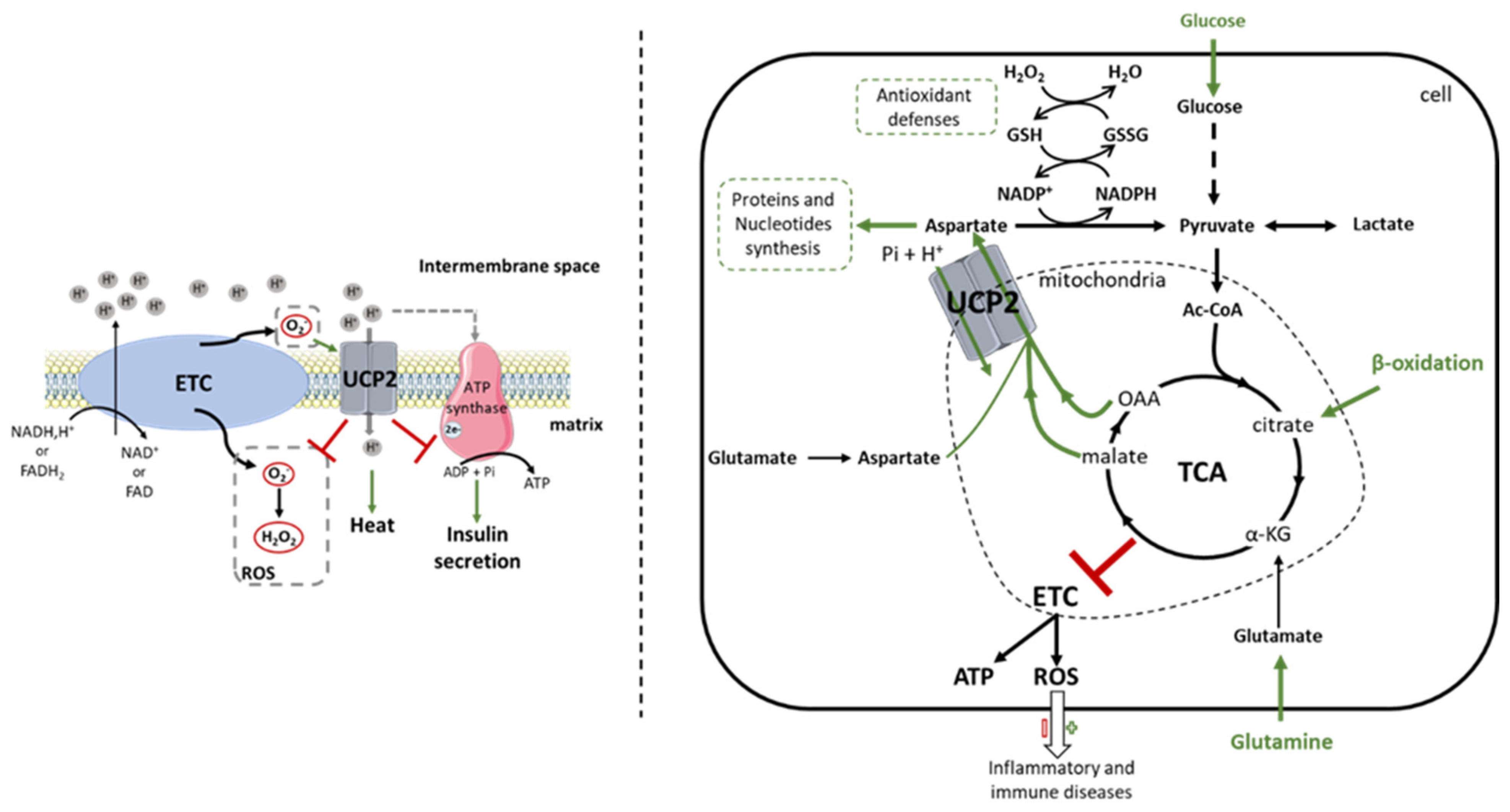
2.1. Biochemical Function
2.1.1. Uncoupling?
2.1.2. Transport of Metabolites
2.2. UCP2 a Metabolic Regulator
2.3. UCP2 and Oxidative Stress: A Link toward Immunity
2.3.1. Insulin Regulation—Type 2 Diabetes (T2D)—Cardiovascular Diseases
2.3.2. Infectious Diseases
2.3.3. Autoimmune Diseases
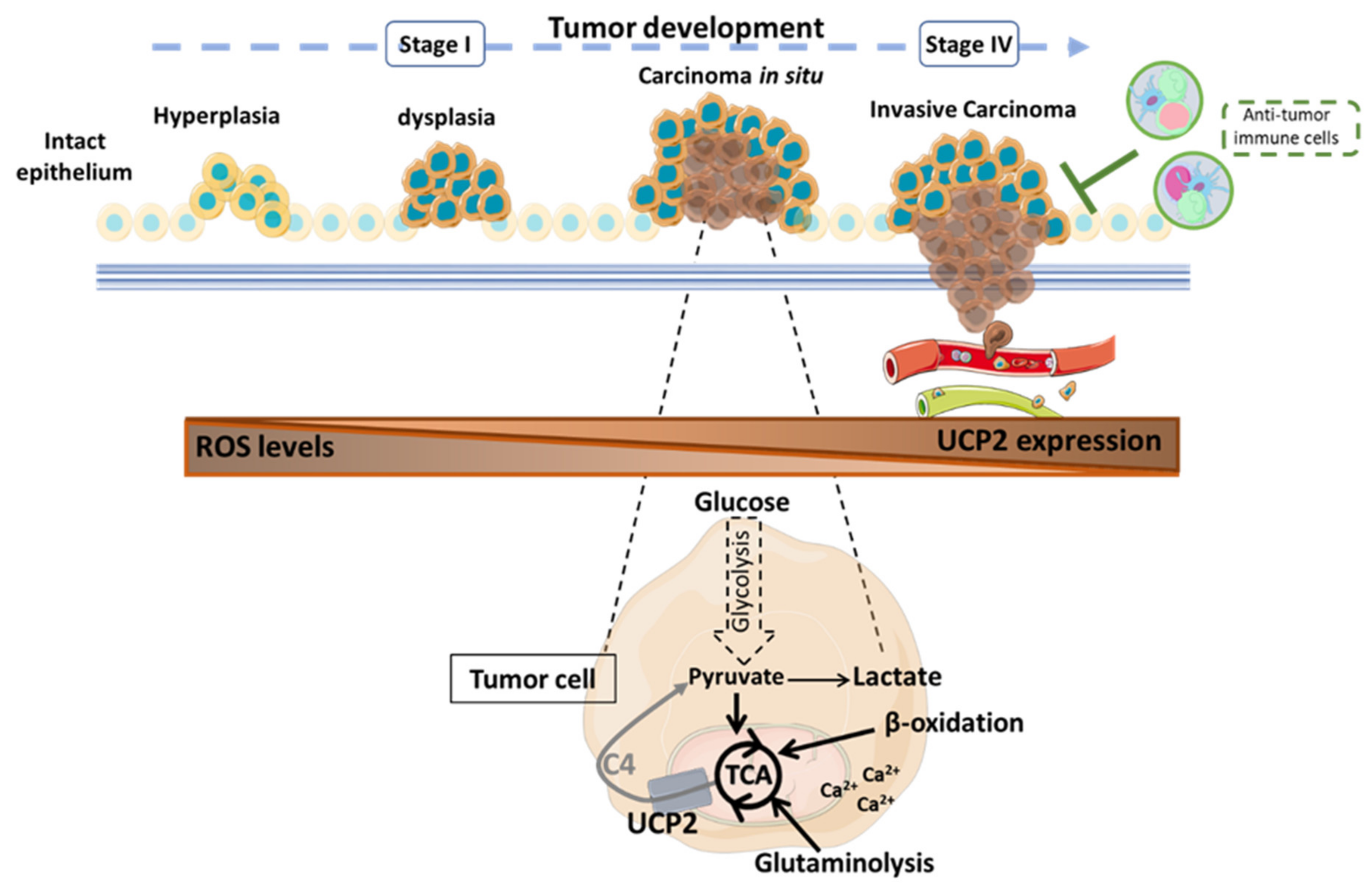
3. UCP2 and Cancer
3.1. UCP2: A Double-Edged Fight against ROS
3.2. UCP2 and Tumoral Metabolic Reprogramming
3.2.1. Glycolysis
3.2.2. Glutaminolysis
3.2.3. Ca2+ Signaling
3.3. UCP2-Activated Antitumor Immunity
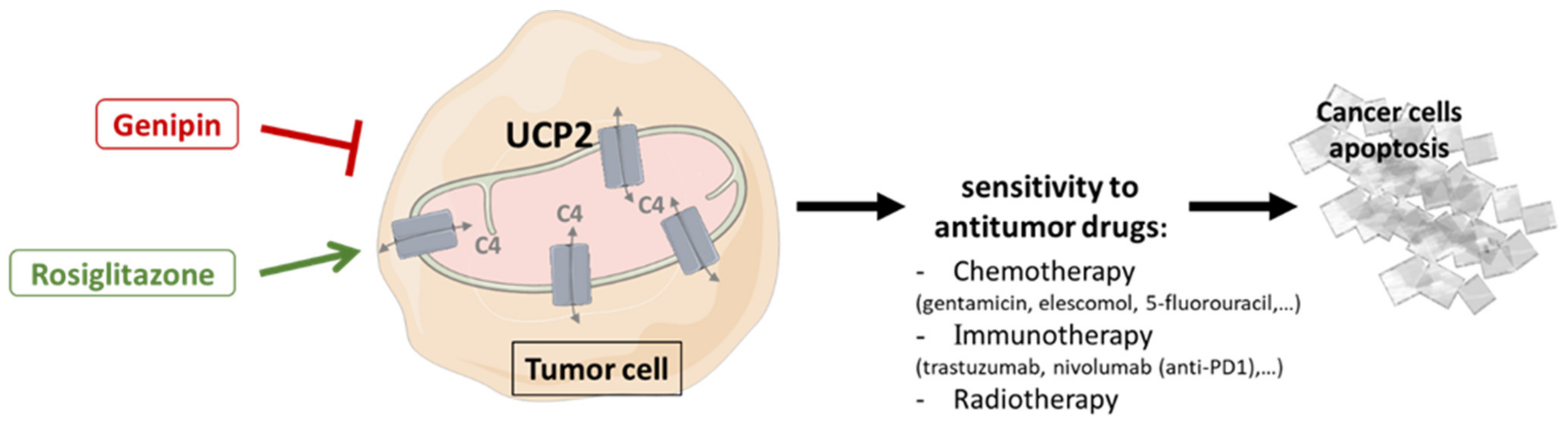
3.4. Drug Sensitivity and Therapeutic Improvement
3.4.1. Genipin
3.4.2. Rosiglitazone
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as Biosynthetic Factories for Cancer Proliferation. Cancer Metab. 2015, 3, 1–10. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Amoedo, N.D.; Sarlak, S.; Obre, E.; Esteves, P.; Bégueret, H.; Kieffer, Y.; Rousseau, B.; Dupis, A.; Izotte, J.; Bellance, N.; et al. Targeting the Mitochondrial Trifunctional Protein Restrains Tumor Growth in Oxidative Lung Carcinomas. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial Reactive Oxygen Species and Cancer. Cancer Metab. 2014, 2, 1–12. [Google Scholar] [CrossRef]
- Bouillaud, F.; Alves-Guerra, M.-C.; Ricquier, D. UCPs, at the Interface between Bioenergetics and Metabolism. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2443–2456. [Google Scholar] [CrossRef]
- Heaton, G.M.; Wagenvoord, R.J.; Kemp, A.; Nicholls, D.G. Brown-Adipose-Tissue Mitochondria: Photoaffinity Labelling of the Regulatory Site of Energy Dissipation. Eur. J. Biochem. 1978, 82, 515–521. [Google Scholar] [CrossRef]
- Fleury, C.; Neverova, M.; Collins, S.; Raimbault, S.; Champigny, O.; Levi-Meyrueis, C.; Bouillaud, F.; Seldin, M.F.; Surwit, R.S.; Ricquier, D.; et al. Uncoupling Protein-2: A Novel Gene Linked to Obesity and Hyperinsulinemia. Nat. Genet. 1997, 15, 269–272. [Google Scholar] [CrossRef]
- Mao, W.; Yu, X.X.; Zhong, A.; Li, W.; Brush, J.; Sherwood, S.W.; Adams, S.H.; Pan, G. UCP4, a Novel Brain-Specific Mitochondrial Protein that Reduces Membrane Potential in Mammalian Cells. FEBS Lett. 1999, 443, 326–330. [Google Scholar] [CrossRef]
- Sanchis, D.; Fleury, C.; Chomiki, N.; Goubern, M.; Huang, Q.; Neverova, M.; Grégoire, F.; Easlick, J.; Raimbault, S.; Lévi-Meyrueis, C.; et al. BMCP1, a Novel Mitochondrial Carrier with High Expression in the Central Nervous System of Humans and Rodents, and Respiration Uncoupling Activity in Recombinant Yeast. J. Biol. Chem. 1998, 273, 34611–34615. [Google Scholar] [CrossRef]
- Enerbäck, S.; Jacobsson, A.; Simpson, E.M.; Guerra, C.; Yamashita, H.; Harper, M.-E.; Kozak, L.P. Mice Lacking Mitochondrial Uncoupling Protein Are Cold-Sensitive but not Obese. Nature 1997, 387, 90–94. [Google Scholar] [CrossRef]
- Pecqueur, C.; Alves-Guerra, M.-C.; Gelly, C.; Lévi-Meyrueis, C.; Couplan, E.; Collins, S.; Ricquier, D.; Bouillaud, F.; Miroux, B. Uncoupling Protein 2, in vivo Distribution, Induction upon Oxidative Stress, and Evidence for Translational Regulation. J. Biol. Chem. 2001, 276, 8705–8712. [Google Scholar] [CrossRef]
- Hurtaud, C.; Gelly, C.; Bouillaud, F.; Lévi-Meyrueis, C. Translation Control of UCP2 Synthesis by the Upstream Open Reading Frame. Cell. Mol. Life Sci. 2006, 63, 1780. [Google Scholar] [CrossRef]
- Couplan, E.; del Mar Gonzalez-Barroso, M.; Alves-Guerra, M.C.; Ricquier, D.; Goubern, M.; Bouillaud, F. No Evidence for a Basal, Retinoic, or Superoxide-Induced Uncoupling Activity of the Uncoupling Protein 2 Present in Spleen or Lung Mitochondria. J. Biol. Chem. 2002, 277, 26268–26275. [Google Scholar] [CrossRef]
- Vozza, A.; Parisi, G.; De Leonardis, F.; Lasorsa, F.M.; Castegna, A.; Amorese, D.; Marmo, R.; Calcagnile, V.M.; Palmieri, L.; Ricquier, D.; et al. UCP2 Transports C4 Metabolites out of Mitochondria, Regulating Glucose and Glutamine Oxidation. Proc. Natl. Acad. Sci. USA 2014, 111, 960–965. [Google Scholar] [CrossRef]
- Bai, Y.; Onuma, H.; Bai, X.; Medvedev, A.V.; Misukonis, M.; Weinberg, J.B.; Cao, W.; Robidoux, J.; Floering, L.M.; Daniel, K.W.; et al. Persistent Nuclear Factor-ΚB Activation in Ucp2-/- Mice Leads to Enhanced Nitric Oxide and Inflammatory Cytokine Production. J. Biol. Chem. 2005, 280, 19062–19069. [Google Scholar] [CrossRef]
- Krauss, S.; Zhang, C.-Y.; Scorrano, L.; Dalgaard, L.T.; St-Pierre, J.; Grey, S.T.; Lowell, B.B. Superoxide-Mediated Activation of Uncoupling Protein 2 Causes Pancreatic β Cell Dysfunction. J. Clin. Investig. 2003, 112, 1831–1842. [Google Scholar] [CrossRef]
- Alves-Guerra, M.-C.; Rousset, S.; Pecqueur, C.; Mallat, Z.; Blanc, J.; Tedgui, A.; Bouillaud, F.; Cassard-Doulcier, A.-M.; Ricquier, D.; Miroux, B. Bone Marrow Transplantation Reveals the in vivo Expression of the Mitochondrial Uncoupling Protein 2 in Immune and Nonimmune Cells during Inflammation. J. Biol. Chem. 2003, 278, 42307–42312. [Google Scholar] [CrossRef]
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling Protein-2 Controls Proliferation by Promoting Fatty Acid Oxidation and Limiting Glycolysis-Derived Pyruvate Utilization. FASEB J. 2007, 22, 9–18. [Google Scholar] [CrossRef]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.-C.; Goubern, M.; Surwit, R.; et al. Disruption of the Uncoupling Protein-2 Gene in Mice Reveals a Role in Immunity and Reactive Oxygen Species Production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- Rousset, S.; Emre, Y.; Join-Lambert, O.; Hurtaud, C.; Ricquier, D.; Cassard-Doulcier, A.-M. The Uncoupling Protein 2 Modulates the Cytokine Balance in Innate Immunity. Cytokine 2006, 35, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Emre, Y.; Hurtaud, C.; Karaca, M.; Nubel, T.; Zavala, F.; Ricquier, D. Role of Uncoupling Protein UCP2 in Cell-Mediated Immunity: How Macrophage-Mediated Insulitis Is Accelerated in a Model of Autoimmune Diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 19085–19090. [Google Scholar] [CrossRef]
