
B7 Family Members in Pancreatic Ductal Adenocarcinoma: Attractive Targets for Cancer Immunotherapy
Abstract
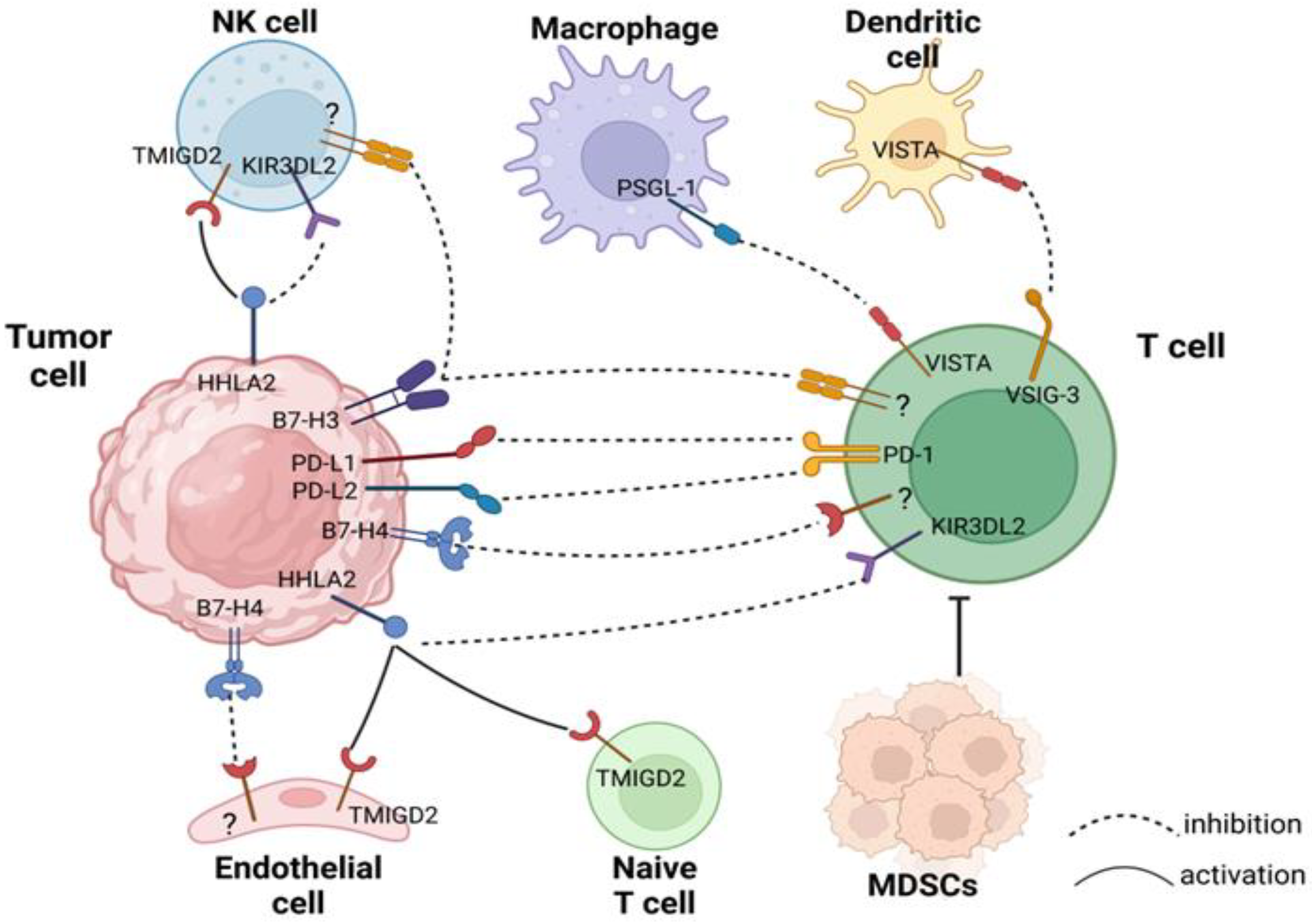
:1. Introduction
2. PD-L1
3. PD-L2
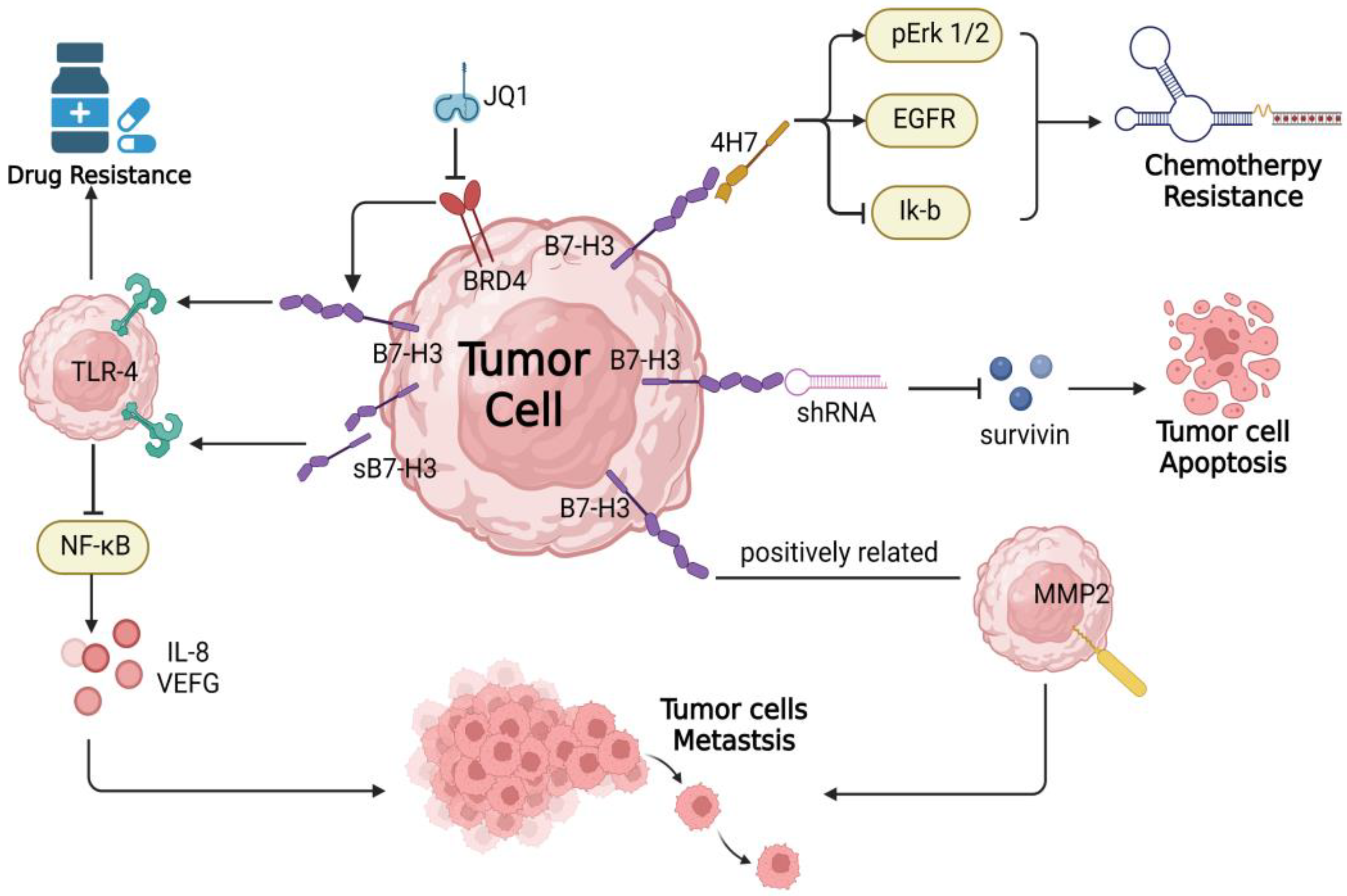
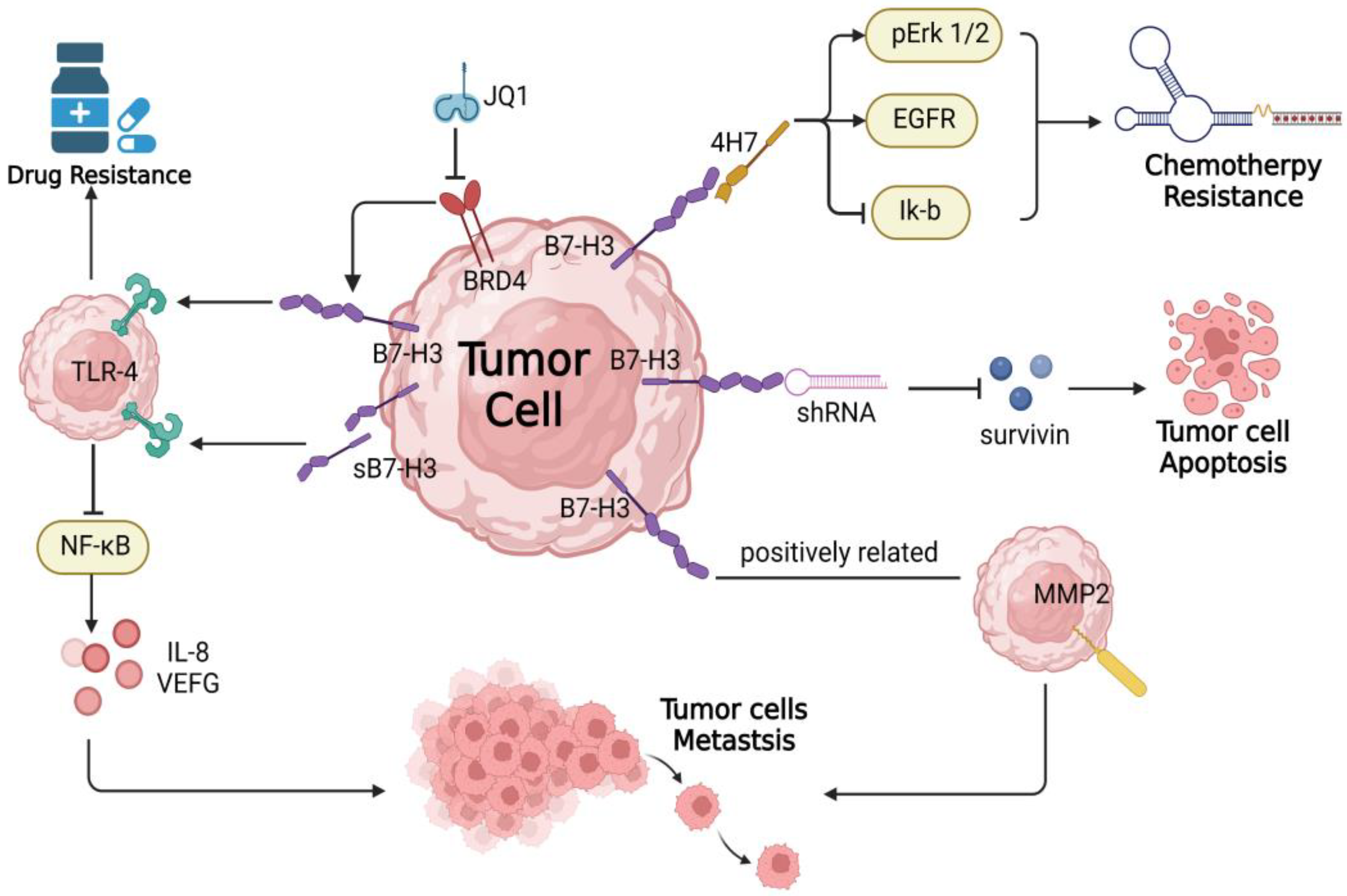
4. B7-H3
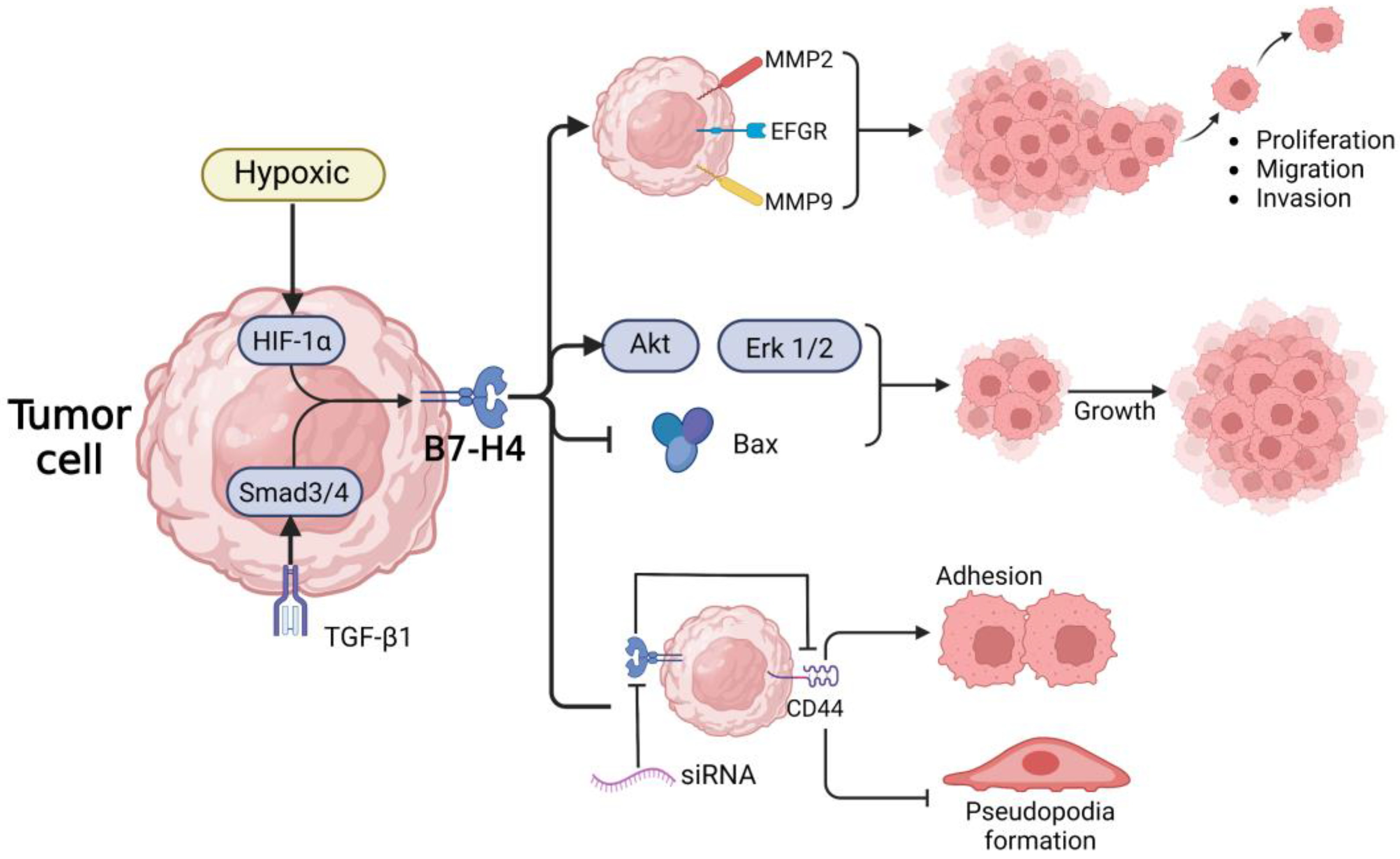
5. B7-H4
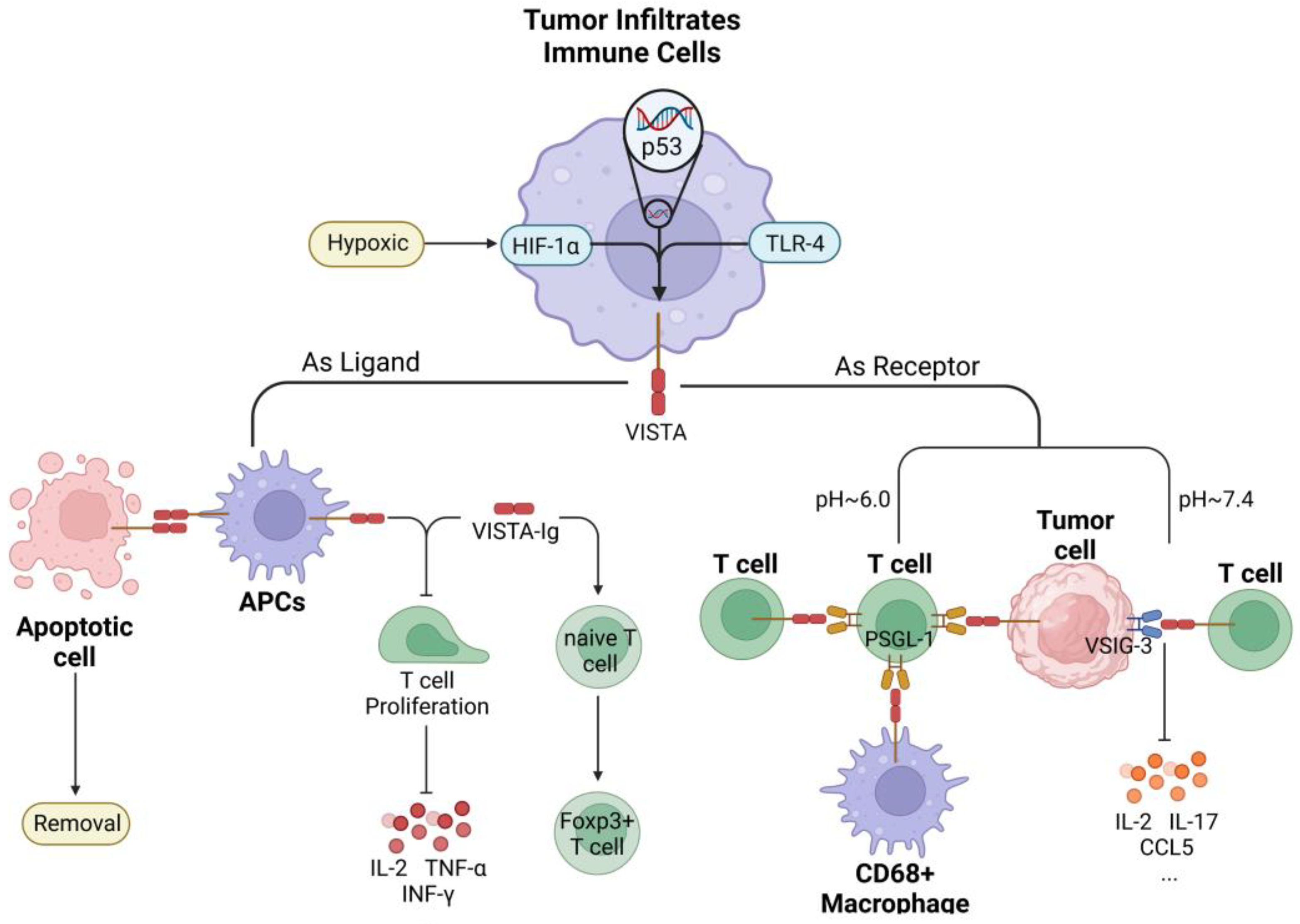
6. VISTA
7. HHLA2
8. Conclusions
9. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Alqahtani, A.; Noel, M.S. The Next Frontier in Pancreatic Cancer: Targeting the Tumor Immune Milieu and Molecular Pathways. Cancers 2022, 14, 2619. [Google Scholar] [CrossRef]
- Ullman, N.A.; Burchard, P.R.; Dunne, R.F.; Linehan, D.C. Immunologic Strategies in Pancreatic Cancer: Making Cold Tumors Hot. J. Clin. Oncol. 2022, 40, 2789–2805. [Google Scholar] [CrossRef]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental Determinants of Pancreatic Cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Blando, J.; Sharma, A.; Higa, M.G.; Zhao, H.; Vence, L.; Yadav, S.S.; Kim, J.; Sepulveda, A.M.; Sharp, M.; Maitra, A.; et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 1692–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, L.; Dong, C. New B7 Family Checkpoints in Human Cancers. Mol. Cancer Ther. 2017, 16, 1203–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolandi, N.; Derakhshani, A.; Hemmat, N.; Baghbanzadeh, A.; Asadzadeh, Z.; Afrashteh Nour, M.; Brunetti, O.; Bernardini, R.; Silvestris, N.; Baradaran, B. The Positive and Negative Immunoregulatory Role of B7 Family: Promising Novel Targets in Gastric Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 10719. [Google Scholar] [CrossRef]
- Di Federico, A.; Mosca, M.; Pagani, R.; Carloni, R.; Frega, G.; De Giglio, A.; Rizzo, A.; Ricci, D.; Tavolari, S.; Di Marco, M.; et al. Immunotherapy in Pancreatic Cancer: Why Do We Keep Failing? A Focus on Tumor Immune Microenvironment, Predictive Biomarkers and Treatment Outcomes. Cancers 2022, 14, 2429. [Google Scholar] [CrossRef] [PubMed]
- Sally, A.; McGowan, R.; Finn, K.; Moran, B.M. Current and Future Therapies for Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 2417. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, X.; Li, E.; Zhang, G.; Wang, X.; Tang, T.; Bai, X.; Liang, T. VISTA: An immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Kontos, F.; Michelakos, T.; Kurokawa, T.; Sadagopan, A.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. B7-H3: An Attractive Target for Antibody-based Immunotherapy. Clin. Cancer Res. 2021, 27, 1227–1235. [Google Scholar] [CrossRef]
- Qian, Y.; Hong, B.; Shen, L.; Wu, Z.; Yao, H.; Zhang, L. B7-H4 enhances oncogenicity and inhibits apoptosis in pancreatic cancer cells. Cell Tissue Res. 2013, 353, 139–151. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Hua, J.; Liu, J.; Liang, C.; Meng, Q.; Wei, M.; Zhang, B.; Yu, X.; Shi, S. A PD-L2-based immune marker signature helps to predict survival in resected pancreatic ductal adenocarcinoma. J. Immunother. Cancer 2019, 7, 233. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, L.; Huang, D.; Liu, J.; Qian, Y.; Deng, J.; Li, D.; Hu, Z.; Zhang, J.; Jiang, G.; Zheng, S. B7-H1 up-regulated expression in human pancreatic carcinoma tissue associates with tumor progression. J. Cancer Res. Clin. Oncol. 2008, 134, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Kruger, S.; Legenstein, M.L.; Rosgen, V.; Haas, M.; Modest, D.P.; Westphalen, C.B.; Ormanns, S.; Kirchner, T.; Heinemann, V.; Holdenrieder, S.; et al. Serum levels of soluble programmed death protein 1 (sPD-1) and soluble programmed death ligand 1 (sPD-L1) in advanced pancreatic cancer. Oncoimmunology 2017, 6, e1310358. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Bang, J.H.; Nam, A.R.; Eun Park, J.; Hua Jin, M.; Bang, Y.J.; Oh, D.Y. Prognostic implications of soluble programmed death-ligand 1 and its dynamics during chemotherapy in unresectable pancreatic cancer. Sci. Rep. 2019, 9, 11131. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.K.; Vandsemb, E.N.; Herbst, R.S.; Chen, L. Adaptive immune resistance at the tumour site: Mechanisms and therapeutic opportunities. Nat. Rev. Drug Discov. 2022, 21, 529–540. [Google Scholar] [CrossRef]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Song, X.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett. 2017, 407, 57–65. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Nagaraju, G.P.; Malla, R.R.; Basha, R.; Motofei, I.G. Contemporary clinical trials in pancreatic cancer immunotherapy targeting PD-1 and PD-L1. Semin. Cancer Biol. 2022, 86, 616–621. [Google Scholar] [CrossRef]
