
SecA2 Associates with Translating Ribosomes and Contributes to the Secretion of Potent IFN-β Inducing RNAs

 , , and
, , and
Abstract
:1. Introduction
2. Results
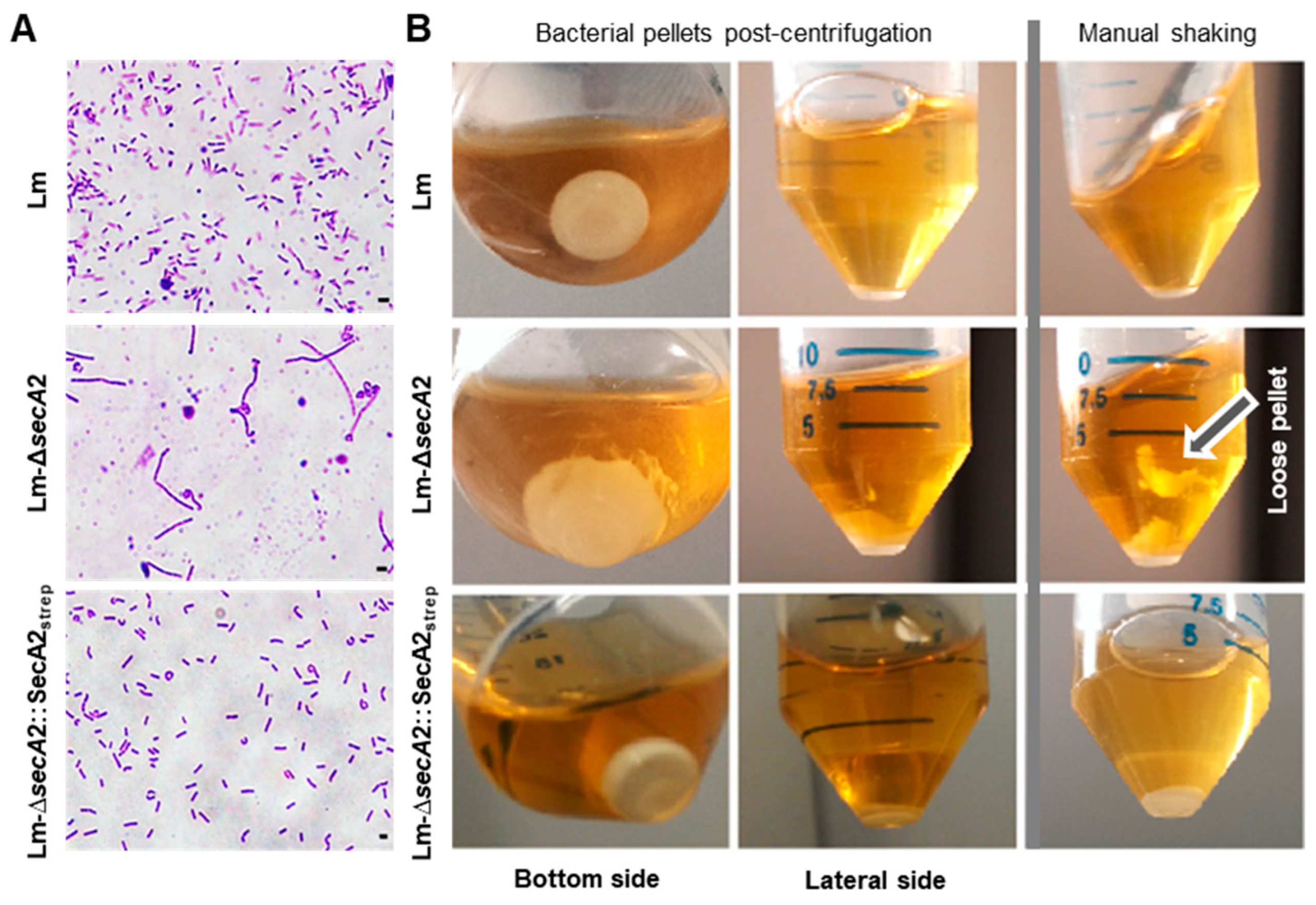
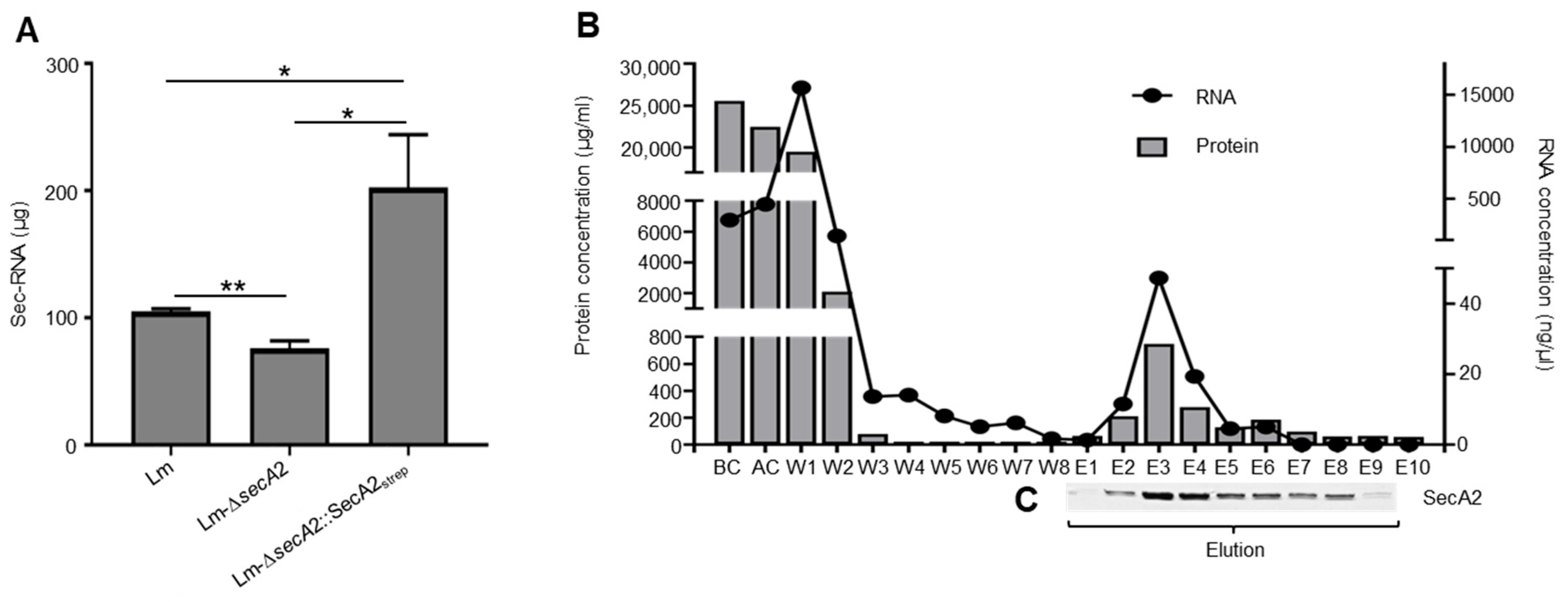
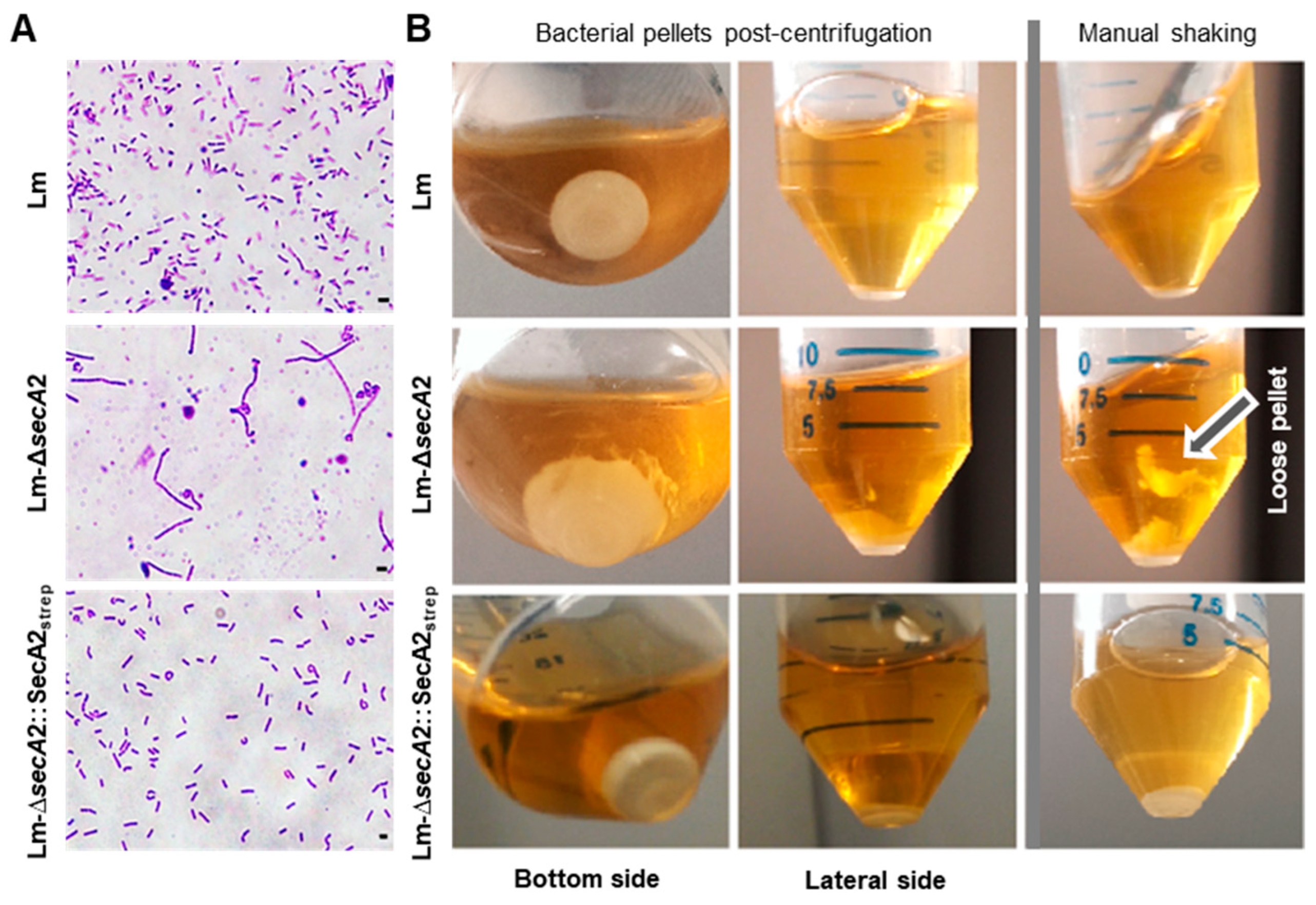
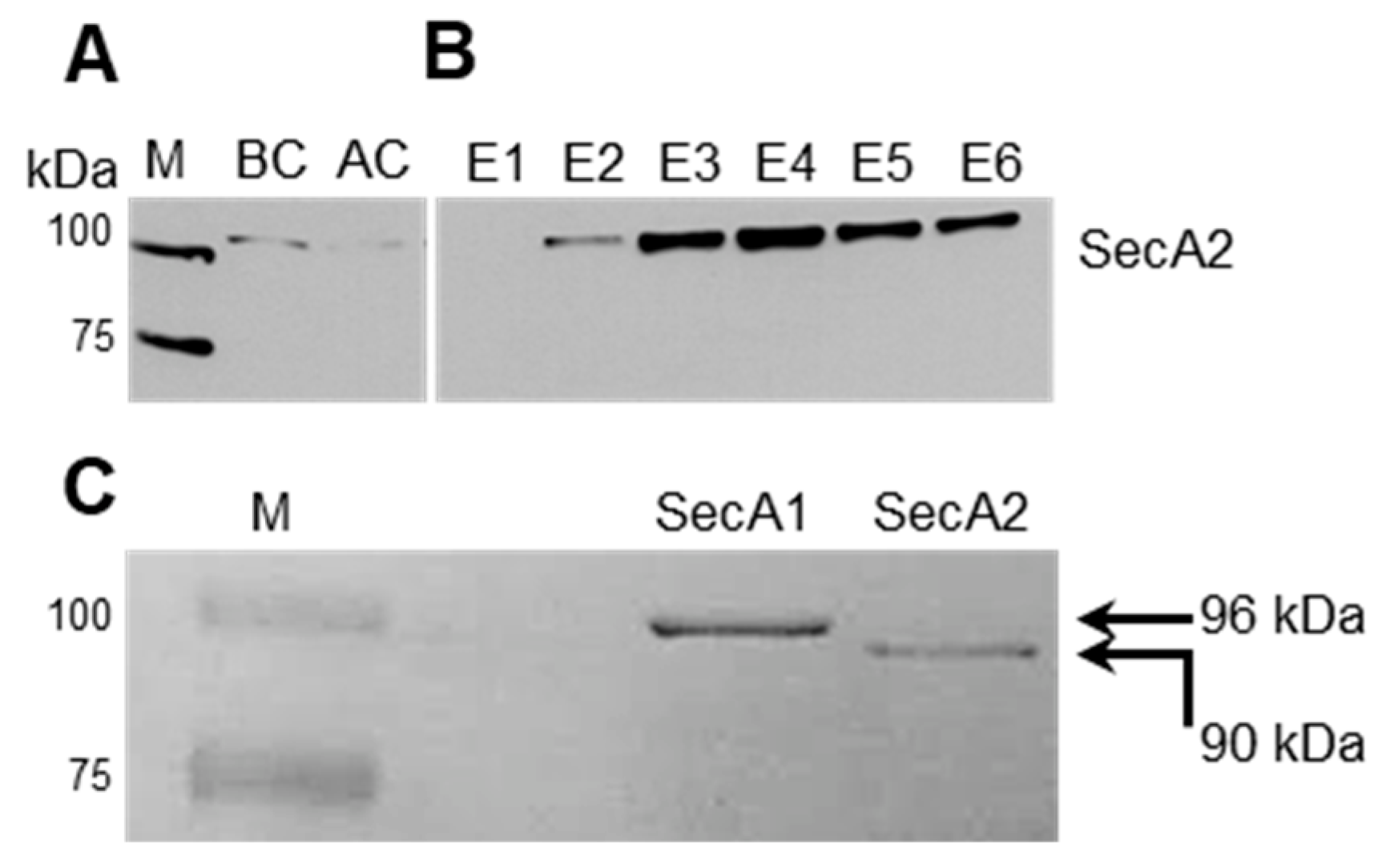
2.1. Production of Tagged and Functional SecA2 in ∆secA2 Deletion Mutant
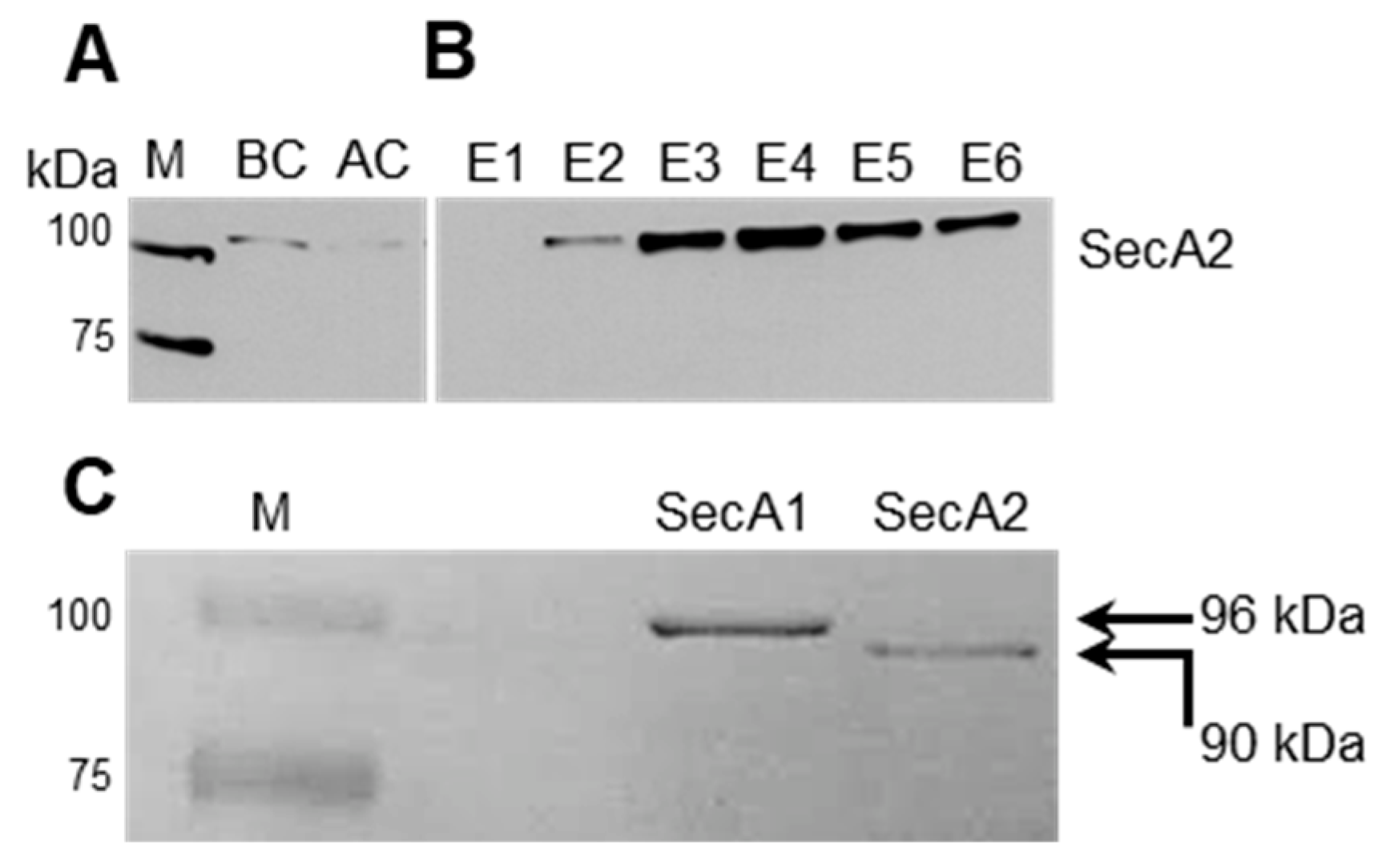
2.2. Affinity-Based Purification of Tagged SecA2
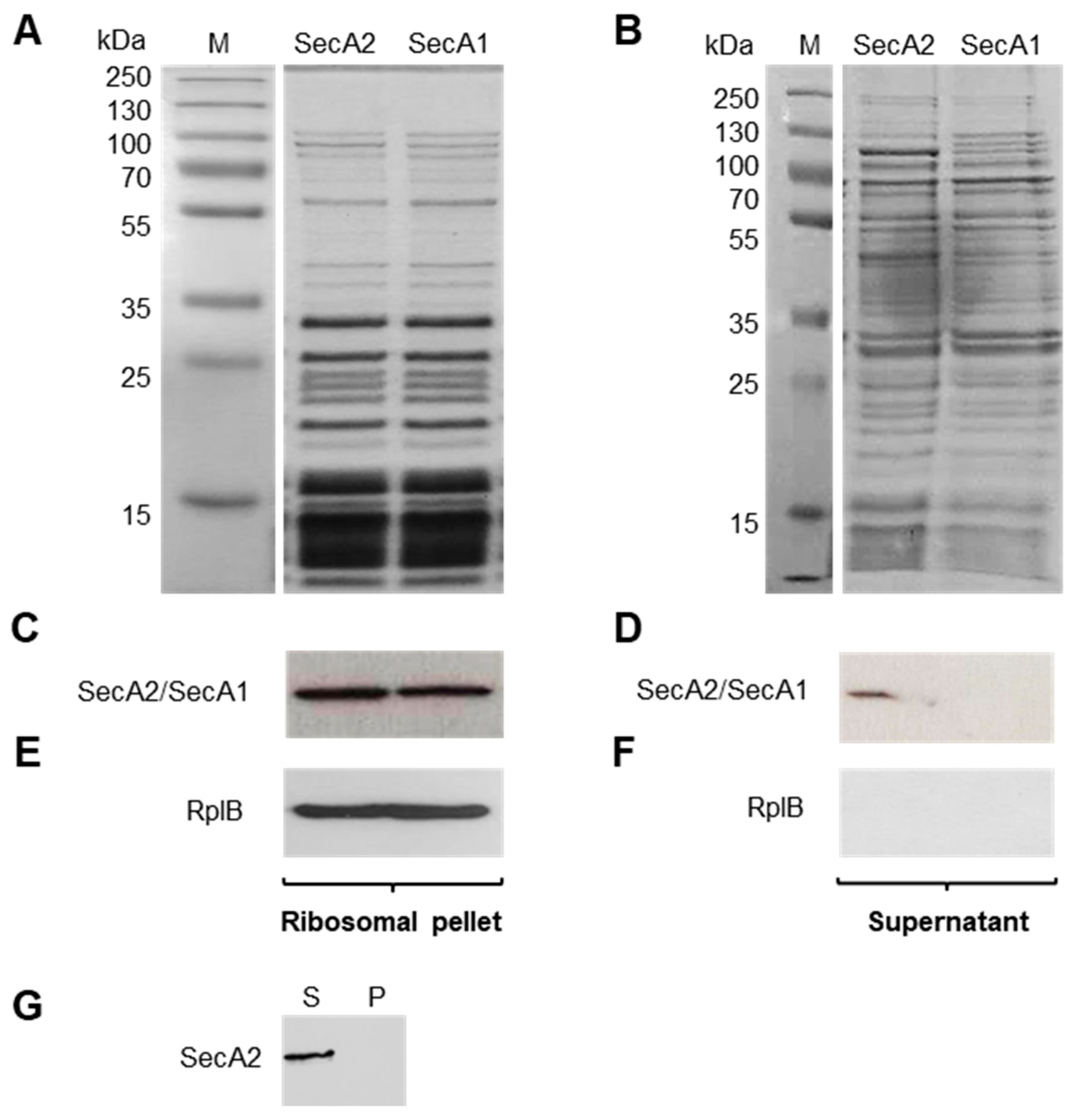
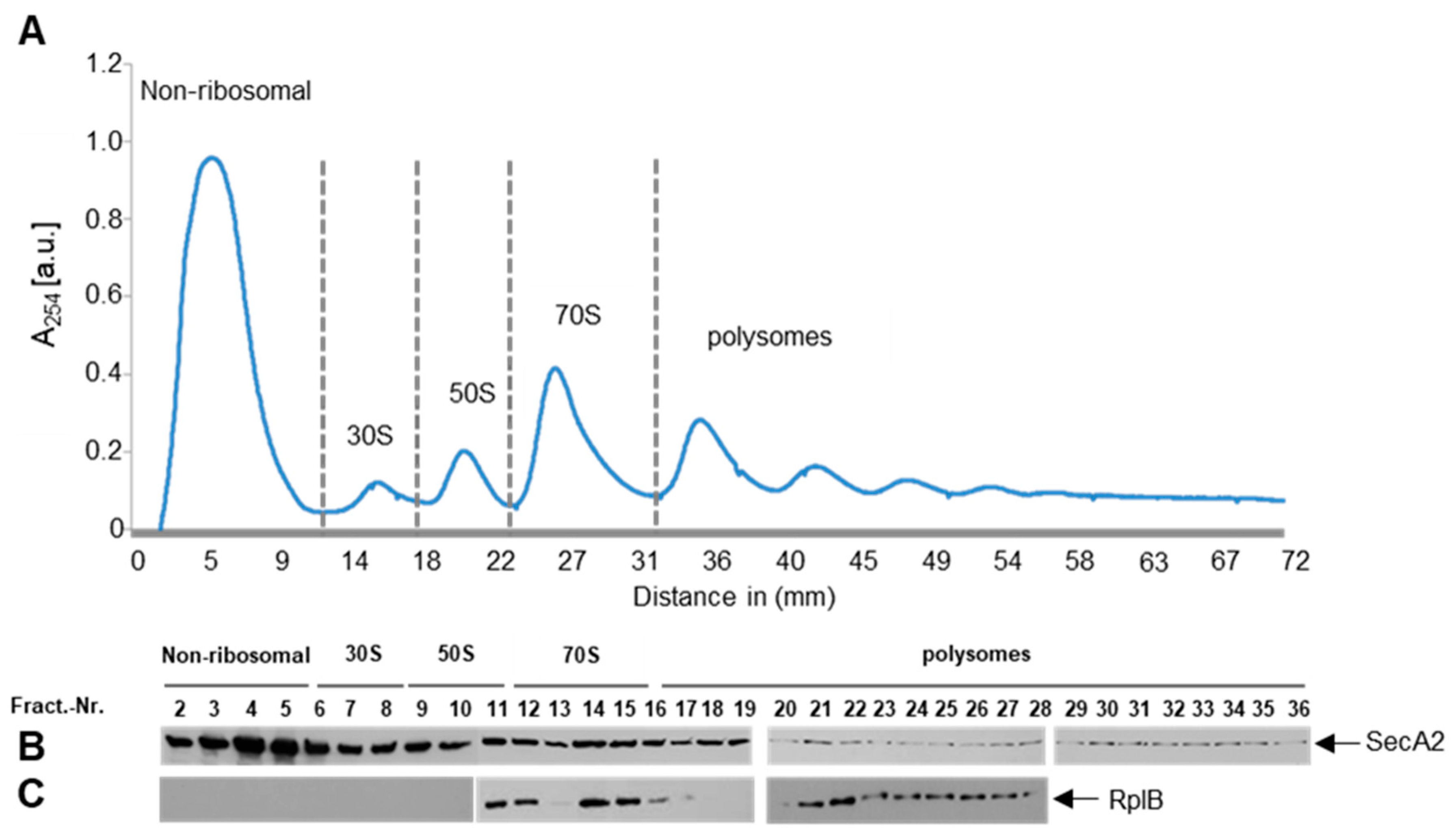
2.3. SecA2 Co-Sediments with Ribosomes
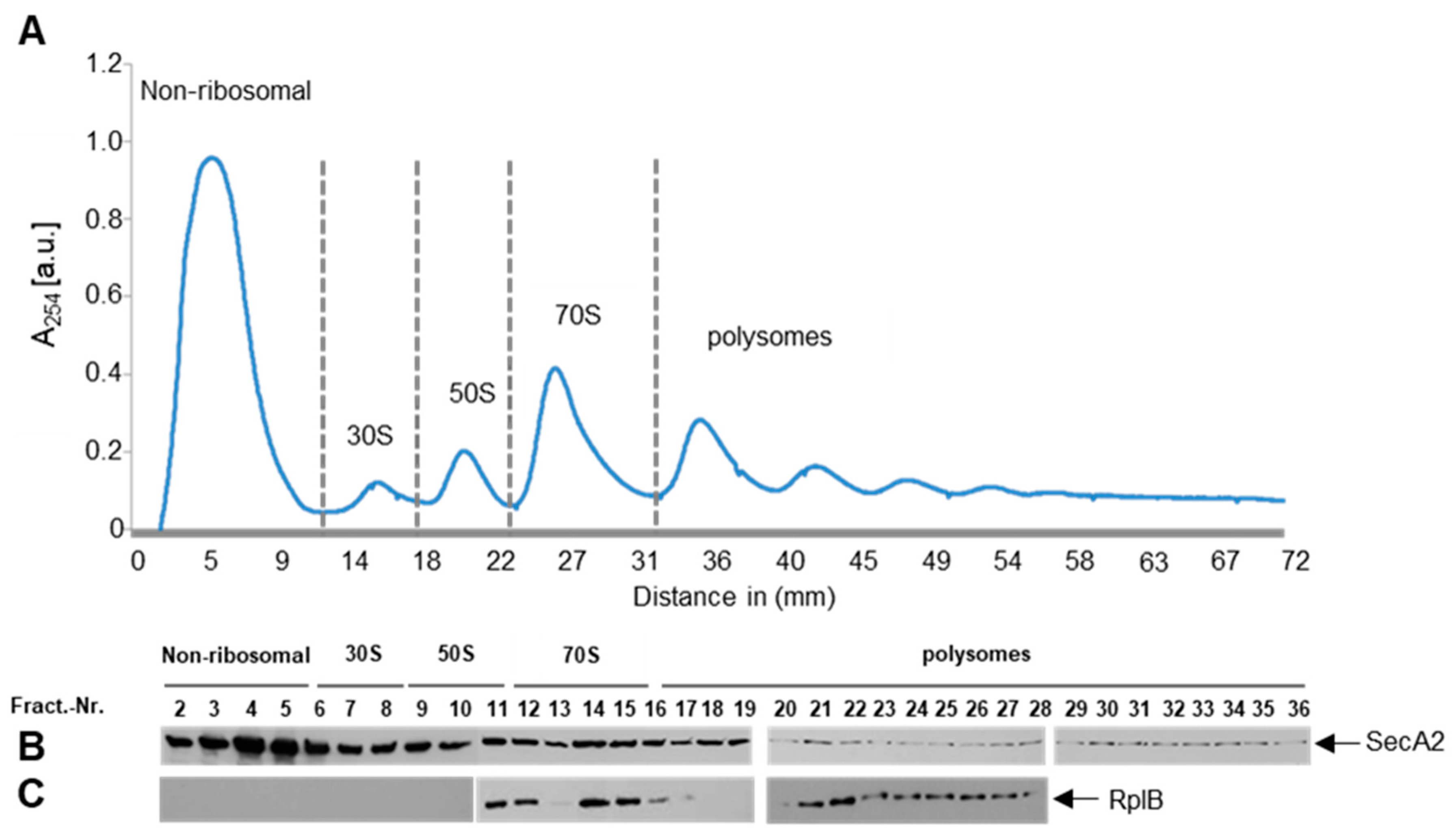
2.4. SecA2 Interacts with Translating Ribosomes
2.5. Identification of SecA2 Co-Eluted Proteins
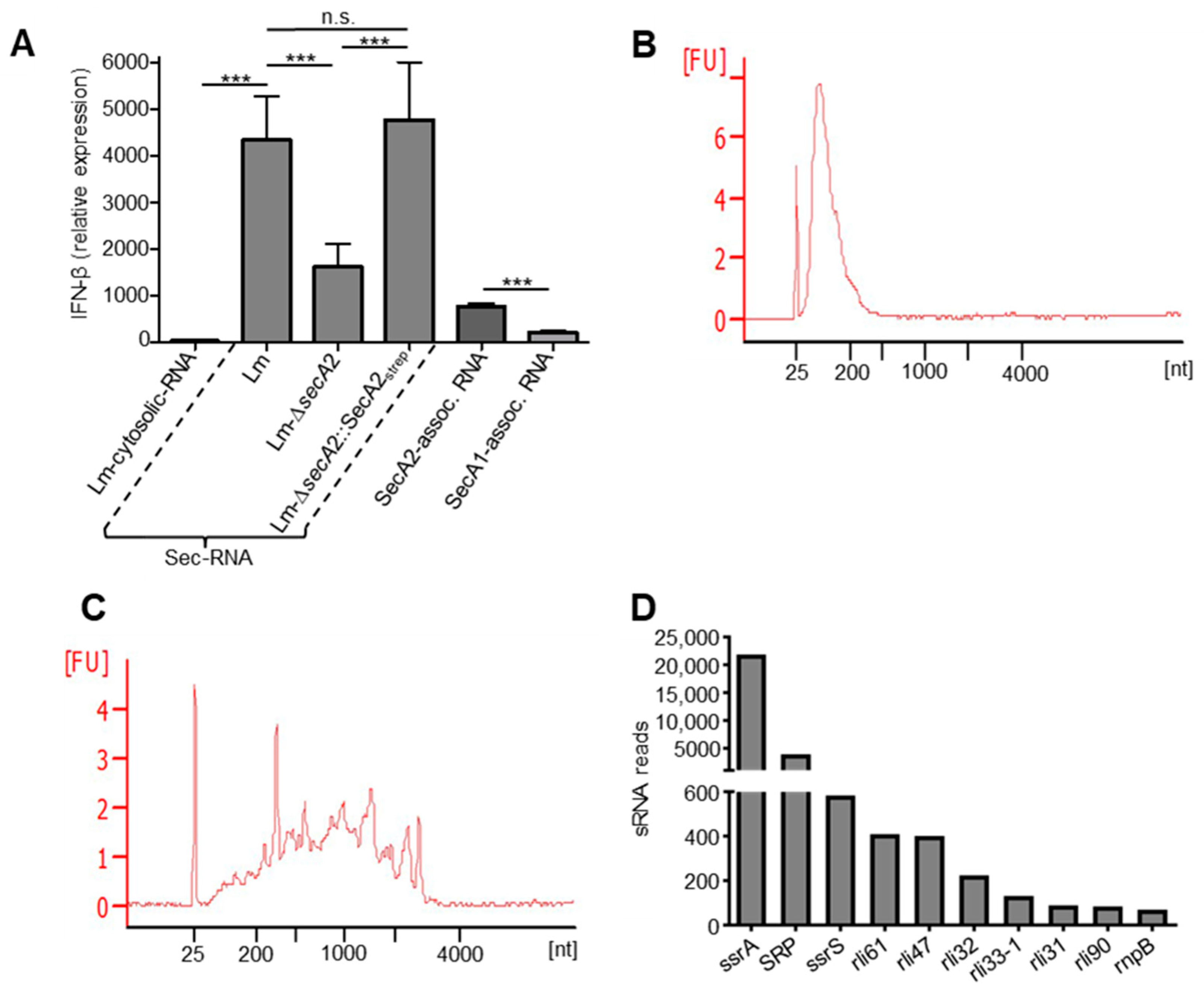
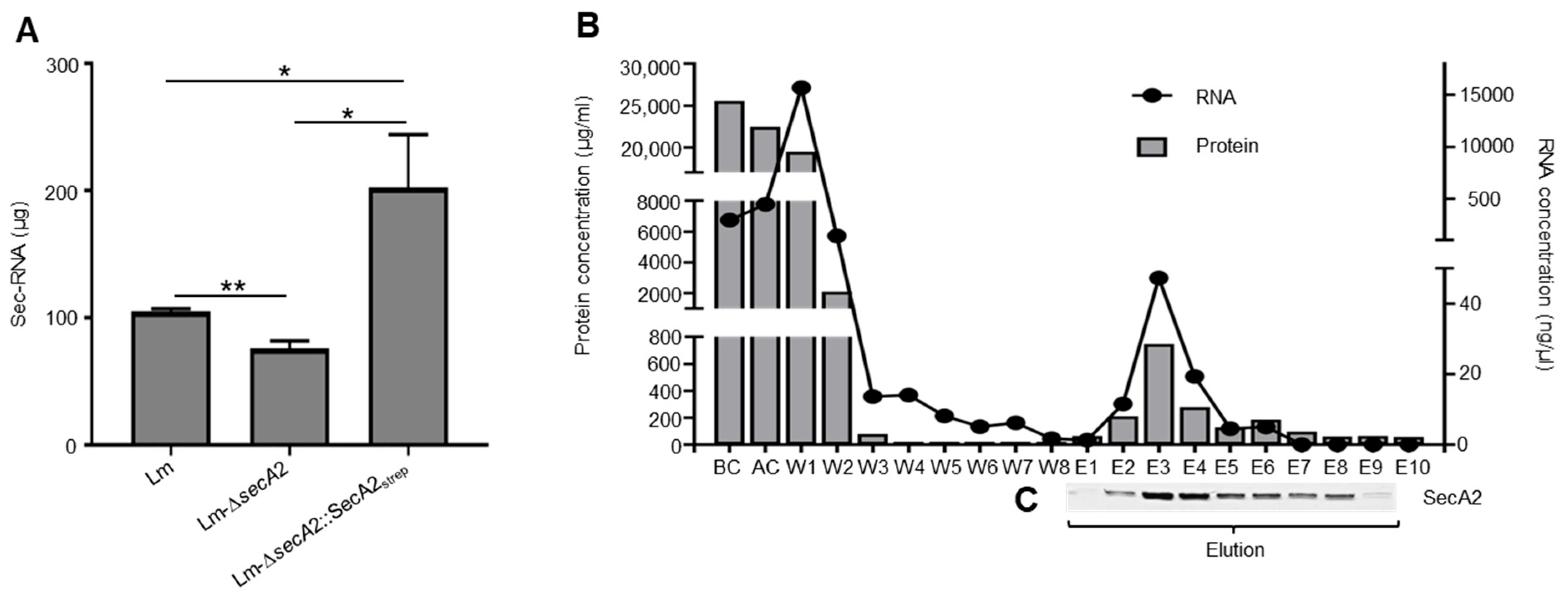
2.6. Association of RNA with SecA2
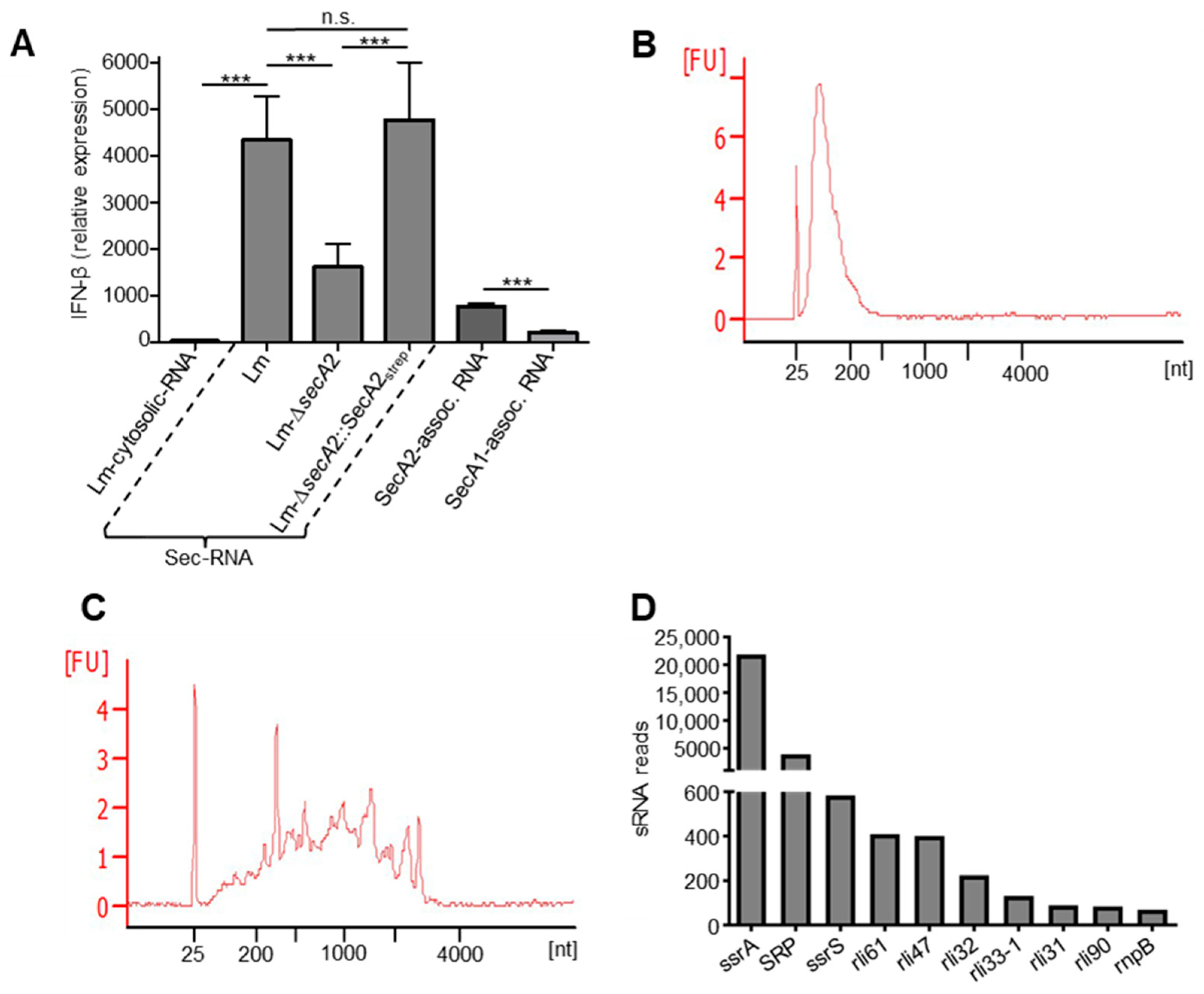
2.7. SecA2-Associated RNAs Are Potent Inducers of IFN-β
2.8. Detection of Small Non-Coding RNAs in SecA2-Associated RNAs
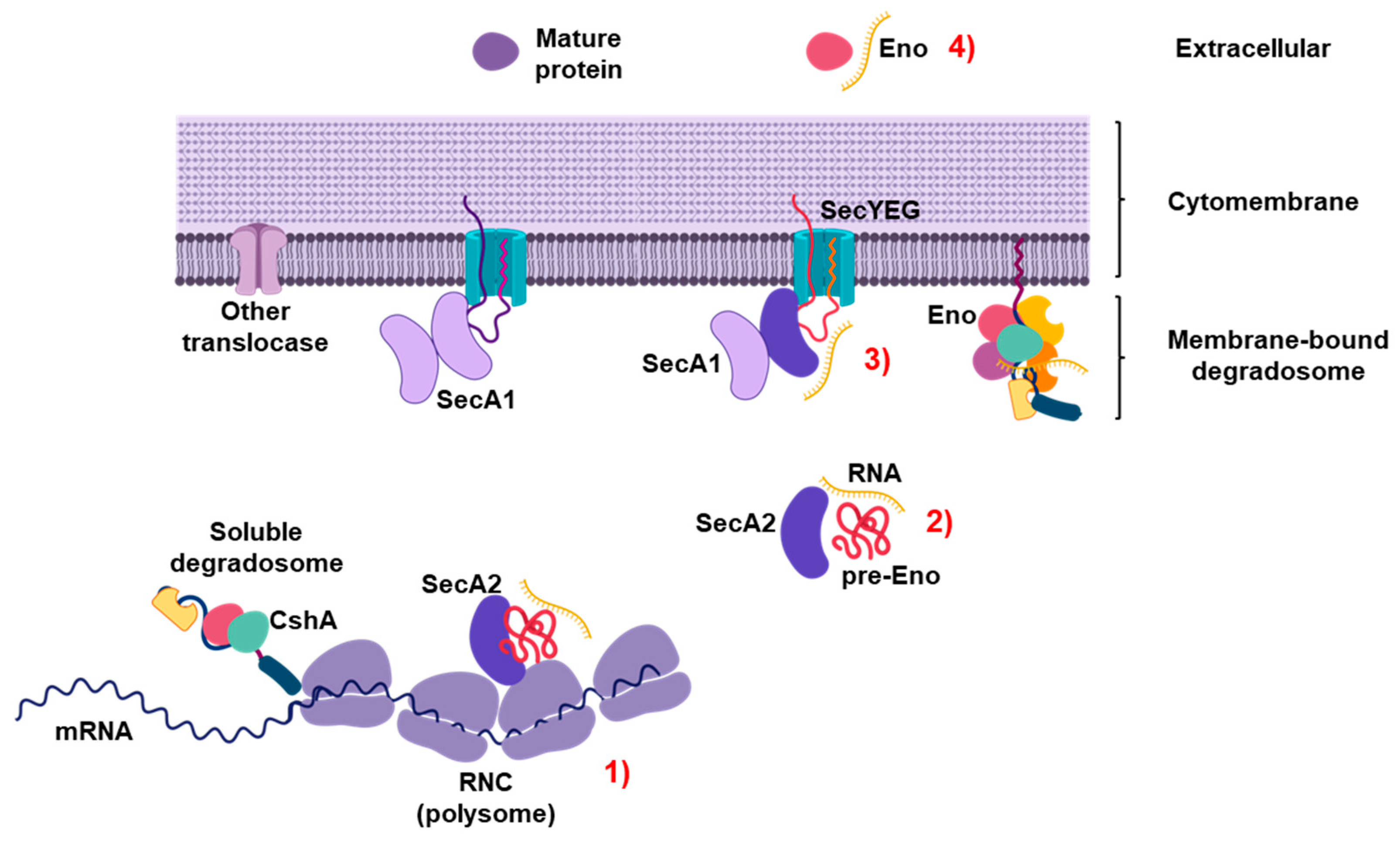
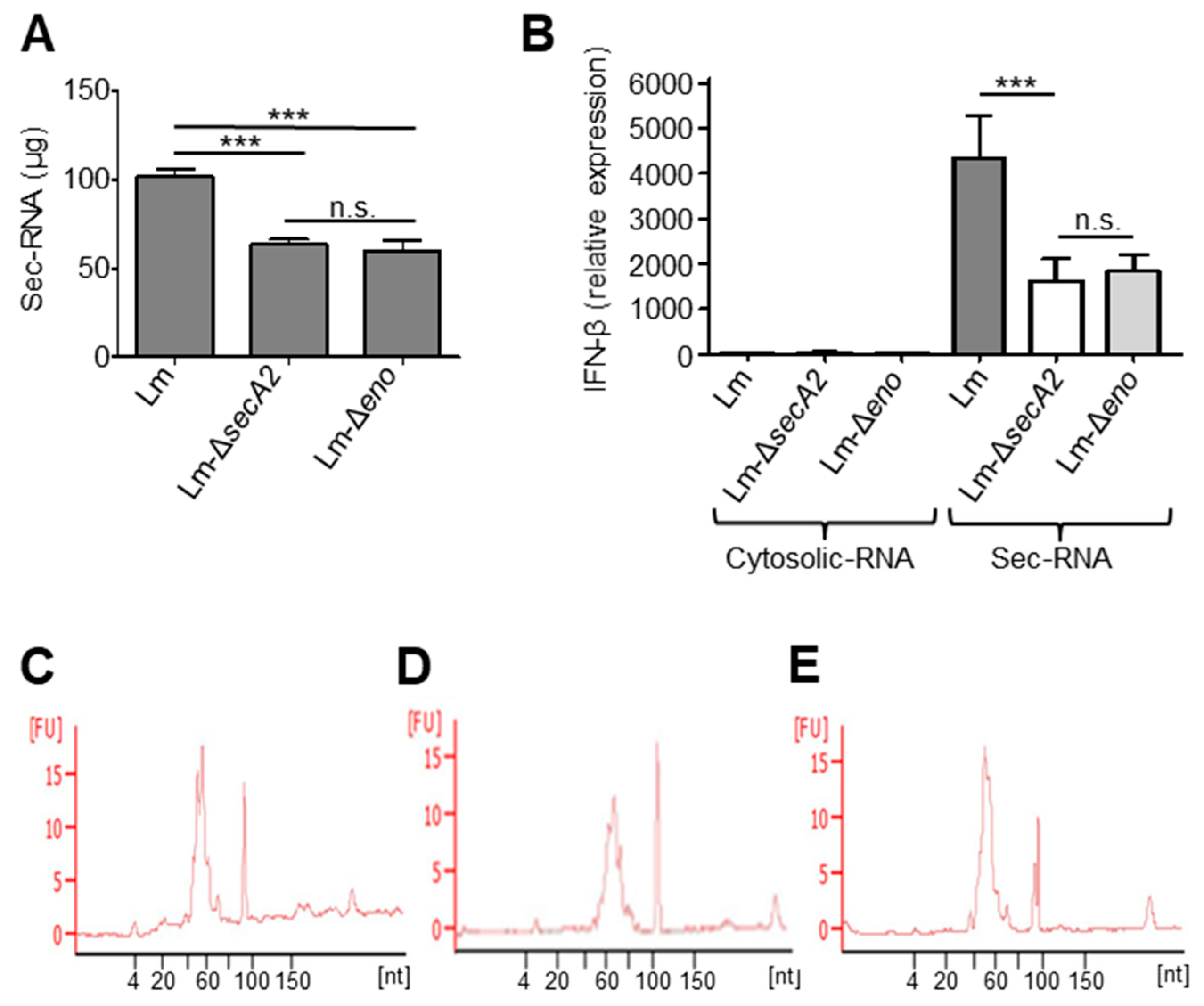
2.9. Deletion of Enolase Affects the Sec-RNA Amount and Composition in the Sec-RNA Fraction
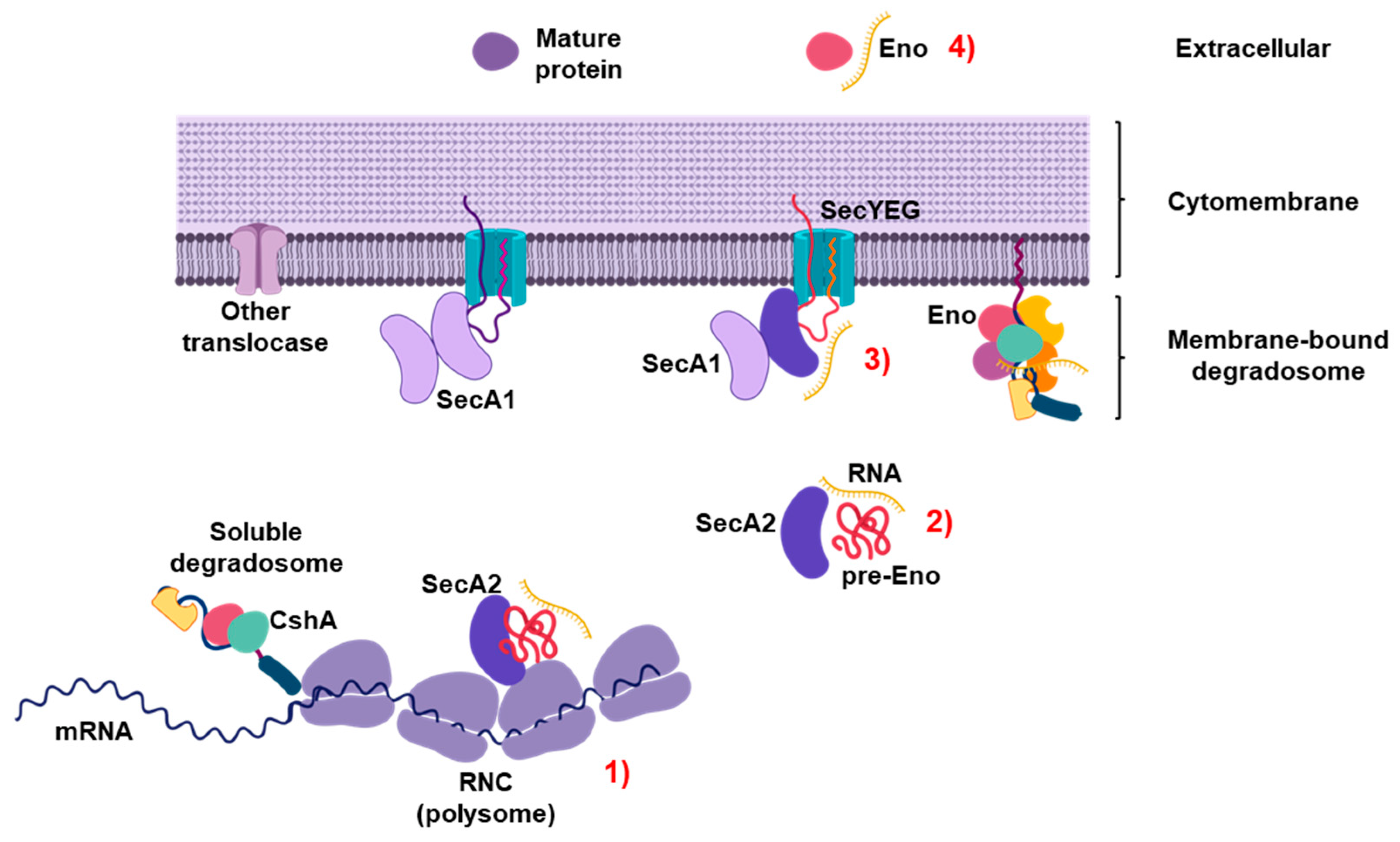
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Construction of Deletion Mutants
4.3. Construction of Affinity-Tagged SecA2 Expression Plasmid
4.4. Protein Purification
4.5. Matrix-Assisted Laser-Desorption Ionization Time-Of-Flight Mass Spectrometry (MALDI-TOF-MS)
4.6. Ribosome Purification
4.7. Co-Sedimentation Experiments
4.8. Western Blotting
4.9. Polysome Profiling Using Sucrose Gradient Centrifugation
4.10. Isolation of Cytosolic RNA
4.11. Isolation of Sec-RNA and SecA2/SecA1-Associated RNAs
4.12. cDNA Synthesis and RNA-Sequencing
4.13. Cell Culture and Transfection Assay
4.14. Quantitative Real-Time PCR
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Desvaux, M.; Hébraud, M. The protein secretion systems in Listeria. FEMS Microbiol. Rev. 2006, 30, 774–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desvaux, M.; Dumas, E.; Chafsey, I.; Chambon, C.; Hébraud, M. Comprehensive Appraisal of the Extracellular Proteins from a Monoderm Bacterium: Theoretical and Empirical Exoproteomes of Listeria monocytogenes EGD-e by Secretomics. J. Proteome Res. 2010, 9, 5076–5092. [Google Scholar] [CrossRef] [PubMed]
- Halbedel, S.; Reiss, S.; Hahn, B.; Albrecht, D.; Mannala, G.K.; Chakraborty, T.; Hain, T. A Systematic Proteomic Analysis of Listeria monocytogenes House-keeping Protein Secretion Systems. Mol. Cell. Proteom. 2014, 13, 3063–3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renier, S.; Micheau, P.; Talon, R.; Hébraud, M.; Desvaux, M. Subcellular localization of extracytoplasmic proteins in monoderm bacteria. Rational secretomics-based strategy for genomic and proteomic analyses. PLoS ONE 2012, 7, e42982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renier, S.; Chagnot, C.; Deschamps, J.; Caccia, N.; Szlavik, J.; Joyce, S.A.; Popowska, M.; Hill, C.; Knøchel, S.; Briandet, R.; et al. Inactivation of the SecA2 protein export pathway in Listeria monocytogenes promotes cell aggregation, impacts biofilm architecture and induces biofilm formation in environmental condition. Environ. Microbiol. 2014, 16, 1176–1192. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, H.; Fives-Taylor, P.M. Investigating the role of secA2 in secretion and glycosylation of a fimbrial adhesin in Streptococcus parasanguis FW213. Mol. Microbiol. 2004, 53, 843–856. [Google Scholar] [CrossRef]