- Blanc, J.; Alves-Guerra, M.C.; Esposito, B.; Rousset, S.; Gourdy, P.; Ricquier, D.; Tedgui, A.; Miroux, B.; Mallat, Z. Protective Role of Uncoupling Protein 2 in Atherosclerosis. Circulation 2003, 107, 388–390. [Google Scholar] [CrossRef]
- Brand, M.D.; Esteves, T.C. Physiological Functions of the Mitochondrial Uncoupling Proteins UCP2 and UCP3. Cell Metab. 2005, 2, 85–93. [Google Scholar] [CrossRef]
- Aguilar, E.; Esteves, P.; Sancerni, T.; Lenoir, V.; Aparicio, T.; Bouillaud, F.; Dentin, R.; Prip-Buus, C.; Ricquier, D.; Pecqueur, C.; et al. UCP2 Deficiency Increases Colon Tumorigenesis by Promoting Lipid Synthesis and Depleting NADPH for Antioxidant Defenses. Cell Rep. 2019, 28, 2306–2316.e5. [Google Scholar] [CrossRef]
- Pons, D.G.; Nadal-Serrano, M.; Torrens-Mas, M.; Valle, A.; Oliver, J.; Roca, P. UCP2 Inhibition Sensitizes Breast Cancer Cells to Therapeutic Agents by Increasing Oxidative Stress. Free Radic. Biol. Med. 2015, 86, 67–77. [Google Scholar] [CrossRef]
- Raho, S.; Capobianco, L.; Malivindi, R.; Vozza, A.; Piazzolla, C.; De Leonardis, F.; Gorgoglione, R.; Scarcia, P.; Pezzuto, F.; Agrimi, G.; et al. KRAS-Regulated Glutamine Metabolism Requires UCP2-Mediated Aspartate Transport to Support Pancreatic Cancer Growth. Nat. Metab. 2020, 2, 1373–1381. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 377, 1345–1356. [Google Scholar] [CrossRef]
- Bozic, I.; Allen, B.; Nowak, M.A. Dynamics of Targeted Cancer Therapy. Trends Mol. Med. 2012, 18, 311–316. [Google Scholar] [CrossRef][Green Version]
- Wang, K.; Jiang, J.; Lei, Y.; Zhou, S.; Wei, Y.; Huang, C. Targeting Metabolic–Redox Circuits for Cancer Therapy. Trends Biochem. Sci. 2019, 44, 401–414. [Google Scholar] [CrossRef]
- Leary, M.; Heerboth, S.; Lapinska, K.; Sarkar, S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. [Google Scholar] [CrossRef]
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial Uncoupling Proteins: Subtle Regulators of Cellular Redox Signaling Reviewing Editors: Jerzy Beltowski, Joseph Burgoyne, Gabor Csanyi, Sergey Dikalov, Frank Krause, Anibal Vercesi, and Jeremy Ward. Antioxid. Redox Signal. 2018, 29, 667–714. [Google Scholar] [CrossRef]
- Jabůrek, M.; Vařecha, M.; Gimeno, R.E.; Dembski, M.; Ježek, P.; Zhang, M.; Burn, P.; Tartaglia, L.A.; Garlid, K.D. Transport Function and Regulation of Mitochondrial Uncoupling Proteins 2 and 3 *. J. Biol. Chem. 1999, 274, 26003–26007. [Google Scholar] [CrossRef]
- Rial, E.; González-Barroso, M.; Fleury, C.; Iturrizaga, S.; Sanchis, D.; Jiménez-Jiménez, J.; Ricquier, D.; Goubern, M.; Bouillaud, F. Retinoids Activate Proton Transport by the Uncoupling Proteins UCP1 and UCP2. EMBO J. 1999, 18, 5827–5833. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Baffy, G.; Perret, P.; Krauss, S.; Peroni, O.; Grujic, D.; Hagen, T.; Vidal-Puig, A.J.; Boss, O.; Kim, Y.-B.; et al. Uncoupling Protein-2 Negatively Regulates Insulin Secretion and Is a Major Link between Obesity, β Cell Dysfunction, and Type 2 Diabetes. Cell 2001, 105, 745–755. [Google Scholar] [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide Activates Mitochondrial Uncoupling Proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Conti, B.; Sanchez-Alavez, M.; Winsky-Sommerer, R.; Morale, M.C.; Lucero, J.; Brownell, S.; Fabre, V.; Huitron-Resendiz, S.; Henriksen, S.; Zorrilla, E.P.; et al. Transgenic Mice with a Reduced Core Body Temperature Have an Increased Life Span. Science 2006, 314, 825–828. [Google Scholar] [CrossRef]
- Harper, J.A.; Dickinson, K.; Brand, M.D. Mitochondrial Uncoupling as a Target for Drug Development for the Treatment of Obesity. Obes. Rev. 2001, 2, 255–265. [Google Scholar] [CrossRef]
- Chan, C.B.; De Leo, D.; Joseph, J.W.; McQuaid, T.S.; Ha, X.F.; Xu, F.; Tsushima, R.G.; Pennefather, P.S.; Salapatek, A.M.F.; Wheeler, M.B. Increased Uncoupling Protein-2 Levels in β-Cells Are Associated with Impaired Glucose-Stimulated Insulin Secretion: Mechanism of Action. Diabetes 2001, 50, 1302–1310. [Google Scholar] [CrossRef]
- Bienengraeber, M.; Echtay, K.S.; Klingenberg, M. H+ Transport by Uncoupling Protein (UCP-1) Is Dependent on a Histidine Pair, Absent in UCP-2 and UCP-3. Biochemistry 1998, 37, 3–8. [Google Scholar] [CrossRef]
- Gimeno, R.E.; Dembski, M.; Weng, X.; Deng, N.; Shyjan, A.W.; Gimeno, C.J.; Iris, F.; Ellis, S.J.; Woolf, E.A.; Tartaglia, L.A. Cloning and Characterization of an Uncoupling Protein Homolog: A Potential Molecular Mediator of Human Thermogenesis. Diabetes 1997, 46, 900–906. [Google Scholar] [CrossRef]
- Stuart, J.A.; Harper, J.A.; Brindle, K.M.; Jekabsons, M.B.; Brand, M.D. A Mitochondrial Uncoupling Artifact Can Be Caused by Expression of Uncoupling Protein 1 in Yeast. Biochem. J. 2001, 356, 779–789. [Google Scholar] [CrossRef]
- Stuart, J.A.; Harper, J.A.; Brindle, K.M.; Jekabsons, M.B.; Brand, M.D. Physiological Levels of Mammalian Uncoupling Protein 2 Do Not Uncouple Yeast Mitochondria. J. Biol. Chem. 2001, 276, 18633–18639. [Google Scholar] [CrossRef]