- Liu, L.; Huang, X.; Shi, F.; Song, J.; Guo, C.; Yang, J.; Liang, T.; Bai, X. Combination therapy for pancreatic cancer: Anti-PD-(L)1-based strategy. J. Exp. Clin. Cancer Res. 2022, 41, 56. [Google Scholar] [CrossRef]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Renouf, D.J.; Loree, J.M.; Knox, J.J.; Topham, J.T.; Kavan, P.; Jonker, D.; Welch, S.; Couture, F.; Lemay, F.; Tehfe, M.; et al. The CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nat. Commun. 2022, 13, 5020. [Google Scholar] [CrossRef]
- Wei, S.C.; Anang, N.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melisi, D.; Oh, D.Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and activity of the TGFbeta receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Immunother. Cancer 2021, 9, e002068. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, A.; Dyhl-Polk, A.; Chen, I.; Nielsen, D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat. Rev. 2019, 78, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Macherla, S.; Laks, S.; Naqash, A.R.; Bulumulle, A.; Zervos, E.; Muzaffar, M. Emerging Role of Immune Checkpoint Blockade in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3505. [Google Scholar] [CrossRef] [Green Version]
- Ahmad-Nielsen, S.A.; Bruun Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Frequency of mismatch repair deficiency in pancreatic ductal adenocarcinoma. Pathol. Res. Pract. 2020, 216, 152985. [Google Scholar] [CrossRef]
- Okudaira, K.; Hokari, R.; Tsuzuki, Y.; Okada, Y.; Komoto, S.; Watanabe, C.; Kurihara, C.; Kawaguchi, A.; Nagao, S.; Azuma, M.; et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int. J. Oncol. 2009, 35, 741–749. [Google Scholar]
- Deng, J.; Qian, Y.; Geng, L.; Xie, H.; Wang, Y.; Jiang, G.; Zhou, L.; Zhang, M.; Zheng, S. Involvement of ERK and JNK pathways in IFN-gamma-induced B7-DC expression on tumor cells. J. Cancer Res. Clin. Oncol. 2011, 137, 243–250. [Google Scholar] [CrossRef]
- Loos, M.; Giese, N.A.; Kleeff, J.; Giese, T.; Gaida, M.M.; Bergmann, F.; Laschinger, M.; Buchler, M.W.; Friess, H. Clinical significance and regulation of the costimulatory molecule B7-H1 in pancreatic cancer. Cancer Lett. 2008, 268, 98–109. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Mo, S.; Ma, H.; Lu, Z.; Yu, S.; Chen, J. PD-L1 and PD-L2 expression in pancreatic ductal adenocarcinoma and their correlation with immune infiltrates and DNA damage response molecules. J. Pathol. Clin. Res. 2022, 8, 257–267. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Shin, T.; Kennedy, G.; Gorski, K.; Tsuchiya, H.; Koseki, H.; Azuma, M.; Yagita, H.; Chen, L.; Powell, J.; Pardoll, D.; et al. Cooperative B7-1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. J. Exp. Med. 2003, 198, 31–38. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, S.; Zhu, B.; Bedoret, D.; Bu, X.; Francisco, L.M.; Hua, P.; Duke-Cohan, J.S.; Umetsu, D.T.; Sharpe, A.H.; et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J. Exp. Med. 2014, 211, 943–959. [Google Scholar] [CrossRef] [Green Version]
- Kenkel, J.A.; Tseng, W.W.; Davidson, M.G.; Tolentino, L.L.; Choi, O.; Bhattacharya, N.; Seeley, E.S.; Winer, D.A.; Reticker-Flynn, N.E.; Engleman, E.G. An Immunosuppressive Dendritic Cell Subset Accumulates at Secondary Sites and Promotes Metastasis in Pancreatic Cancer. Cancer Res. 2017, 77, 4158–4170. [Google Scholar] [CrossRef] [Green Version]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Tian, H. Development of small-molecule immune checkpoint inhibitors of PD-1/PD-L1 as a new therapeutic strategy for tumour immunotherapy. J. Drug Target. 2019, 27, 244–256. [Google Scholar] [CrossRef]