- Sibbald, M.J.J.B.; Ziebandt, A.K.; Engelmann, S.; Hecker, M.; de Jong, A.; Harmsen, H.J.M.; Raangs, G.C.; Stokroos, I.; Arends, J.P.; Dubois, J.Y.F.; et al. Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol. Mol. Biol. Rev. 2006, 70, 755–788. [Google Scholar] [CrossRef] [Green Version]
- Cranford-Smith, T.; Huber, D. The way is the goal: How SecA transports proteins across the cytoplasmic membrane in bacteria. FEMS Microbiol. Lett. 2018, 365, fny093. [Google Scholar] [CrossRef] [Green Version]
- Rigel, N.W.; Braunstein, M. A new twist on an old pathway—Accessory Sec systems. Mol. Microbiol. 2008, 70, 271. [Google Scholar] [CrossRef] [Green Version]
- Feltcher, M.E.; Braunstein, M. Emerging themes in SecA2-mediated protein 690 export. Nat. Rev. Microbiol. 2012, 10, 779–789. [Google Scholar] [CrossRef]
- Bensing, B.A.; Seepersaud, R.; Yen, Y.T.; Sullam, P.M. Selective transport by SecA2. An expanding family of customized motor proteins. Biochim. Biophys. Acta 2014, 1843, 1674–1686. [Google Scholar] [CrossRef] [Green Version]
- Caspers, M.; Freudl, R. Corynebacterium glutamicum possesses two secA homologous genes that are essential for viability. Arch. Microbiol. 2008, 189, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Fagan, R.P.; Fairweather, N.F. Clostridium difficile Has Two Parallel and Essential Sec Secretion Systems. J. Biol. Chem. 2011, 286, 27483–27493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunstein, M.; Brown, A.M.; Kurtz, S.; Jacobs, W.R. Two nonredundant SecA homologues function in mycobacteria. J. Bacteriol. 2001, 183, 6979–6990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, L.L.; Portnoy, D.A. Identification of a second Listeria secA gene associated with protein secretion and the rough phenotype. Mol. Microbiol. 2002, 45, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Lenz, L.L.; Mohammadi, S.; Geissler, A.; Portnoy, D.A. SecA2-dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 12432–12437. [Google Scholar] [CrossRef] [Green Version]
- Machata, S.; Hain, T.; Rohde, M.; Chakraborty, T. Simultaneous deficiency of both MurA and p60 proteins generates a rough phenotype in Listeria monocytogenes. J. Bacteriol. 2005, 187, 8385–8394. [Google Scholar] [CrossRef] [Green Version]
- Chandrabos, C.; M’Homa Soudja, S.; Weinrick, B.; Gros, M.; Frangaj, A.; Rahmoun, M.; Jacobs, W.R.; Lauvau, G. The p60 and NamA autolysins from Listeria monocytogenes contribute to host colonization and induction of protective memory. Cell. Microbiol. 2015, 17, 147–163. [Google Scholar] [CrossRef] [Green Version]
- Pilgrim, S.; Kolb-Mäurer, A.; Gentschev, I.; Goebel, W.; Kuhn, M. Deletion of the gene encoding p60 in Listeria monocytogenes leads to abnormal cell division and loss of actin-based motility. Infect. Immun. 2003, 71, 3473–3484. [Google Scholar] [CrossRef] [Green Version]