- Harper, J.A.; Stuart, J.A.; Jekabsons, M.B.; Roussel, D.; Brindle, K.M.; Dickinson, K.; Jones, R.B.; Brand, M.D. Artifactual Uncoupling by Uncoupling Protein 3 in Yeast Mitochondria at the Concentrations Found in Mouse and Rat Skeletal-Muscle Mitochondria. Biochem. J. 2002, 361, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Murphy, M.P.; Smith, R.A.J.; Talbot, D.A.; Brand, M.D. Superoxide Activates Mitochondrial Uncoupling Protein 2 from the Matrix Side: Studies using targeted antioxidants. J. Biol. Chem. 2002, 277, 47129–47135. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qian, H.; Huang, X.; Li, J.; Zhang, J.; Zhu, N.; Chen, H.; Zhu, C.; Wang, J.; Zhang, P.; et al. UCP2 Mitigates the Loss of Human Spermatozoa Motility by Promoting MROS Elimination. CPB 2018, 50, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Vogler, S.; Pahnke, J.; Rousset, S.; Ricquier, D.; Moch, H.; Miroux, B.; Ibrahim, S.M. Uncoupling Protein 2 Has Protective Function during Experimental Autoimmune Encephalomyelitis. Am. J. Pathol. 2006, 168, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Sweetlove, L.J.; Lytovchenko, A.; Morgan, M.; Nunes-Nesi, A.; Taylor, N.L.; Baxter, C.J.; Eickmeier, I.; Fernie, A.R. Mitochondrial Uncoupling Protein Is Required for Efficient Photosynthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 19587–19592. [Google Scholar] [CrossRef]
- Stuart, J.A.; Harper, J.A.; Brindle, K.M.; Brand, M.D. Uncoupling Protein 2 from Carp and Zebrafish, Ectothermic Vertebrates. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1413, 50–54. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Kunji, E.R.S. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci. 2020, 45, 244–258. [Google Scholar] [CrossRef]
- Mozo, J.; Ferry, G.; Studeny, A.; Pecqueur, C.; Rodriguez, M.; Boutin, J.A.; Bouillaud, F. Expression of UCP3 in CHO Cells Does Not Cause Uncoupling, but Controls Mitochondrial Activity in the Presence of Glucose. Biochem. J. 2006, 393, 431–439. [Google Scholar] [CrossRef]
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling Proteins 2 and 3 Are Fundamental for Mitochondrial Ca2+ Uniport. Nat. Cell Biol. 2007, 9, 445–452. [Google Scholar] [CrossRef]
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 Controls Cristae Junction and Spatially Anchors Mitochondrial Ca2+ Uniporter Complex. Nat. Commun. 2019, 10, 3732. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Klec, C.; Parichatikanond, W.; Stryeck, S.; Gottschalk, B.; Pulido, S.; Rost, R.; Eroglu, E.; Hofmann, N.A.; Bondarenko, A.I.; et al. PRMT1-Mediated Methylation of MICU1 Determines the UCP2/3 Dependency of Mitochondrial Ca2+ Uptake in Immortalized Cells. Nat. Commun. 2016, 7, 12897. [Google Scholar] [CrossRef] [PubMed]
- Koshenov, Z.; Oflaz, F.E.; Hirtl, M.; Bachkoenig, O.A.; Rost, R.; Osibow, K.; Gottschalk, B.; Madreiter-Sokolowski, C.T.; Waldeck-Weiermair, M.; Malli, R.; et al. The Contribution of Uncoupling Protein 2 to Mitochondrial Ca2+ Homeostasis in Health and Disease—A Short Revisit. Mitochondrion 2020, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Barati, D.; Walters, J.D.; Pajoum Shariati, S.R.; Moeinzadeh, S.; Jabbari, E. Effect of Organic Acids on Calcium Phosphate Nucleation and Osteogenic Differentiation of Human Mesenchymal Stem Cells on Peptide Functionalized Nanofibers. Langmuir 2015, 31, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Reinwald, S.; Weaver, C.M.; Kester, J.J. Chapter 6—The Health Benefits of Calcium Citrate Malate: A Review of the Supporting Science. In Advances in Food and Nutrition Research; Taylor, S.L., Ed.; Academic Press: Cambridge, MA, USA, 2008; Volume 54, pp. 219–346. [Google Scholar]
- Diano, S.; Horvath, T.L. Mitochondrial Uncoupling Protein 2 (UCP2) in Glucose and Lipid Metabolism. Trends Mol. Med. 2012, 18, 52–58. [Google Scholar] [CrossRef]
- Toda, C.; Diano, S. Mitochondrial UCP2 in the Central Regulation of Metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 757–764. [Google Scholar] [CrossRef]
- Pecqueur, C.; Alves-Guerra, C.; Ricquier, D.; Bouillaud, F. UCP2, a Metabolic Sensor Coupling Glucose Oxidation to Mitochondrial Metabolism? IUBMB Life 2009, 61, 762–767. [Google Scholar] [CrossRef]
- Bouillaud, F. UCP2, Not a Physiologically Relevant Uncoupler but a Glucose Sparing Switch Impacting ROS Production and Glucose Sensing. Biochim. Biophys. Acta 2009, 1787, 377–383. [Google Scholar] [CrossRef]
- Heinitz, S.; Piaggi, P.; Yang, S.; Bonfiglio, S.; Steel, J.; Krakoff, J.; Votruba, S.B. Response of Skeletal Muscle UCP2-Expression during Metabolic Adaptation to Caloric Restriction. Int. J. Obes. 2018, 42, 974–984. [Google Scholar] [CrossRef]
- Sheets, A.R.; Fülöp, P.; Derdák, Z.; Kassai, A.; Sabo, E.; Mark, N.M.; Paragh, G.; Wands, J.R.; Baffy, G. Uncoupling Protein-2 Modulates the Lipid Metabolic Response to Fasting in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1017–G1024. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Parton, L.E.; Ye, C.P.; Krauss, S.; Shen, R.; Lin, C.-T.; Porco, J.A.; Lowell, B.B. Genipin Inhibits UCP2-Mediated Proton Leak and Acutely Reverses Obesity- and High Glucose-Induced β Cell Dysfunction in Isolated Pancreatic Islets. Cell Metab. 2006, 3, 417–427. [Google Scholar] [CrossRef]
- He, Y.; Wang, N.; Shen, Y.; Zheng, Z.; Xu, X. Inhibition of High Glucose-Induced Apoptosis by Uncoupling Protein 2 in Human Umbilical Vein Endothelial Cells. Int. J. Mol. Med. 2014, 33, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Liu, Z.-W.; Walllingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschöp, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.I.; et al. UCP2 Mediates Ghrelin’s Action on NPY/AgRP Neurons by Lowering Free Radicals. Nature 2008, 454, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 Regulates Energy Metabolism and Differentiation Potential of Human Pluripotent Stem Cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [PubMed]
- Kukat, A.; Dogan, S.A.; Edgar, D.; Mourier, A.; Jacoby, C.; Maiti, P.; Mauer, J.; Becker, C.; Senft, K.; Wibom, R.; et al. Loss of UCP2 Attenuates Mitochondrial Dysfunction without Altering ROS Production and Uncoupling Activity. PLOS Genet. 2014, 10, e1004385. [Google Scholar] [CrossRef]