- Zong, L.; Mo, S.; Sun, Z.; Lu, Z.; Yu, S.; Chen, J.; Xiang, Y. Analysis of the immune checkpoint V-domain Ig-containing suppressor of T-cell activation (VISTA) in endometrial cancer. Mod. Pathol. 2022, 35, 266–273. [Google Scholar] [CrossRef]
- Ling, V.; Wu, P.W.; Spaulding, V.; Kieleczawa, J.; Luxenberg, D.; Carreno, B.M.; Collins, M. Duplication of primate and rodent B7-H3 immunoglobulin V- and C-like domains: Divergent history of functional redundancy and exon loss. Genomics 2003, 82, 365–377. [Google Scholar] [CrossRef]
- Langbein, T.; Weber, W.A.; Eiber, M. Future of Theranostics: An Outlook on Precision Oncology in Nuclear Medicine. J. Nucl. Med. 2019, 60 (Suppl. 2), 13S–19S. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [Green Version]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef] [Green Version]
- Kanchan, R.K.; Doss, D.; Khan, P.; Nasser, M.W.; Mahapatra, S. To kill a cancer: Targeting the immune inhibitory checkpoint molecule, B7-H3. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188783. [Google Scholar] [CrossRef]
- Xu, L.; Ding, X.; Tan, H.; Qian, J. Correlation between B7-H3 expression and matrix metalloproteinases 2 expression in pancreatic cancer. Cancer Cell Int. 2013, 13, 81. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Liu, D.; Chen, Q.; Yang, C.; Wang, B.; Wu, H. Soluble B7-H3 promotes the invasion and metastasis of pancreatic carcinoma cells through the TLR4/NF-kappaB pathway. Sci. Rep. 2016, 6, 27528. [Google Scholar] [CrossRef] [Green Version]
- Yamato, I.; Sho, M.; Nomi, T.; Akahori, T.; Shimada, K.; Hotta, K.; Kanehiro, H.; Konishi, N.; Yagita, H.; Nakajima, Y. Clinical importance of B7-H3 expression in human pancreatic cancer. Br. J. Cancer 2009, 101, 1709–1716. [Google Scholar] [CrossRef]
- Li, D.; Wang, J.; Zhou, J.; Zhan, S.; Huang, Y.; Wang, F.; Zhang, Z.; Zhu, D.; Zhao, H.; Li, D.; et al. B7-H3 combats apoptosis induced by chemotherapy by delivering signals to pancreatic cancer cells. Oncotarget 2017, 8, 74856–74868. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, D.C.; Zhu, X.G.; Gan, W.J.; Li, Z.; Xiong, F.; Zhang, Z.X.; Zhang, G.B.; Zhang, X.G.; Zhao, H. B7-H3 overexpression in pancreatic cancer promotes tumor progression. Int. J. Mol. Med. 2013, 31, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Vigdorovich, V.; Ramagopal, U.A.; Lazar-Molnar, E.; Sylvestre, E.; Lee, J.S.; Hofmeyer, K.A.; Zang, X.; Nathenson, S.G.; Almo, S.C. Structure and T cell inhibition properties of B7 family member, B7-H3. Structure 2013, 21, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Takazawa, Y.; Inoue, Y.; Yokouchi, Y.; Kobayashi, M.; Saiura, A.; Shibutani, T.; Ishikawa, Y. Tumor B7-H3 (CD276) Expression and Survival in Pancreatic Cancer. J. Clin. Med. 2018, 7, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhang, Y.; Mo, S.; Ma, H.; Lu, Z.; Yu, S.; Chen, J. Prognostic Value of Programmed Death Ligand-1 in Discriminating Patients With Lymph Node-Negative, p53-Wild-Type, or Low-BRCA1/2-Expression Pancreatic Ductal Adenocarcinoma. Arch. Pathol. Lab. Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, G.B.; Gan, W.J.; Xiong, F.; Li, Z.; Zhao, H.; Zhu, D.M.; Zhang, B.; Zhang, X.G.; Li, D.C. Silencing of B7-H3 increases gemcitabine sensitivity by promoting apoptosis in pancreatic carcinoma. Oncol. Lett. 2013, 5, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Jiang, B.; Zou, S.T.; Liu, F.; Hua, D. Overexpression of B7-H3 augments anti-apoptosis of colorectal cancer cells by Jak2-STAT3. World J. Gastroenterol. 2015, 21, 1804–1813. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Meng, Z.; Xie, C.; Yang, C.; Liu, Z.; Wu, S.; Wang, B.; Fan, P.; Jin, X.; Wu, H. B7-H3 is regulated by BRD4 and promotes TLR4 expression in pancreatic ductal adenocarcinoma. Int. J. Biochem. Cell Biol. 2019, 108, 84–91. [Google Scholar] [CrossRef]
- Wang, L.; Kang, F.B.; Shan, B.E. B7-H3-mediated tumor immunology: Friend or foe? Int. J. Cancer 2014, 134, 2764–2771. [Google Scholar] [CrossRef]