- Muraille, E.; Narni-Mancinelli, E.; Gounon, P.; Bassand, D.; Glaichenhaus, N.; Lenz, L.L.; Lauvau, G. Cytosolic expression of SecA2 is a prerequisite for long-term protective immunity. Cell. Microbiol. 2007, 9, 1445–1454. [Google Scholar] [CrossRef]
- Chafsey, I.; Ostrowski, R.; Guilbaud, M.; Teixeira, P.; Herry, J.M.; Caccia, N.; Chambon, C.; Hébraud, M.; Azeredo, J.; Bellon-Fontaine, M.N.; et al. Deep impact of the inactivation of the SecA2-only protein export pathway on the proteosurfaceome of Listeria monocytogenes. J. Proteom. 2022, 250, 104388. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, Z.; Schlee, M.; Roth, S.; Mraheil, M.A.; Barchet, W.; Böttcher, J.; Hain, T.; Geiger, S.; Hayakawa, Y.; Fritz, J.H.; et al. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J. 2012, 31, 4153–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, R.; Teubner, L.; Schultze, T.; La Pietra, L.; Müller, C.; Gwozdzinski, K.; Pillich, H.; Hain, T.; Weber-Gerlach, M.; Panagiotidis, G.D.; et al. The secRNome of Listeria monocytogenes Harbors Small Noncoding RNAs That Are Potent Inducers of Beta Interferon. mBio 2019, 10, e01223-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stifter, S.A.; Feng, C.G. Interfering with immunity: Detrimental role of type I IFNs during infection. J. Immunol. 2015, 194, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Leimeister-Wächter, M.; Haffner, C.; Domann, E.; Goebel, W.; Chakraborty, T. Identification of a gene that positively regulates expression of listeriolysin, the major virulence factor of listeria monocytogenes. Proc. Natl. Acad. Sci. USA 1990, 87, 8336–8340. [Google Scholar] [CrossRef] [Green Version]
- Riedel, C.U.; Monk, I.R.; Casey, P.G.; Morrissey, D.; O’Sullivan, G.C.; Tangney, M.; Hill, C.; Gahan, C.G.M. Improved luciferase tagging system for Listeria monocytogenes allows real-time monitoring in vivo and in vitro. Appl. Environ. Microbiol. 2007, 73, 3091–3094. [Google Scholar] [CrossRef] [Green Version]
- Huber, D.; Rajagopalan, N.; Preissler, S.; Rocco, M.A.; Merz, F.; Kramer, G.; Bukau, B. SecA interacts with ribosomes in order to facilitate posttranslational translocation in bacteria. Mol. Cell 2011, 41, 343–353. [Google Scholar] [CrossRef]
- Huber, D.; Jamshad, M.; Hanmer, R.; Schibich, D.; Döring, K.; Marcomini, I.; Kramer, G.; Bukau, B. SecA Cotranslationally Interacts with Nascent Substrate Proteins In Vivo. J. Bcteriol. 2017, 199, e00622-16. [Google Scholar] [CrossRef] [Green Version]
- Henderson, B.; Martin, A. Bacterial virulence in the moonlight. Multitasking bacterial moonlighting proteins are virulence determinants in infectious disease. Infect. Immun. 2011, 79, 3476–3491. [Google Scholar] [CrossRef] [Green Version]
- Renier, S.; Chambon, C.; Viala, D.; Chagnot, C.; Hébraud, M.; Desvaux, M. Exoproteomic analysis of the SecA2-dependent secretion in Listeria monocytogenes EGD-e. J. Proteom. 2013, 80, 183–195. [Google Scholar] [CrossRef]