- Nübel, T.; Emre, Y.; Rabier, D.; Chadefaux, B.; Ricquier, D.; Bouillaud, F. Modified Glutamine Catabolism in Macrophages of Ucp2 Knock-out Mice. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1777, 48–54. [Google Scholar] [CrossRef]
- Hurtaud, C.; Gelly, C.; Chen, Z.; Lévi-Meyrueis, C.; Bouillaud, F. Glutamine Stimulates Translation of Uncoupling Protein 2mRNA. Cell. Mol. Life Sci. 2007, 64, 1853–1860. [Google Scholar] [CrossRef]
- Millet, L.; Vidal, H.; Andreelli, F.; Larrouy, D.; Riou, J.P.; Ricquier, D.; Laville, M.; Langin, D. Increased Uncoupling Protein-2 and -3 MRNA Expression during Fasting in Obese and Lean Humans. J. Clin. Investig. 1997, 100, 2665–2670. [Google Scholar] [CrossRef]
- Schrauwen, P.; Hoppeler, H.; Billeter, R.; Bakker, A.H.; Pendergast, D.R. Fiber Type Dependent Upregulation of Human Skeletal Muscle UCP2 and UCP3 MRNA Expression by High-Fat Diet. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 449–456. [Google Scholar] [CrossRef]
- Nègre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Pénicaud, L.; Casteilla, L. A Role for Uncoupling Protein-2 as a Regulator of Mitochondrial Hydrogen Peroxide Generation. FASEB J. 1997, 11, 809–815. [Google Scholar] [CrossRef]
- Horimoto, M.; Fülöp, P.; Derdák, Z.; Wands, J.R.; Baffy, G. Uncoupling Protein-2 Deficiency Promotes Oxidant Stress and Delays Liver Regeneration in Mice. Hepatology 2004, 39, 386–392. [Google Scholar] [CrossRef]
- Teshima, Y.; Akao, M.; Jones, S.P.; Marbán, E. Uncoupling Protein-2 Overexpression Inhibits Mitochondrial Death Pathway in Cardiomyocytes. Circ. Res. 2003, 93, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-U.; Lee, I.K.; Han, J.; Song, D.-K.; Kim, Y.M.; Song, H.S.; Kim, H.S.; Lee, W.J.; Koh, E.H.; Song, K.-H.; et al. Effects of Recombinant Adenovirus-Mediated Uncoupling Protein 2 Overexpression on Endothelial Function and Apoptosis. Circ. Res. 2005, 96, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1859, 940–950. [CrossRef]
- Monteiro, B.S.; Freire-Brito, L.; Carrageta, D.F.; Oliveira, P.F.; Alves, M.G. Mitochondrial Uncoupling Proteins (UCPs) as Key Modulators of ROS Homeostasis: A Crosstalk between Diabesity and Male Infertility? Antioxidants 2021, 10, 1746. [Google Scholar] [CrossRef]
- Hirschenson, J.; Melgar-Bermudez, E.; Mailloux, R.J. The Uncoupling Proteins: A Systematic Review on the Mechanism Used in the Prevention of Oxidative Stress. Antioxidants 2022, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Hass, D.T.; Barnstable, C.J. Uncoupling Proteins in the Mitochondrial Defense against Oxidative Stress. Prog. Retin. Eye Res. 2021, 83, 100941. [Google Scholar] [CrossRef]
- Broche, B.; Fradj, S.B.; Aguilar, E.; Sancerni, T.; Bénard, M.; Makaci, F.; Berthault, C.; Scharfmann, R.; Alves-Guerra, M.-C.; Duvillié, B. Mitochondrial Protein UCP2 Controls Pancreas Development. Diabetes 2018, 67, 78–84. [Google Scholar] [CrossRef]
- DeSouza, C.T.; Araújo, E.P.; Stoppiglia, L.F.; Pauli, J.R.; Ropelle, E.; Rocco, S.A.; Marin, R.M.; Franchini, K.G.; Carvalheira, J.B.; Saad, M.J.; et al. Inhibition of UCP2 Expression Reverses Diet-Induced Diabetes Mellitus by Effects on both Insulin Secretion and Action. FASEB J. 2007, 21, 1153–1163. [Google Scholar] [CrossRef]
- Saleh, M.C.; Wheeler, M.B.; Chan, C.B. Endogenous Islet Uncoupling Protein-2 Expression and Loss of Glucose Homeostasis in Ob/Ob Mice. J. Endocrinol. 2006, 190, 659–667. [Google Scholar] [CrossRef]
- Produit-Zengaffinen, N.; Davis-Lameloise, N.; Perreten, H.; Bécard, D.; Gjinovci, A.; Keller, P.A.; Wollheim, C.B.; Herrera, P.; Muzzin, P.; Assimacopoulos-Jeannet, F. Increasing Uncoupling Protein-2 in Pancreatic Beta Cells Does Not Alter Glucose-Induced Insulin Secretion but Decreases Production of Reactive Oxygen Species. Diabetologia 2007, 50, 84–93. [Google Scholar] [CrossRef]
- Pi, J.; Bai, Y.; Daniel, K.W.; Liu, D.; Lyght, O.; Edelstein, D.; Brownlee, M.; Corkey, B.E.; Collins, S. Persistent Oxidative Stress Due to Absence of Uncoupling Protein 2 Associated with Impaired Pancreatic β-Cell Function. Endocrinology 2009, 150, 3040–3048. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Collins, S. Reactive Oxygen Species and Uncoupling Protein 2 in Pancreatic β-Cell Function. Diabetes Obes. Metab. 2010, 12, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Robson-Doucette, C.A.; Sultan, S.; Allister, E.M.; Wikstrom, J.D.; Koshkin, V.; Bhattacharjee, A.; Prentice, K.J.; Sereda, S.B.; Shirihai, O.S.; Wheeler, M.B. β-Cell Uncoupling Protein 2 Regulates Reactive Oxygen Species Production, Which Influences Both Insulin and Glucagon Secretion. Diabetes 2011, 60, 2710–2719. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Karaca, M.; Maechler, P. Upregulation of UCP2 in Beta-Cells Confers Partial Protection against Both Oxidative Stress and Glucotoxicity. Redox Biol. 2017, 13, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Martín-Timón, I.; Sevillano-Collantes, C.; Segura-Galindo, A.; del Cañizo-Gómez, F.J. Type 2 Diabetes and Cardiovascular Disease: Have all Risk Factors the Same Strength? World J. Diabetes 2014, 5, 444–470. [Google Scholar] [CrossRef]
- Moukdar, F.; Robidoux, J.; Lyght, O.; Pi, J.; Daniel, K.W.; Collins, S. Reduced Antioxidant Capacity and Diet-Induced Atherosclerosis in Uncoupling Protein-2-Deficient Mice S⃞. J. Lipid Res. 2009, 50, 59–70. [Google Scholar] [CrossRef]
- Luo, J.-Y.; Cheng, C.K.; He, L.; Pu, Y.; Zhang, Y.; Lin, X.; Xu, A.; Lau, C.W.; Tian, X.Y.; Ma, R.C.W.; et al. Endothelial UCP2 Is a Mechanosensitive Suppressor of Atherosclerosis. Circ. Res. 2022, 131, 424–441. [Google Scholar] [CrossRef]
- Emre, Y.; Hurtaud, C.; Nübel, T.; Criscuolo, F.; Ricquier, D.; Cassard-Doulcier, A.-M. Mitochondria Contribute to LPS-Induced MAPK Activation via Uncoupling Protein UCP2 in Macrophages. Biochem. J. 2007, 402, 271–278. [Google Scholar] [CrossRef]