- Sun, M.; Richards, S.; Prasad, D.V.; Mai, X.M.; Rudensky, A.; Dong, C. Characterization of mouse and human B7-H3 genes. J. Immunol. 2002, 168, 6294–6297. [Google Scholar] [CrossRef] [Green Version]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, J.; Kelly, J.; Gu, G.; Hou, J.; Zhou, Y.; Redmond, H.P.; Wang, J.H.; Zhang, X. B7-H3 augments the inflammatory response and is associated with human sepsis. J. Immunol. 2010, 185, 3677–3684. [Google Scholar] [CrossRef] [Green Version]
- Loos, M.; Hedderich, D.M.; Ottenhausen, M.; Giese, N.A.; Laschinger, M.; Esposito, I.; Kleeff, J.; Friess, H. Expression of the costimulatory molecule B7-H3 is associated with prolonged survival in human pancreatic cancer. BMC Cancer 2009, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Hofmeyer, K.A.; Ray, A.; Zang, X. The contrasting role of B7-H3. Proc. Natl. Acad. Sci. USA 2008, 105, 10277–10278. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237.e8. [Google Scholar] [CrossRef] [Green Version]
- Durlanik, S.; Fundel-Clemens, K.; Viollet, C.; Huber, H.J.; Lenter, M.; Kitt, K.; Pflanz, S. CD276 is an important player in macrophage recruitment into the tumor and an upstream regulator for PAI-1. Sci. Rep. 2021, 11, 14849. [Google Scholar] [CrossRef]
- Chen, C.; Shen, Y.; Qu, Q.X.; Chen, X.Q.; Zhang, X.G.; Huang, J.A. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. Exp. Cell Res. 2013, 319, 96–102. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Augugliaro, R.; Cantoni, C.; Carnemolla, B.; Sementa, A.R.; Negri, F.; Conte, R.; Corrias, M.V.; Moretta, L.; et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc. Natl. Acad. Sci. USA 2004, 101, 12640–12645. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Chen, X.; Tao, M.; Chen, K.; Chen, C.; Xu, G.; Li, W.; Yuan, S.; Mao, Y. B7-H3 and B7-H4 are independent predictors of a poor prognosis in patients with pancreatic cancer. Oncol. Lett. 2016, 11, 1841–1846. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, J.; Liu, Y.; Zheng, X.; Feng, J.; Chen, X.; Jiang, T.; Li, Y.; Chen, L. Prognostic values of B7-H3, B7-H4, and HHLA2 expression in human pancreatic cancer tissues based on mIHC and spatial distribution analysis. Pathol. Res. Pract. 2022, 234, 153911. [Google Scholar] [CrossRef]
- Yonesaka, K.; Haratani, K.; Takamura, S.; Sakai, H.; Kato, R.; Takegawa, N.; Takahama, T.; Tanaka, K.; Hayashi, H.; Takeda, M.; et al. B7-H3 Negatively Modulates CTL-Mediated Cancer Immunity. Clin. Cancer Res. 2018, 24, 2653–2664. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, C.; Prawira, A.; Antonia, S.; Rahma, O.; Tolcher, A.; Cohen, R.B.; Lou, Y.; Hauke, R.; Vogelzang, N.; Zandberg, D.P.; et al. Dual checkpoint targeting of B7-H3 and PD-1 with enoblituzumab and pembrolizumab in advanced solid tumors: Interim results from a multicenter phase I/II trial. J. Immunother. Cancer 2022, 10, e004424. [Google Scholar] [CrossRef]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef]
- Li, G.; Wang, H.; Wu, H.; Chen, J. B7-H3-targeted CAR-T cell therapy for solid tumors. Int. Rev. Immunol. 2022, 41, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Seaman, S.; Zhu, Z.; Saha, S.; Zhang, X.M.; Yang, M.Y.; Hilton, M.B.; Morris, K.; Szot, C.; Morris, H.; Swing, D.A.; et al. Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer Cell 2017, 31, 501–515.e8. [Google Scholar] [CrossRef] [Green Version]
- Rasic, P.; Jovanovic-Tucovic, M.; Jeremic, M.; Djuricic, S.M.; Vasiljevic, Z.V.; Milickovic, M.; Savic, D. B7 homologue 3 as a prognostic biomarker and potential therapeutic target in gastrointestinal tumors. World J. Gastrointest. Oncol. 2021, 13, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Mews, E.A.; Beckmann, P.; Patchava, M.; Wang, Y.; Largaespada, D.A.; Wagner, C.R. Multivalent, Bispecific alphaB7-H3-alphaCD3 Chemically Self-Assembled Nanorings Direct Potent T Cell Responses against Medulloblastoma. ACS Nano 2022, 16, 12185–12201. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xie, K.; Yin, Y.; Li, B.; Pi, C.; Xu, X.; Huang, T.; Zhang, J.; Wang, B.; Gu, H.; et al. A Novel Anti-B7-H3 x Anti-CD3 Bispecific Antibody with Potent Antitumor Activity. Life 2022, 12, 157. [Google Scholar] [CrossRef]