- Mraheil, M.A.; Billion, A.; Mohamed, W.; Mukherjee, K.; Kuenne, C.; Pischimarov, J.; Krawitz, C.; Retey, J.; Hartsch, T.; Chakraborty, T.; et al. The intracellular sRNA transcriptome of Listeria monocytogenes during growth in macrophages. Nucleic Acids Res. 2011, 39, 4235–4248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Pérez, L.; Depardón, F.; Fernández-Ramírez, F.; Sánchez-Trujillo, A.; Bermúdez-Crúz, R.M.; Dangott, L.; Montañez, C. α-Enolase binds to RNA. Biochimie 2011, 93, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Hentze, M.W.; Preiss, T. Metabolic Enzymes Enjoying New Partnerships as RNA-Binding Proteins. Trends Endocrinol. Metab. 2015, 26, 746–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewicz, D.; Bergmann, S.; Didiasova, M.; Giaimo, B.D.; Borggrefe, T.; Mieth, M.; Hocke, A.C.; Lochnit, G.; Schaefer, L.; Hammerschmidt, S.; et al. Host-derived extracellular RNA promotes adhesion of Streptococcus pneumoniae to endothelial and epithelial cells. Sci. Rep. 2016, 6, 2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasara, T.; Stephan, R. Evaluation of housekeeping genes in Listeria monocytogenes as potential internal control references for normalizing mRNA expression levels in stress adaptation models using real-time PCR. FEMS Microbiol. Lett. 2007, 269, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Schorey, J.S. Mycobacterium tuberculosis-induced IFN-β production requires cytosolic DNA and RNA sensing pathways. J. Exp. Med. 2018, 215, 2919–2935. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, R.M.; Saha, S.K.; Vaidya, S.A.; Bruhn, K.W.; Miranda, G.A.; Zarnegar, B.; Perry, A.K.; Nguyen, B.O.; Lane, T.F.; Taniguchi, T.; et al. Type I Interferon Production Enhances Susceptibility to Listeria monocytogenes Infection. J. Exp. Med. 2004, 200, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Auerbuch, V.; Brockstedt, D.G.; Meyer-Morse, N.; O’Riordan, M.; Portnoy, D.A. Mice Lacking the Type I Interferon Receptor Are Resistant to Listeria monocytogenes. J. Exp. Med. 2004, 200, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Osborne, S.E.; Sit, B.; Shaker, A.; Currie, E.; Tan, J.M.J.; van Rijn, J.; Higgins, D.E.; Brumell, J.H. Type I interferon promotes cell-to-cell spread of Listeria monocytogenes. Cell. Microbiol. 2017, 19, e12660. [Google Scholar] [CrossRef] [Green Version]
- Pagliuso, A.; Tham, T.N.; Allemand, E.; Robertin, S.; Dupuy, B.; Bertrand, Q.; Bécavin, C.; Koutero, M.; Najburg, V.; Nahori, M.A.; et al. An RNA-Binding Protein Secreted by a Bacterial Pathogen Modulates RIG-I Signaling. Cell Host 2019, 81, 823–835.e11. [Google Scholar] [CrossRef]
- Skerra, A.; Schmidt, T.G. Use of the Strep-Tag and streptavidin for detection and purification of recombinant proteins. Methods Enzymol. 2000, 326, 271–304. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.G.M.; Skerra, A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007, 2, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Py, B.; Higgins, C.F.; Krisch, H.M.; Carpousis, A.J. A DEAD-box RNA helicase in 789 the Escherichia coli RNA degradosome. Nature 1996, 381, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Miczak, A.; Kaberdin, V.R.; Wei, C.L.; Lin-Chao, S. Proteins associated with RNase E in a multicomponent ribonucleolytic complex. Proc. Natl. Acad. Sci. USA 1996, 93, 3865–3869. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Kawamoto, H.; Mizota, T.; Inada, T.; Aiba, H. Enolase in the RNA degradosome plays a crucial role in the rapid decay of glucose transporter mRNA in the response to phosphosugar stress in Escherichia coli. Mol. Microbiol. 2004, 54, 1063–1075. [Google Scholar] [CrossRef]