- Ball, W.B.; Kar, S.; Mukherjee, M.; Chande, A.G.; Mukhopadhyaya, R.; Das, P.K. Uncoupling Protein 2 Negatively Regulates Mitochondrial Reactive Oxygen Species Generation and Induces Phosphatase-Mediated Anti-Inflammatory Response in Experimental Visceral Leishmaniasis. J. Immunol. 2011, 187, 1322–1332. [Google Scholar] [CrossRef]
- Carrión, J.; Abengozar, M.A.; Fernández-Reyes, M.; Sánchez-Martín, C.; Rial, E.; Domínguez-Bernal, G.; González-Barroso, M.M. UCP2 Deficiency Helps to Restrict the Pathogenesis of Experimental Cutaneous and Visceral Leishmaniosis in Mice. PLOS Negl. Trop. Dis. 2013, 7, e2077. [Google Scholar] [CrossRef][Green Version]
- Gupta, A.K.; Ghosh, K.; Palit, S.; Barua, J.; Das, P.K.; Ukil, A. Leishmania Donovani Inhibits Inflammasome-Dependent Macrophage Activation by Exploiting the Negative Regulatory Proteins A20 and UCP2. FASEB J. 2017, 31, 5087–5101. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-S.; Lee, S.; Park, M.-A.; Siempos, I.I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W.; et al. UCP2-Induced Fatty Acid Synthase Promotes NLRP3 Inflammasome Activation during Sepsis. J. Clin. Investig. 2015, 125, 665–680. [Google Scholar] [CrossRef]
- Jiang, Z.-M.; Yang, Q.-H.; Zhu, C.-Q. UCP2 in Early Diagnosis and Prognosis of Sepsis. Eur. Rev. Med. Pharm. Sci. 2017, 21, 549–553. [Google Scholar]
- Lee, S.C.; Robson-Doucette, C.A.; Wheeler, M.B. Uncoupling Protein 2 Regulates Reactive Oxygen Species Formation in Islets and Influences Susceptibility to Diabetogenic Action of Streptozotocin. J. Endocrinol. 2009, 203, 33–43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aheng, C.; Ly, N.; Kelly, M.; Ibrahim, S.; Ricquier, D.; Alves-Guerra, M.-C.; Miroux, B. Deletion of UCP2 in iNOS Deficient Mice Reduces the Severity of the Disease during Experimental Autoimmune Encephalomyelitis. PLoS ONE 2011, 6, e22841. [Google Scholar] [CrossRef]
- Smorodchenko, A.; Schneider, S.; Rupprecht, A.; Hilse, K.; Sasgary, S.; Zeitz, U.; Erben, R.G.; Pohl, E.E. UCP2 Up-Regulation within the Course of Autoimmune Encephalomyelitis Correlates with T-Lymphocyte Activation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1002–1012. [Google Scholar] [CrossRef]
- Derdák, Z.; Fülöp, P.; Sabo, E.; Tavares, R.; Berthiaume, E.P.; Resnick, M.B.; Paragh, G.; Wands, J.R.; Baffy, G. Enhanced Colon Tumor Induction in Uncoupling Protein-2 Deficient Mice Is Associated with NF-KappaB Activation and Oxidative Stress. Carcinogenesis 2006, 27, 956–961. [Google Scholar] [CrossRef]
- Deng, S.; Yang, Y.; Han, Y.; Li, X.; Wang, X.; Li, X.; Zhang, Z.; Wang, Y. UCP2 Inhibits ROS-Mediated Apoptosis in A549 under Hypoxic Conditions. PLoS ONE 2012, 7, e30714. [Google Scholar] [CrossRef]
- Dando, I.; Fiorini, C.; Pozza, E.D.; Padroni, C.; Costanzo, C.; Palmieri, M.; Donadelli, M. UCP2 Inhibition Triggers ROS-Dependent Nuclear Translocation of GAPDH and Autophagic Cell Death in Pancreatic Adenocarcinoma Cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 672–679. [Google Scholar] [CrossRef]
- Esteves, P.; Pecqueur, C.; Ransy, C.; Esnous, C.; Lenoir, V.; Bouillaud, F.; Bulteau, A.-L.; Lombès, A.; Prip-Buus, C.; Ricquier, D.; et al. Mitochondrial Retrograde Signaling Mediated by UCP2 Inhibits Cancer Cell Proliferation and Tumorigenesis. Cancer Res. 2014, 74, 3971–3982. [Google Scholar] [CrossRef]
- Yu, J.; Shi, L.; Shen, X.; Zhao, Y. UCP2 Regulates Cholangiocarcinoma Cell Plasticity via Mitochondria-to-AMPK Signals. Biochem. Pharmacol. 2019, 166, 174–184. [Google Scholar] [CrossRef]
- Brandi, J.; Cecconi, D.; Cordani, M.; Torrens-Mas, M.; Pacchiana, R.; Dalla Pozza, E.; Butera, G.; Manfredi, M.; Marengo, E.; Oliver, J.; et al. The Antioxidant Uncoupling Protein 2 Stimulates HnRNPA2/B1, GLUT1 and PKM2 Expression and Sensitizes Pancreas Cancer Cells to Glycolysis Inhibition. Free Radic. Biol. Med. 2016, 101, 305–316. [Google Scholar] [CrossRef]
- Sancerni, T. UCP2 Silencing Restrains Leukemia Cell Proliferation through Glutamine Metabolic Remodeling. Front. Immunol. 2022, 13, 960226. [Google Scholar] [CrossRef]
- Sreedhar, A.; Lefort, J.; Petruska, P.; Gu, X.; Shi, R.; Miriyala, S.; Panchatcharam, M.; Zhao, Y. UCP2 Upregulation Promotes PLCγ-1 Signaling during Skin Cell Transformation. Mol. Carcinog. 2017, 56, 2290–2300. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Gottschalk, B.; Sokolowski, A.A.; Malli, R.; Graier, W.F. Dynamic Control of Mitochondrial Ca2+ Levels as a Survival Strategy of Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 614668. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-C.; Tsui, Y.-C.; Ragusa, S.; Koelzer, V.H.; Mina, M.; Franco, F.; Läubli, H.; Tschumi, B.; Speiser, D.; Romero, P.; et al. Uncoupling Protein 2 Reprograms the Tumor Microenvironment to Support the Anti-Tumor Immune Cycle. Nat. Immunol. 2019, 20, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Horimoto, M.; Resnick, M.B.; Konkin, T.A.; Routhier, J.; Wands, J.R.; Baffy, G. Expression of Uncoupling Protein-2 in Human Colon Cancer. Clin. Cancer Res. 2004, 10, 6203–6207. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nichols, K.; Nathan, C.-A.; Zhao, Y. Mitochondrial uncoupling protein 2 is up-regulated in human head and neck, skin, pancreatic, and prostate tumors. Cancer Biomark. 2013, 13, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, A.; Meng, Z.; Luciani, G.; Chen, L.-C.; Bennington, J.L.; Dairkee, S.H. Negative Regulation of UCP2 by TGFβ Signaling Characterizes Low and Intermediate-Grade Primary Breast Cancer. Cell Death Dis. 2010, 1, e53. [Google Scholar] [CrossRef]