- Kaur, G.; Janakiram, M. B7x-from bench to bedside. ESMO Open 2019, 4, e000554. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Qu, Q.X.; Shen, Y.; Lv, Y.T.; Zhu, Y.B.; Zhang, X.G.; Huang, J.A. Overexpression of B7-H4 in tumor infiltrated dendritic cells. J. Immunoass. Immunochem. 2011, 32, 353–364. [Google Scholar] [CrossRef]
- Jeon, H.; Ohaegbulam, K.C.; Abadi, Y.M.; Zang, X. B7x and myeloid-derived suppressor cells in the tumor microenvironment: A tale of two cities. Oncoimmunology 2013, 2, e24744. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tao, L.; Yuan, C.; Xiu, D. The prognostic value of B7-H4 in pancreatic cancer: Systematic review and meta-analysis. Medicine 2018, 97, e0088. [Google Scholar] [CrossRef]
- Zang, X.; Loke, P.; Kim, J.; Murphy, K.; Waitz, R.; Allison, J.P. B7x: A widely expressed B7 family member that inhibits T cell activation. Proc. Natl. Acad. Sci. USA 2003, 100, 10388–10392. [Google Scholar] [CrossRef]
- Altan, M.; Pelekanou, V.; Schalper, K.A.; Toki, M.; Gaule, P.; Syrigos, K.; Herbst, R.S.; Rimm, D.L. B7-H3 Expression in NSCLC and Its Association with B7-H4, PD-L1 and Tumor-Infiltrating Lymphocytes. Clin. Cancer Res. 2017, 23, 5202–5209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tringler, B.; Zhuo, S.; Pilkington, G.; Torkko, K.C.; Singh, M.; Lucia, M.S.; Heinz, D.E.; Papkoff, J.; Shroyer, K.R. B7-h4 is highly expressed in ductal and lobular breast cancer. Clin. Cancer Res. 2005, 11, 1842–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Lee, Y.; Li, Y.; Jiang, Y.; Lu, H.; Zang, W.; Zhao, X.; Liu, L.; Chen, Y.; Tan, H.; et al. Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells. Immunity 2018, 48, 773–786.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, G.L.; Choi, I.H.; Zhu, G.; Tamada, K.; Wang, S.D.; Tamura, H.; Chapoval, A.I.; Flies, D.B.; Bajorath, J.; Chen, L. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity 2003, 18, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Prasad, D.V.; Richards, S.; Mai, X.M.; Dong, C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity 2003, 18, 863–873. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Dong, C. New checkpoints in cancer immunotherapy. Immunol. Rev. 2017, 276, 52–65. [Google Scholar] [CrossRef]
- Diao, W.; Lu, L.; Li, S.; Chen, J.; Zen, K.; Li, L. MicroRNA-125b-5p modulates the inflammatory state of macrophages via targeting B7-H4. Biochem. Biophys. Res. Commun. 2017, 491, 912–918. [Google Scholar] [CrossRef]
- Zhou, L.; Ruan, M.; Liu, Y.; Zhu, Y.; Fu, D.; Wu, K.; Zhang, Q. B7H4 expression in tumor cells impairs CD8 T cell responses and tumor immunity. Cancer Immunol. Immunother. 2020, 69, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Awadallah, N.S.; Shroyer, K.R.; Langer, D.A.; Torkko, K.C.; Chen, Y.K.; Bentz, J.S.; Papkoff, J.; Liu, W.; Nash, S.R.; Shah, R.J. Detection of B7-H4 and p53 in pancreatic cancer: Potential role as a cytological diagnostic adjunct. Pancreas 2008, 36, 200–206. [Google Scholar] [CrossRef]
- Abadi, Y.M.; Jeon, H.; Ohaegbulam, K.C.; Scandiuzzi, L.; Ghosh, K.; Hofmeyer, K.A.; Lee, J.S.; Ray, A.; Gravekamp, C.; Zang, X. Host b7x promotes pulmonary metastasis of breast cancer. J. Immunol. 2013, 190, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Heng, X.; Lu, Y.; Cai, Z.; Yi, Q.; Che, F. Could B7-H4 serve as a target to activate anti-cancer immunity? Int. Immunopharmacol. 2016, 38, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Feng, L.; Wu, W.; Weng, T.; Hu, C.; Hong, B.; Wang, F.X.C.; Shen, L.; Wang, Q.; Jin, X.; et al. MicroRNA Expression Profiling of Pancreatic Cancer Cell Line L3.6p1 Following B7-H4 Knockdown. Cell. Physiol. Biochem. 2017, 44, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podojil, J.R.; Miller, S.D. Potential targeting of B7-H4 for the treatment of cancer. Immunol. Rev. 2017, 276, 40–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Y.; Sang, Y.; Wang, F.X.; Hong, B.; Wang, Q.; Zhou, X.; Weng, T.; Wu, Z.; Zheng, M.; Zhang, H.; et al. Prognostic significance of B7-H4 expression in matched primary pancreatic cancer and liver metastases. Oncotarget 2016, 7, 72242–72249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Y.; Wang, W.P. B7-H4, a promising target for immunotherapy. Cell. Immunol. 