- Worrall, J.A.R.; Górna, M.; Crump, N.T.; Phillips, L.G.; Tuck, A.C.; Price, A.J.; Bavro, V.N.; Luisi, B.F. Reconstitution and Analysis of the Multienzyme Escherichia coli RNA Degradosome. J. Mol. Biol. 2008, 382, 870–883. [Google Scholar] [CrossRef]
- Roux, C.M.; DeMuth, J.P.; Dunman, P.M. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J. Bacteriol. 2011, 193, 5520–5526. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.J.G.; Moore, J.C.; Lane, J.D.; Wilson, R.; Pribul, P.K.; Younes, Z.N.; Dobson, R.J.; Everest, P.; Reason, A.J.; Redfern, J.M.; et al. Identification of major outer surface proteins of Streptococcus agalactiae. Infect. Immun. 2002, 70, 1254–1259. [Google Scholar] [CrossRef] [Green Version]
- Ling, E.; Feldman, G.; Portnoi, M.; Dagan, R.; Overweg, K.; Mulholland, F.; Chalifa- Caspi, V.; Wells, J.; Mizrachi-Nebenzahl, Y. Glycolytic enzymes associated with the cell surface of Streptococcus pneumoniae are antigenic in humans and elicit protective immune responses in the mouse. Clin. Exp. Immunol. 2004, 138, 290–298. [Google Scholar] [CrossRef]
- Dos Santos, P.T.; Menendez-Gil, P.; Sabharwal, D.; Christensen, J.H.; Brunhede, M.Z.; Lillebæk, E.M.S.; Kallipolitis, B.H. The Small Regulatory RNAs LhrC1–5 Contribute to the Response of Listeria monocytogenes to Heme Toxicity. Front. Microbiol. 2018, 9, 2023. [Google Scholar] [CrossRef]
- Toledo-Arana, A.; Dussurget, O.; Nikitas, G.; Sesto, N.; Guet-Revillet, H.; Balestrino, D.; Loh, E.; Gripenland, J.; Tiensuu, T.; Vaitkevicius, K.; et al. The Listeria transcriptional landscape from saprophytism to virulence. Nature 2009, 459, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Premaratne, R.J.; Lin, W.J.; Johnson, E.A. Development of an improved chemically defined minimal medium for Listeria monocytogenes. Appl. Environ. Microbiol. 1991, 57, 3046–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, T.; Leimeister-Wächter, M.; Domann, E.; Hartl, M.; Goebel, W.; Nichterlein, T.; Notermans, S. Coordinate regulation of virulence genes in Listeria monocytogenes requires the product of the prfA gene. J. Bacteriol. 1992, 174, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [Green Version]
- Kaval, K.G.; Hahn, B.; Tusamda, N.; Albrecht, D.; Halbedel, S. The PadR-like transcriptional regulator LftR ensures efficient invasion of Listeria monocytogenes into human host cells. Front. Microbiol. 2015, 6, 772. [Google Scholar] [CrossRef] [Green Version]
- Becker, A.H.; Oh, E.; Weissman, J.S.; Kramer, G.; Bukau, B. Selective ribosome profiling as a tool for studying the interaction of chaperones and targeting factors with nascent polypeptide chains and ribosomes. Nat. Protoc. 2013, 8, 2212–2239. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lmo Nr. | Gene Name | Protein Name | Function (COGs) * | emPAI * | mol % * |