- Yu, J.; Shi, L.; Lin, W.; Lu, B.; Zhao, Y. UCP2 Promotes Proliferation and Chemoresistance through Regulating the NF-ΚB/β-Catenin Axis and Mitochondrial ROS in Gallbladder Cancer. Biochem. Pharmacol. 2020, 172, 113745. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant P53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant P53 Blocks SESN1/AMPK/PGC-1α/UCP2 Axis Increasing Mitochondrial O2−· Production in Cancer Cells. Br. J. Cancer 2018, 119, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Donadelli, M.; Dando, I.; Pozza, E.D.; Palmieri, M. Mitochondrial Uncoupling Protein 2 and Pancreatic Cancer: A New Potential Target Therapy. World J. Gastroenterol. 2015, 21, 3232–3238. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The Role of ROS in Tumour Development and Progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Wu, S.; Luo, C.; Hameed, N.U.F.; Wang, Y.; Zhuang, D. UCP2 Silencing in Glioblastoma Reduces Cell Proliferation and Invasiveness by Inhibiting P38 MAPK Pathway. Exp. Cell Res. 2020, 394, 112110. [Google Scholar] [CrossRef]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The P38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Esteves, P.; Pecqueur, C.; Alves-Guerra, M.-C. UCP2 Induces Metabolic Reprogramming to Inhibit Proliferation of Cancer Cells. Mol. Cell. Oncol. 2015, 2, e975024. [Google Scholar] [CrossRef]
- Klaus, A.; Polge, C.; Zorman, S.; Auchli, Y.; Brunisholz, R.; Schlattner, U. A Two-Dimensional Screen for AMPK Substrates Identifies Tumor Suppressor Fumarate Hydratase as a Preferential AMPKα2 Substrate. J. Proteom. 2012, 75, 3304–3313. [Google Scholar] [CrossRef]
- Tong, W.-H.; Sourbier, C.; Kovtunovych, G.; Jeong, S.Y.; Vira, M.; Ghosh, M.; Romero, V.V.; Sougrat, R.; Vaulont, S.; Viollet, B.; et al. The Glycolytic Shift in Fumarate-Hydratase-Deficient Kidney Cancer Lowers AMPK Levels, Increases Anabolic Propensities and Lowers Cellular Iron Levels. Cancer Cell 2011, 20, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Stancliffe, E.; Fowle-Grider, R.; Wang, R.; Wang, C.; Schwaiger-Haber, M.; Shriver, L.P.; Patti, G.J. Saturation of the Mitochondrial NADH Shuttles Drives Aerobic Glycolysis in Proliferating Cells. Mol. Cell 2022, 82, 3270–3283. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.; Cassell, T.; Smith, P.; Lu, D.; Nam, H.W.; Lane, A.N.; Zhao, Y. UCP2 Overexpression Redirects Glucose into Anabolic Metabolic Pathways. Proteomics 2019, 19, 1800353. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-C.; Chou, C.-C.; Kulp, S.K.; Chen, C.-S. AMPK as a Potential Anticancer Target—Friend or Foe? Curr. Pharm. Des. 2014, 20, 2607–2618. [Google Scholar] [CrossRef]
- Sreedhar, A.; Petruska, P.; Miriyala, S.; Panchatcharam, M.; Zhao, Y. UCP2 Overexpression Enhanced Glycolysis via Activation of PFKFB2 during Skin Cell Transformation. Oncotarget 2017, 8, 95504–95515. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Rupprecht, A. Glutamine regulates mitochondrial uncoupling protein 2 to promote glutaminolysis in neuroblastoma cells. Biochim. Biophys. Acta (BBA) Bioenerg. 2019, 1860, 391–401. [Google Scholar] [CrossRef]
- Gorgoglione, R.; Impedovo, V.; Riley, C.L.; Fratantonio, D.; Tiziani, S.; Palmieri, L.; Dolce, V.; Fiermonte, G. Glutamine-Derived Aspartate Biosynthesis in Cancer Cells: Role of Mitochondrial Transporters and New Therapeutic Perspectives. Cancers 2022, 14, 245. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ Signalling Checkpoints in Cancer: Remodelling Ca2+ for Cancer Cell Proliferation and Survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Patterson, R.L.; van Rossum, D.B.; Nikolaidis, N.; Gill, D.L.; Snyder, S.H. Phospholipase C-γ: Diverse Roles in Receptor-Mediated Calcium Signaling. Trends Biochem. Sci. 2005, 30, 688–697. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Győrffy, B.; Klec, C.; Sokolowski, A.A.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. UCP2 and PRMT1 Are Key Prognostic Markers for Lung Carcinoma Patients. Oncotarget 2017, 8, 80278–80285. [Google Scholar] [CrossRef] [PubMed]
- Luby, A.; Alves-Guerra, M.-C. Targeting Metabolism to Control Immune Responses in Cancer and Improve Checkpoint Blockade Immunotherapy. Cancers 2021, 13, 5912. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shi, M.; Wu, Q.; Wei, W.; Sun, S.; Zhu, S. Identification of UCP1 and UCP2 as Potential Prognostic Markers in Breast Cancer: A Study Based on Immunohistochemical Analysis and Bioinformatics. Front. Cell Dev. Biol. 2022, 10, 891731. [Google Scholar] [CrossRef]
- Rupprecht, A.; Bräuer, A.U.; Smorodchenko, A.; Goyn, J.; Hilse, K.E.; Shabalina, I.G.; Infante-Duarte, C.; Pohl, E.E. Quantification of Uncoupling Protein 2 Reveals Its Main Expression in Immune Cells and Selective Up-Regulation during T-Cell Proliferation. PLoS ONE 2012, 7, e41406. [Google Scholar] [CrossRef]
- Chaudhuri, L.; Srivastava, R.K.; Kos, F.; Shrikant, P.A. Uncoupling Protein 2 Regulates Metabolic Reprogramming and Fate of Antigen-Stimulated CD8+ T Cells. Cancer Immunol. Immunother. 2016, 65, 869–874. [Google Scholar] [CrossRef]
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 Inhibition Induces ROS/Akt/MTOR Axis: Role of GAPDH Nuclear Translocation in Genipin/Everolimus Anticancer Synergism. Free Radic. Biol. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef]
- Katzung, B.G. (Ed.) Basic & Clinical Pharmacology, 14th ed.; A Lange Medical Book; McGraw-Hill Education: New York, NY, USA, 2018; ISBN 978-1-259-64115-2. [Google Scholar]
- Kachalaki, S.; Ebrahimi, M.; Mohamed Khosroshahi, L.; Mohammadinejad, S.; Baradaran, B. Cancer Chemoresistance; Biochemical and Molecular Aspects: A Brief Overview. Eur. J. Pharm. Sci. 2016, 89, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, M.; Fukuda, T.; Shimomura, M.; Inoue, Y.; Wada, T.; Tasaka, R.; Yasui, T.; Sumi, T. Expression of UCP2 Is Associated with Sensitivity to Platinum-Based Chemotherapy for Ovarian Serous Carcinoma. Oncol. Lett. 2018, 15, 9923–9928. [Google Scholar] [CrossRef]