2020, 347, 104008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, H.; Lu, D.; Li, G.; Sun, C.; Song, H.; Li, J.; Zhai, T.; Huang, L.; Hou, C.; et al. The costimulatory molecule B7-H4 promote tumor progression and cell proliferation through translocating into nucleus. Oncogene 2013, 32, 5347–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, G.B.; Song, H.; Kim, Y.S.; Sung, M.; Ryu, J.W.; Lee, H.K.; Cho, D.H.; Kim, D.; Lee, W.J.; Hur, D.Y. Cell cycle arrest induced by engagement of B7-H4 on Epstein-Barr virus-positive B-cell lymphoma cell lines. Immunology 2009, 128, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, Y.; Zhao, J.; Gu, M.; Giscombe, R.; Lefvert, A.K.; Wang, X. B7-H3 and B7-H4 expression in non-small-cell lung cancer. Lung Cancer 2006, 53, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Ye, H.; Qi, Z.; Mo, L.; Yue, Q.; Baral, A.; Hoon, D.S.B.; Vera, J.C.; Heiss, J.D.; Chen, C.C.; et al. B7-H4(B7x)-Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin. Cancer Res. 2016, 22, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- John, P.; Wei, Y.; Liu, W.; Du, M.; Guan, F.; Zang, X. The B7x Immune Checkpoint Pathway: From Discovery to Clinical Trial. Trends Pharmacol. Sci. 2019, 40, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Gardiner, J.C.; Maggi, E.C.; Huang, S.; Adem, A.; Bagdasarov, S.; Li, G.; Lee, S.; Slegowski, D.; Exarchakis, A.; et al. B7 immune-checkpoints as targets for the treatment of neuroendocrine tumors. Endocr. Relat. Cancer 2021, 28, 135–149. [Google Scholar] [CrossRef] [PubMed]
- John, P.; Pulanco, M.C.; Galbo, P.M., Jr.; Wei, Y.; Ohaegbulam, K.C.; Zheng, D.; Zang, X. The immune checkpoint B7x expands tumor-infiltrating Tregs and promotes resistance to anti-CTLA-4 therapy. Nat. Commun. 2022, 13, 2506. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Liu, W.; Jeon, H.; Almo, S.C.; Zang, X. Tumor-expressed immune checkpoint B7x promotes cancer progression and antigen-specific CD8 T cell exhaustion and suppressive innate immune cells. Oncotarget 2017, 8, 82740–82753. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Mao, Y.; Zhu, J.; Meng, F.; Chen, Q.; Tao, L.; Li, R.; Fu, F.; Liu, C.; Hu, Y.; et al. TGF-beta1 promotes colorectal cancer immune escape by elevating B7-H3 and B7-H4 via the miR-155/miR-143 axis. Oncotarget 2016, 7, 67196–67211. [Google Scholar] [CrossRef] [Green Version]
- Leong, S.R.; Liang, W.C.; Wu, Y.; Crocker, L.; Cheng, E.; Sampath, D.; Ohri, R.; Raab, H.; Hass, P.E.; Pham, T.; et al. An anti-B7-H4 antibody-drug conjugate for the treatment of breast cancer. Mol. Pharm. 2015, 12, 1717–1729. [Google Scholar] [CrossRef]
- Smith, J.B.; Lanitis, E.; Dangaj, D.; Buza, E.; Poussin, M.; Stashwick, C.; Scholler, N.; Powell, D.J., Jr. Tumor Regression and Delayed Onset Toxicity Following B7-H4 CAR T Cell Therapy. Mol. Ther. 2016, 24, 1987–1999. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, A.; Nonomura, C.; Ashizawa, T.; Kondou, R.; Ohshima, K.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; Maruyama, K.; et al. A T-cell-engaging B7-H4/CD3-bispecific Fab-scFv Antibody Targets Human Breast Cancer. Clin. Cancer Res. 2019, 25, 2925–2934. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Tatineni, J.; Mahoney, K.M.; Freeman, G.J. VISTA: A Mediator of Quiescence and a Promising Target in Cancer Immunotherapy. Trends Immunol. 2021, 42, 209–227. [Google Scholar] [CrossRef]
- ElTanbouly, M.A.; Schaafsma, E.; Noelle, R.J.; Lines, J.L. VISTA: Coming of age as a multi-lineage immune checkpoint. Clin. Exp. Immunol. 2020, 200, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hieu, T.; Malarkannan, S.; Wang, L. The structure, expression, and multifaceted role of immune-checkpoint protein VISTA as a critical regulator of anti-tumor immunity, autoimmunity, and inflammation. Cell. Mol. Immunol. 2018, 15, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Feng, H.; Cheng, X.; Liu, K.; Cai, D.; Zhao, R. Potential Therapeutic Targets of B7 Family in Colorectal Cancer. Front. Immunol. 2020, 11, 681. [Google Scholar] [CrossRef] [PubMed]