|---|---|---|---|---|---|
| lmo0583 | secA2 | Protein translocase subunit SecA 2 | Intracellular trafficking and secretion | 5.81 | 6.75 |
| lmo2632 | rplC | 50S ribosomal protein L3 | Translation | 4.75 | 4.69 |
| lmo1934 | hup | DNA-binding protein HU | Replication. recombination and repair | 3.54 | 4.69 |
| lmo2629 | rplB | 50S ribosomal protein L2 | Translation | 3.20 | 3.87 |
| lmo2653 * | tuf | Elongation factor Tu | Translation | 2.78 | 3.69 |
| lmo1830 | Short-chain dehydrogenase | Lipid transport and metabolism; Secondary metabolites biosynthesis. transport and catabolism; General function prediction only; | 1.73 | 2.41 | |
| lmo1783 | rplT | 50S ribosomal protein L20 | Translation | 2.08 | 2.26 |
| lmo1072 | pycA | Pyruvate carboxylase | Energy production and conversion | 2.08 | 2.20 |
| lmo1356 | Acetyl-CoA carboxylase biotin carboxyl carrier protein | Lipid transport and metabolism | 1.70 | 2.16 | |
| lmo1330 | rpsO | 30S ribosomal protein S15 | Translation | 2.32 | 2.15 |
| lmo2624 | rpmC | 50S ribosomal protein L29 | Translation | 1.60 | 2.09 |
| lmo2016 | cspB | Cold shock-like protein CspLB | Transcription | 1.51 | 1.98 |
| lmo1473 * | dnaK | Chaperone protein DnaK | Posttranslational modification. protein turnover. chaperones | 1.72 | 1.96 |
| lmo2631 | rplD | 50S ribosomal protein L4 | Translation | 1.53 | 1.78 |
| lmo2459 | gap | Glyceraldehyde-3-phosphate dehydrogenase | Carbohydrate transport and metabolism | 1.25 | 1.60 |
| lmo1596 | rpsD | 30S ribosomal protein S4 | Translation | 1.47 | 1.37 |
| lmo1055 | PdhD | Dihydrolipoamide dehydrogenase | Energy production and conversion | 0.91 | 1.33 |
| lmo2625 | rplP | 50S ribosomal protein L16 | Translation | 1.00 | 1.29 |
| lmo1542 | rplU | 50S ribosomal protein L21 | Translation | 0.93 | 1.13 |
| lmo2455 * | eno | Enolase | Carbohydrate transport and metabolism | 0.77 | 1.10 |
| lmo1658 | rpsB | 30S ribosomal protein S2 | Translation | 0.94 | 1.09 |
| lmo0972 | dltC | D-alanine--poly(phosphoribitol) ligase subunit 2 | Cell wall/membrane biogenesis | 0.98 | 1.02 |
| lmo2613 | rplO | 50S ribosomal protein L15 | Translation | 0.75 | 0.90 |
| lmo2785 | kat | Catalase | Inorganic ion transport and metabolism | 0.65 | 0.90 |
| lmo1657 | tsf | Elongation factor Ts | Translation | 0.70 | 0.88 |
| lmo2654 | fus | Elongation factor G | Translation | 0.71 | 0.78 |
| lmo2630 | rplW | 50S ribosomal protein L23 | Translation | 0.62 | 0.77 |
| lmo2969 | groES | 10 kDa chaperonin | not in COGs | 0.46 | 0.75 |
| lmo2483 | hprK | Phosphocarrier protein HPr | Signal transduction mechanisms | 0.70 | 0.73 |
| lmo1920 | Copper chaperone CopZ | Function unknown | 0.52 | 0.64 | |
| lmo1364 | cspL | Cold shock protein CspA | Transcription | 0.47 | 0.61 |
| lmo0044 | rpsF | 30S ribosomal protein S6 | Translation | 0.47 | 0.61 |
| lmo2608 | rpsM | 30S ribosomal protein S13 | Translation | 0.39 | 0.51 |
| lmo2068 * | groEL | 60 kDa chaperonin | Posttranslational modification. protein turnover. chaperones | 0.40 | 0.50 |
| lmo2703 | similar to B. subtilis YaaK protein | Function unknown | 0.39 | 0.48 | |