- Derdak, Z.; Mark, N.M.; Beldi, G.; Robson, S.C.; Wands, J.R.; Baffy, G. The Mitochondrial Uncoupling Protein-2 Promotes Chemoresistance in Cancer Cells. Cancer Res. 2008, 68, 2813–2819. [Google Scholar] [CrossRef] [PubMed]
- Dalla Pozza, E.; Fiorini, C.; Dando, I.; Menegazzi, M.; Sgarbossa, A.; Costanzo, C.; Palmieri, M.; Donadelli, M. Role of Mitochondrial Uncoupling Protein 2 in Cancer Cell Resistance to Gemcitabine. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2012, 1823, 1856–1863. [Google Scholar] [CrossRef]
- Wang, M.; Li, G.; Yang, Z.; Wang, L.; Zhang, L.; Wang, T.; Zhang, Y.; Zhang, S.; Han, Y.; Jia, L. Uncoupling Protein 2 Downregulation by Hypoxia through Repression of Peroxisome Proliferator-Activated Receptor γ Promotes Chemoresistance of Non-Small Cell Lung Cancer. Oncotarget 2016, 8, 8083–8094. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-P.; Lo, Y.-C.; Yan, J.-J.; Liao, I.-C.; Tsai, P.-J.; Wang, H.-C.; Yeh, H.-H.; Lin, C.-C.; Chen, H.H.W.; Lai, W.-W.; et al. Mitochondrial uncoupling protein 2 regulates the effects of paclitaxel on Stat3 activation and cellular survival in lung cancer cells. Carcinogenesis 2012, 33, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, J.; Rupprecht, A.; Zimmermann, L.; Moschinger, M.; Rokitskaya, T.I.; Antonenko, Y.N.; Gille, L.; Fedorova, M.; Pohl, E.E. Molecular Mechanisms Responsible for Pharmacological Effects of Genipin on Mitochondrial Proteins. Biophys. J. 2019, 117, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Shen, H.; Tang, F.R.; Arfuso, F.; Rajesh, M.; Wang, L.; Kumar, A.P.; Bian, J.; Goh, B.C.; Bishayee, A.; et al. Potential Role of Genipin in Cancer Therapy. Pharmacol. Res. 2018, 133, 195–200. [Google Scholar] [CrossRef]
- Cho, Y.S. Genipin, an Inhibitor of UCP2 as a Promising New Anticancer Agent: A Review of the Literature. Int. J. Mol. Sci. 2022, 23, 5637. [Google Scholar] [CrossRef]
- Ayyasamy, V.; Owens, K.M.; Desouki, M.M.; Liang, P.; Bakin, A.; Thangaraj, K.; Buchsbaum, D.J.; LoBuglio, A.F.; Singh, K.K. Cellular Model of Warburg Effect Identifies Tumor Promoting Function of UCP2 in Breast Cancer and Its Suppression by Genipin. PLoS ONE 2011, 6, e24792. [Google Scholar] [CrossRef]
- Ko, H.; Kim, J.M.; Kim, S.-J.; Shim, S.H.; Ha, C.H.; Chang, H.I. Induction of Apoptosis by Genipin Inhibits Cell Proliferation in AGS Human Gastric Cancer Cells via Egr1/P21 Signaling Pathway. Bioorg. Med. Chem. Lett. 2015, 25, 4191–4196. [Google Scholar] [CrossRef]
- Feng, Q.; Cao, H.; Xu, W.; Li, X.; Ren, Y.; Du, L. Apoptosis Induced by Genipin in Human Leukemia K562 Cells: Involvement of c-Jun N-Terminal Kinase in G2/M Arrest. Acta Pharm. Sin. 2011, 32, 519–527. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, M.; Tsao, S.-W.; Man, K.; Zhang, Z.; Feng, Y. Up-Regulation of TIMP-1 by Genipin Inhibits MMP-2 Activities and Suppresses the Metastatic Potential of Human Hepatocellular Carcinoma. PLoS ONE 2012, 7, e46318. [Google Scholar] [CrossRef]
- Cho, Y.S.; Lee, J.H.; Jung, K.-H.; Park, J.-W.; Moon, S.H.; Choe, Y.S.; Lee, K.-H. Molecular Mechanism of 18F-FDG Uptake Reduction Induced by Genipin in T47D Cancer Cell and Role of Uncoupling Protein-2 in Cancer Cell Glucose Metabolism. Nucl. Med. Biol. 2016, 43, 587–592. [Google Scholar] [CrossRef]
- Tan, H.-Y.; Wang, N.; Tsao, S.-W.; Che, C.-M.; Yuen, M.-F.; Feng, Y. Inhibition by Natural Compound Genipin on Tumour Associated Macrophages Reduces Growth of Hepatocellular Carcinoma. Oncotarget 2016, 7, 43792–43804. [Google Scholar] [CrossRef]
- Hua, J.; Zhang, Z.; Zhang, L.; Sun, Y.; Yuan, Y. UCP-2 Inhibitor Enhanced the Efficacy of Trastuzumab against HER2 Positive Breast Cancer Cells. Cancer Chemother. Pharm. 2021, 88, 633–642. [Google Scholar] [CrossRef]
- Lee, J.H.; Cho, Y.S.; Jung, K.-H.; Park, J.W.; Lee, K.-H. Genipin Enhances the Antitumor Effect of Elesclomol in A549 Lung Cancer Cells by Blocking Uncoupling Protein-2 and Stimulating Reactive Oxygen Species Production. Oncol. Lett. 2020, 20, 374. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Adjeitey, C.N.-K.; Harper, M.-E. Genipin-Induced Inhibition of Uncoupling Protein-2 Sensitizes Drug-Resistant Cancer Cells to Cytotoxic Agents. PLoS ONE 2010, 5, e13289. [Google Scholar] [CrossRef] [PubMed]
- Bugge, A.; Siersbæk, M.; Madsen, M.S.; Göndör, A.; Rougier, C.; Mandrup, S. A Novel Intronic Peroxisome Proliferator-Activated Receptor γ Enhancer in the Uncoupling Protein (UCP) 3 Gene as a Regulator of Both UCP2 and -3 Expression in Adipocytes. J. Biol. Chem. 2010, 285, 17310–17317. [Google Scholar] [CrossRef]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef] [PubMed]
- Laganà, A.S.; Vitale, S.G.; Nigro, A.; Sofo, V.; Salmeri, F.M.; Rossetti, P.; Rapisarda, A.M.C.; La Vignera, S.; Condorelli, R.A.; Rizzo, G.; et al. Pleiotropic Actions of Peroxisome Proliferator-Activated Receptors (PPARs) in Dysregulated Metabolic Homeostasis, Inflammation and Cancer: Current Evidence and Future Perspectives. Int. J. Mol. Sci. 2016, 17, 999. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, P.; Mueller, E.; Jones, D.; King, F.J.; DeAngelo, D.J.; Partridge, J.B.; Holden, S.A.; Chen, L.B.; Singer, S.; Fletcher, C.; et al. Differentiation and Reversal of Malignant Changes in Colon Cancer through PPARγ. Nat. Med. 1998, 4, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Bonofiglio, D.; Cione, E.; Qi, H.; Pingitore, A.; Perri, M.; Catalano, S.; Vizza, D.; Panno, M.L.; Genchi, G.; Fuqua, S.A.W.; et al. Combined Low Doses of PPARγ and RXR Ligands Trigger an Intrinsic Apoptotic Pathway in Human Breast Cancer Cells. Am. J. Pathol. 2009, 175, 1270–1280. [Google Scholar] [CrossRef]