- Lines, J.L.; Pantazi, E.; Mak, J.; Sempere, L.F.; Wang, L.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Su, L.J.; Pinckney, J.; Critton, D.; Boyer, E.; Krishnakumar, A.; Corbett, M.; Rankin, A.L.; Dibella, R.; Campbell, L.; et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019, 574, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, G.; Manick, B.; Hernandez, V.; Renelt, M.; Erickson, C.; Guan, J.; Singh, R.; Rollins, S.; Solorz, A.; et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 2019, 156, 74–85. [Google Scholar] [CrossRef] [Green Version]
- ElTanbouly, M.A.; Croteau, W.; Noelle, R.J.; Lines, J.L. VISTA: A novel immunotherapy target for normalizing innate and adaptive immunity. Semin. Immunol. 2019, 42, 101308. [Google Scholar] [CrossRef]
- Liu, J.; Xie, X.; Xuan, C.; Li, T.; Wang, L.; Teng, L.; Liu, J. High-Density Infiltration of V-domain Immunoglobulin Suppressor of T-cell Activation Up-regulated Immune Cells in Human Pancreatic Cancer. Pancreas 2018, 47, 725–731. [Google Scholar] [CrossRef]
- Topcu, K.S.B.; Korucu, E.N.; Menevse, E.; Kocak, N.; Duran, T. Investigation of the effects of the toll-like receptor 4 pathway on immune checkpoint vista in pancreatic cancer. Investig. New Drugs 2022, 40, 519–528. [Google Scholar] [CrossRef]
- Yoon, K.W.; Byun, S.; Kwon, E.; Hwang, S.Y.; Chu, K.; Hiraki, M.; Jo, S.H.; Weins, A.; Hakroush, S.; Cebulla, A.; et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science 2015, 349, 1261669. [Google Scholar] [CrossRef] [PubMed]
- ElTanbouly, M.A.; Zhao, Y.; Nowak, E.; Li, J.; Schaafsma, E.; Le Mercier, I.; Ceeraz, S.; Lines, J.L.; Peng, C.; Carriere, C.; et al. VISTA is a checkpoint regulator for naive T cell quiescence and peripheral tolerance. Science 2020, 367, eaay0524. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Pan, Y.; Fei, Q.; Lin, Y.; Zhou, Y.; Liu, Y.; Guan, H.; Yu, X.; Lin, X.; Lu, F.; et al. Prognostic significance and therapeutic potential of the immune checkpoint VISTA in pancreatic cancer. J. Cancer Res. Clin. Oncol. 2021, 147, 517–531. [Google Scholar] [CrossRef]
- Popp, F.C.; Capino, I.; Bartels, J.; Damanakis, A.; Li, J.; Datta, R.R.; Loser, H.; Zhao, Y.; Quaas, A.; Lohneis, P.; et al. Expression of Immune Checkpoint Regulators IDO, VISTA, LAG3, and TIM3 in Resected Pancreatic Ductal Adenocarcinoma. Cancers 2021, 13, 2689. [Google Scholar] [CrossRef]
- Janakiram, M.; Chinai, J.M.; Fineberg, S.; Fiser, A.; Montagna, C.; Medavarapu, R.; Castano, E.; Jeon, H.; Ohaegbulam, K.C.; Zhao, R.; et al. Expression, Clinical Significance, and Receptor Identification of the Newest B7 Family Member HHLA2 Protein. Clin. Cancer Res. 2015, 21, 2359–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, R.S.; Berjis, A.; Konge, J.C.; Mahoney, K.M.; Klee, A.N.; Freeman, S.S.; Chen, C.H.; Jegede, O.A.; Catalano, P.J.; Pignon, J.C.; et al. KIR3DL3 Is an Inhibitory Receptor for HHLA2 that Mediates an Alternative Immunoinhibitory Pathway to PD1. Cancer Immunol. Res. 2021, 9, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yao, S.; Iliopoulou, B.P.; Han, X.; Augustine, M.M.; Xu, H.; Phennicie, R.T.; Flies, S.J.; Broadwater, M.; Ruff, W.; et al. B7-H5 costimulates human T cells via CD28H. Nat. Commun. 2013, 4, 2043. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Chinai, J.M.; Buhl, S.; Scandiuzzi, L.; Ray, A.; Jeon, H.; Ohaegbulam, K.C.; Ghosh, K.; Zhao, A.; Scharff, M.D.; et al. HHLA2 is a member of the B7 family and inhibits human CD4 and CD8 T-cell function. Proc. Natl. Acad. Sci. USA 2013, 110, 9879–9884. [Google Scholar] [CrossRef] [Green Version]
- Janakiram, M.; Chinai, J.M.; Zhao, A.; Sparano, J.A.; Zang, X. HHLA2 and TMIGD2: New immunotherapeutic targets of the B7 and CD28 families. Oncoimmunology 2015, 4, e1026534. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Xu, J.; Zhang, X.; Liang, T.; Bai, X. Human endogenous retrovirus-H long terminal repeat-associating 2: The next immune checkpoint for antitumour therapy. EBioMedicine 2022, 79, 103987. [Google Scholar] [CrossRef]