| lmo2182 | Iron ABC transporter ATP-binding protein | Inorganic ion transport and metabolism; Coenzyme transport and metabolism; | 0.37 | 0.46 | |
| lmo1797 | rpsP | 30S ribosomal protein S16 | Translation | 0.39 | 0.44 |
| lmo0866 | cshA | DEAD-box ATP-dependent RNA helicase CshA | Replication. recombination and repair; Transcription; Translation; | 0.32 | 0.41 |
| lmo2219 | prsA2 | Foldase protein PrsA 2 | Posttranslational modification. protein turnover. chaperones | 0.30 | 0.34 |
| lmo1398 | recA | Protein RecA | Replication. recombination and repair | 0.30 | 0.34 |
| lmo0249 | rplA | Ribosomal protein (Fragment) | Translation | 0.26 | 0.34 |
| lmo2511 | hpf | Ribosome hibernation promoting factor | Translation | 0.26 | 0.32 |
| lmo1373 | 2-oxoisovalerate dehydrogenase | Energy production and conversion | 0.30 | 0.31 | |
| lmo1388 * | tcsA | CD4+ T-cell-stimulating antigen | General function prediction only | 0.25 | 0.30 |
| lmo0237 | gltX | Glutamate--tRNA ligase | Translation | 0.20 | 0.29 |
| lmo1785 | infC | Initiation factor IF-3 | Translation | 0.28 | 0.29 |
| lmo1267 | tig | Trigger factor | Posttranslational modification. protein turnover. chaperones | 0.19 | 0.28 |
| lmo1054 * | pdhC | Pyruvate dehydrogenase E2 component (Dihydrolipoamide acetyltransferase) | Energy production and conversion | 0.20 | 0.26 |
| lmo2615 | rpsE | 30S ribosomal protein S5 | Translation | 0.19 | 0.25 |
| lmo0135 | Putative oligopeptide ABC transporter. OppA | Amino acid transport and metabolism | 0.21 | 0.24 | |
| lmo2457 | tpiA | Triosephosphate isomerase | Carbohydrate transport and metabolism | 0.21 | 0.24 |
| lmo1579 | Alanine dehydrogenase | Amino acid transport and metabolism | 0.16 | 0.21 | |
| lmo1331 | pnpA | Polyribonucleotide nucleotidyltransferase | Translation | 0.19 | 0.21 |
| lmo2799 | GTP-binding protein YchF | Carbohydrate transport and metabolism | 0.23 | 0.21 | |
| lmo1664 | metK | S-adenosylmethionine synthase | Coenzyme transport and metabolism | 0.20 | 0.20 |
| lmo1439 * | sodA | Superoxide dismutase | Inorganic ion transport and metabolism | 0.13 | 0.20 |
| lmo2597 | rplM | 50S ribosomal protein L13 | Translation | 0.17 | 0.20 |
| lmo2510 | secA | Protein translocase subunit SecA 1 | Intracellular trafficking and secretion | 0.21 | 0.19 |
| lmo2618 | rpsH | 30S ribosomal protein S8 | Translation | 0.16 | 0.19 |
| lmo0539 | Tagatose 1.6-diphosphate aldolase | Carbohydrate transport and metabolism | 0.13 | 0.18 | |
| lmo2637 * | FMN-binding domain protein | Function unknown | 0.13 | 0.16 | |
| lmo2367 | pgi | Glucose-6-phosphate isomerase | Carbohydrate transport and metabolism | 0.11 | 0.16 |
| lmo1571 | pfkA | ATP-dependent 6-phosphofructokinase | Carbohydrate transport and metabolism | 0.12 | 0.15 |
| lmo0223 | cysK | Cysteine synthase | Amino acid transport and metabolism | 0.12 | 0.15 |
| lmo2620 | rplE | 50S ribosomal protein L5 | Translation | 0.12 | 0.15 |
| lmo0416 | Uncharacterized protein. DNA-binding. putative regulator | Transcription | 0.14 | 0.14 | |
| lmo1052 | pdhA | Pyruvate dehydrogenase (Acetyl-transferring) E1 component. alpha subunit | Energy production and conversion | 0.10 | 0.13 |
| lmo2458 | pgk | Phosphoglycerate kinase | Carbohydrate transport and metabolism | 0.09 | 0.13 |