- Motomura, W.; Okumura, T.; Takahashi, N.; Obara, T.; Kohgo, Y. Activation of Peroxisome Proliferator-Activated Receptor Gamma by Troglitazone Inhibits Cell Growth through the Increase of P27KiP1 in Human. Pancreatic Carcinoma Cells. Cancer Res. 2000, 60, 5558–5564. [Google Scholar]
- Bren-Mattison, Y.; Van Putten, V.; Chan, D.; Winn, R.; Geraci, M.W.; Nemenoff, R.A. Peroxisome Proliferator-Activated Receptor-γ (PPARγ) Inhibits Tumorigenesis by Reversing the Undifferentiated Phenotype of Metastatic Non-Small-Cell Lung Cancer Cells (NSCLC). Oncogene 2005, 24, 1412–1422. [Google Scholar] [CrossRef]
- Kubota, T.; Koshizuka, K.; Williamson, E.A.; Asou, H.; Said, J.W.; Holden, S.; Miyoshi, I.; Koeffler, H.P. Ligand for Peroxisome Proliferator-Activated Receptor Gamma (Troglitazone) Has Potent Antitumor Effect against Human Prostate Cancer Both in vitro and in vivo. Cancer Res. 1998, 58, 3344–3352. [Google Scholar] [PubMed]
- Kwon, K.A.; Yun, J.; Oh, S.Y.; Seo, B.-G.; Lee, S.; Lee, J.-H.; Kim, S.-H.; Choi, H.J.; Roh, M.S.; Kim, H.-J. Clinical Significance of Peroxisome Proliferator-Activated Receptor γ and TRAP220 in Patients with Operable Colorectal Cancer. Cancer Res. Treat. 2016, 48, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Iglesias, R.; Giralt, M. PPARs in the Control of Uncoupling Proteins Gene Expression. PPAR Res. 2007, 2007, 74364. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shen, W.; Li, X.; Feng, Y.; Qian, K.; Wang, G.; Gao, Y.; Xu, X.; Zhang, S.; Yue, L.; et al. The PPARγ agonist rosiglitazone enhances the radiosensitivity of human pancreatic cancer cells. Drug Design, Development and Therapy. DDDT 2020, 14, 3099–3110. [Google Scholar] [CrossRef]
- Lau, M.-F.; Chua, K.-H.; Sabaratnam, V.; Kuppusamy, U.R. Rosiglitazone Enhances the Apoptotic Effect of 5-Fluorouracil in Colorectal Cancer Cells with High-Glucose-Induced Glutathione. Sci. Prog. 2020, 103, 0036850419886448. [Google Scholar] [CrossRef]
- Wang, S.; Dougherty, E.J.; Danner, R.L. PPARγ Signaling and Emerging Opportunities for Improved Therapeutics. Pharmacol. Res. 2016, 111, 76–85. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and FDA Drug Safety Communication: Avandia (Rosiglitazone) Labels Now Contain Updated Information about Cardiovascular Risks and Use in Certain Patients. FDA. 2019. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-avandia-rosiglitazone-labels-now-contain-updated-information-about (accessed on 30 November 2022).




{kind=link}
{kind=link}
{kind=link}
| Disease | Experimental Model | UCP2 Status | Impact | Ref. |
|---|---|---|---|---|
| Type 2 diabetes | ob/ob mice | Ucp2−/− | Increased glucose-stimulated insulin secretion | [42] |
| Ucp2 siRNA | Oxidative stress imbalance | [91,92,93] | ||
| Atherosclerosis | Atherogenic diet Ldlr-/-mice | Ucp2−/− Bone marrow | Increased atherosclerotic lesions Increased invasion of macrophages into the intima Oxidative burst | [30] |
| Infections | Toxoplasma gondii | Ucp2−/− | Resistance to infection by increased production of ROS and pro- inflammatory molecules | [27] |
| Listeria | Ucp2−/− | [28] | ||
| Leishmaniasis | Ucp2−/− Ucp2 shRNA | [100,101] | ||
| Autoimmune diseases | Streptozotocin (type 1 diabetes) | Ucp2−/− | Higher disease scores Increased oxidative stress and inflammation | [29] |
| Experimental autoimmune encephalomyelitis | [54,106,107] | |||
| Cancer | Oxidative stress: | |||
| AOM/AOM-DSS / APCmin (colorectal cancer) | Ucp2−/− | Decreased protection against oxidative stress and increased colorectal tumorigenesis | [32,108] | |
| A549 cell line (lung cancer) | UCP2 overexpression | Reduction of ROS accumulation conferring anti-apoptotic properties | [109] | |
| A549 and PaCa44 cell line (lung and pancreatic cancer) | Ucp2 siRNA | ROS stimulate apoptosis derived from autophagy | [109,110] | |
| Glycolysis: | ||||
| B16F10 cell line (melanoma) | UCP2 overexpression | Less tumorigenic cells through down-regulation of glycolytic enzymes | [111] | |
| HuCCT1, TFK-1 and PaCa44 cell lines (bile duct and pancreatic cancer) | Ucp2 siRNA | AMPK activation decreases glycolytic activity and therefore the cell invasiveness | [112,113] | |
| Glutaminolysis: | ||||
| HPB-ALL cell line (leukemia) | Ucp2 CRISPR | Reduction of oxygen consumption Shift of metabolism to glycolysis Low nucleotide synthesis Decreased cell proliferation | [114] | |
| Patu8988T, Panc1 and BxPC3 cell lines (pancreatic cancer) | Ucp2 shRNA | [34] | ||
| Ca2+ signaling: | ||||
| JB6 P+ and HeLa cell lines (skin and cervix cancer) | UCP2 overexpression | Increased calcium activity stimulates ATP production Long-term mitochondrial dysfunction | [115,116] | |
| Immune response: | ||||
| Xenografts in mice with B16-OVA and YUMM1 cell lines (melanoma) | UCP2 overexpression | Better prognosis Infiltration of CD8+ T cells and cDC1 cells Improved response to immunotherapy | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luby, A.; Alves-Guerra, M.-C. UCP2 as a Cancer Target through Energy Metabolism and Oxidative Stress Control. Int. J. Mol. Sci. 2022, 23, 15077. https://doi.org/10.3390/ijms232315077
Luby A, Alves-Guerra M-C. UCP2 as a Cancer Target through Energy Metabolism and Oxidative Stress Control. International Journal of Molecular Sciences. 2022; 23(23):15077. https://doi.org/10.3390/ijms232315077
Chicago/Turabian StyleLuby, Angèle, and Marie-Clotilde Alves-Guerra. 2022. "UCP2 as a Cancer Target through Energy Metabolism and Oxidative Stress Control" International Journal of Molecular Sciences 23, no. 23: 15077. https://doi.org/10.3390/ijms232315077
APA StyleLuby, A., & Alves-Guerra, M.-C. (2022). UCP2 as a Cancer Target through Energy Metabolism and Oxidative Stress Control. International Journal of Molecular Sciences, 23(23), 15077. https://doi.org/10.3390/ijms232315077