| lmo2206 | clpB | Chaperone protein ClpB | Posttranslational modification. protein turnover. chaperones | 0.12 | 0.13 |
| lmo1357 | acetyl-CoA carboxylase biotin carboxylase subunit | Lipid transport and metabolism | 0.12 | 0.11 | |
| lmo1217 | Putative aminopeptidase. M42 family | Carbohydrate transport and metabolism | 0.09 | 0.11 | |
| lmo1928 | aroF | Chorismate synthase | Amino acid transport and metabolism | 0.08 | 0.10 |
| lmo2559 | pyrG | CTP synthase | Nucleotide transport and metabolism | 0.09 | 0.09 |
| lmo2456 | pgm | 2.3-bisphosphoglycerate-independent phosphoglycerate mutase | Carbohydrate transport and metabolism | 0.07 | 0.09 |
| lmo1938 | rpsA | 30S ribosomal protein S1 homolog | Translation | 0.08 | 0.08 |
| lmo1755 | gatA | Glutamyl-tRNA(Gln) amidotransferase subunit A | Translation | 0.07 | 0.08 |
| lmo0727 | glmS | Glutamine--fructose-6-phosphate aminotransferase [isomerizing] | Cell wall/membrane biogenesis | 0.07 | 0.08 |
| lmo1286 | parE | DNA topoisomerase 4 subunit B | Replication. recombination and repair | 0.06 | 0.07 |
| lmo0258 * | rpoB | RNA polymerase beta subunit | Transcription | 0.05 | 0.06 |
| lmo1003 | ptsI | Phosphoenolpyruvate-protein phosphotransferase | Carbohydrate transport and metabolism | 0.05 | 0.06 |
| lmo1634 * | lap | Aldehyde-alcohol dehydrogenase | Energy production and conversion | 0.05 | 0.06 |
| lmo0259 * | rpoC | DNA-directed RNA polymerase subunit beta’ | Transcription | 0.05 | 0.05 |
| lmo1504 | alaS | Alanine—tRNA ligase | Translation | 0.04 | 0.05 |
| lmo2019 | ileS | Isoleucine—tRNA ligase | Translation | 0.03 | 0.04 |
| lmo0007 | gyrA | DNA gyrase subunit A | Replication. recombination and repair | 0.03 | 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teubner, L.; Frantz, R.; La Pietra, L.; Hudel, M.; Bazant, J.; Lochnit, G.; Eismann, L.; Kramer, G.; Chakraborty, T.; Abu Mraheil, M. SecA2 Associates with Translating Ribosomes and Contributes to the Secretion of Potent IFN-β Inducing RNAs. Int. J. Mol. Sci. 2022, 23, 15021. https://doi.org/10.3390/ijms232315021
Teubner L, Frantz R, La Pietra L, Hudel M, Bazant J, Lochnit G, Eismann L, Kramer G, Chakraborty T, Abu Mraheil M. SecA2 Associates with Translating Ribosomes and Contributes to the Secretion of Potent IFN-β Inducing RNAs. International Journal of Molecular Sciences. 2022; 23(23):15021. https://doi.org/10.3390/ijms232315021
Chicago/Turabian StyleTeubner, Lisa, Renate Frantz, Luigi La Pietra, Martina Hudel, Jasmin Bazant, Günter Lochnit, Lena Eismann, Günter Kramer, Trinad Chakraborty, and Mobarak Abu Mraheil. 2022. "SecA2 Associates with Translating Ribosomes and Contributes to the Secretion of Potent IFN-β Inducing RNAs" International Journal of Molecular Sciences 23, no. 23: 15021. https://doi.org/10.3390/ijms232315021
APA StyleTeubner, L., Frantz, R., La Pietra, L., Hudel, M., Bazant, J., Lochnit, G., Eismann, L., Kramer, G., Chakraborty, T., & Abu Mraheil, M. (2022). SecA2 Associates with Translating Ribosomes and Contributes to the Secretion of Potent IFN-β Inducing RNAs. International Journal of Molecular Sciences, 23(23), 15021. https://doi.org/10.3390/ijms232315021