- Luu, K.; Schwarz, H.; Lundqvist, A. B7-H7 Is Inducible on T Cells to Regulate Their Immune Response and Serves as a Marker for Exhaustion. Front. Immunol. 2021, 12, 682627. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Long, E.O. CD28 Homolog Is a Strong Activator of Natural Killer Cells for Lysis of B7H7(+) Tumor Cells. Cancer Immunol. Res. 2019, 7, 939–951. [Google Scholar] [CrossRef]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Qiu, W.; Koehne de Gonzalez, A.K.; Wei, J.S.; Tu, M.; Xi, C.H.; Yang, Y.R.; Peng, Y.P.; Tsai, W.Y.; Remotti, H.E.; et al. HHLA2 is a novel immune checkpoint protein in pancreatic ductal adenocarcinoma and predicts post-surgical survival. Cancer Lett. 2019, 442, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Boor, P.P.C.; Sideras, K.; Biermann, K.; Hosein Aziz, M.; Levink, I.J.M.; Mancham, S.; Erler, N.S.; Tang, X.; van Eijck, C.H.; Bruno, M.J.; et al. HHLA2 is expressed in pancreatic and ampullary cancers and increased expression is associated with better post-surgical prognosis. Br. J. Cancer 2020, 122, 1211–1218. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, J.; Chen, W.; Zhang, Q.; Wei, T.; Zhou, Y.; Xu, X.; Bai, X.; Liang, T. B7-H5/CD28H is a co-stimulatory pathway and correlates with improved prognosis in pancreatic ductal adenocarcinoma. Cancer Sci. 2019, 110, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Ren, X.; Galbo, P.M., Jr.; Moerdler, S.; Wang, H.; Sica, R.A.; Etemad-Gilbertson, B.; Shi, L.; Zhu, L.; Tang, X.; et al. KIR3DL3-HHLA2 is a human immunosuppressive pathway and a therapeutic target. Sci. Immunol. 2021, 6, eabf9792. [Google Scholar] [CrossRef]
- Qian, Z.R.; Rubinson, D.A.; Nowak, J.A.; Morales-Oyarvide, V.; Dunne, R.F.; Kozak, M.M.; Welch, M.W.; Brais, L.K.; Da Silva, A.; Li, T.; et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2018, 4, e173420. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Dias Carvalho, P.; Guimaraes, C.F.; Cardoso, A.P.; Mendonca, S.; Costa, A.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Theate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent/Drug | Combination Regimens | Targets | Phase | Number Enrolled | Trial ID | Primary Outcome(s) |
|---|---|---|---|---|---|---|
| Pembrolizumab | Cyclophosphamide + GVAX + SBR | PD-1 | II | 58 | NCT02648282 | DMFS |
| Defactinib | PD-1 | I/II | 59 | NCT02758587 | AEs, DLTs, MTD | |
| Cyclophosphamide + GVAX + IMC-CS4 | PD-1 | Early I | 12 | NCT03153410 | CD8 T cell density, AEs | |
| Epacadostat + CRS-207 + CY + GVAX | PD-1 | II | 40 | NCT03006302 | DMFS | |
| Olaparib | PD-1 | II | 20 | NCT05093231 | ORR | |
| Cabozantinib | PD-1 | II | 21 | NCT05052723 | PFS | |
| Defactinib | PD-1 | II | 36 | NCT03727880 | pCR rate | |
| ENB003 | PD-1 | I/II | 130 | NCT04205227 | ORR, TRAEs | |
| ONC-392 | PD-1 | I/II | 468 | NCT04140526 | DLT, MTD, TRAEs, RP2D | |
| Tadalafil Ipilimumab CRS-207 | PD-1 | II | 20 | NCT05014776 | ORR | |
| PEGPH20 | PD-1 | II | 35 | NCT03634332 | PFS | |
| Lenvatinib | PD-1 | II | 590 | NCT03797326 | ORR (initial), ORR (expansion), AEs, Discont due to AE | |
| V941 | PD-1 | I | 100 | NCT03948763 | DLTs, AEs | |
| CPI-006 + ciforadenant | PD-1 | I | 378 | NCT03454451 | Incidence of DLTs, TRAEs; MDL | |
| INT230-6 + anti-CTLA-4 antibody | PD-1 | I/II | 110 | NCT03058289 | TRAEs | |
| BCA101 | PD-1 | I | 292 | NCT04429542 | DLTs, safety, tolerability | |
| Spartalizumab | Siltuximab | PD-1 | I/II | 42 | NCT04191421 | MTD |
| Canakinumab injection + nab-paclitaxel + gemcitabine | PD-1 | I | 10 | NCT04581343 | RP2/3D | |
| Dostarlimab | Niraparib | PD-1 | II | 20 | NCT04493060 | DCR |
| Sintilimab | Gemcitabine + Albumin-paclitaxel | PD-1 | II | 20 | NCT05346146 | % R0 resection |
| Surufatinib + AG | PD-1 | II | 32 | NCT05481476 | ORR | |
| Toripalimab | Anlotinib + Nab-paclitaxel | PD-1 | II | 53 | NCT04718701 | PFS |
| YH003 + Nab-paclitaxel + Gemcitabine | PD-1 | II | 129 | NCT05031494 | ORR | |
| Camrelizumab | Nab paclitaxel + Gemcitabine Injection | PD-1 | II | 117 | NCT04498689 | ORR, PFS |
| Chemotherapy + Ablation | PD-1 | Not Applicable | 34 | NCT04420130 | 6-month PFS rate | |
| Surufatinib + nab-paclitaxel + S-1 gemcitabine | PD-1 | II | 68 | NCT05218889 | DLTs, ORR, RP2D | |
| Paclitaxel (Albumin Bound) + Gemcitabine + Placebo | PD-1 | III | 401 | NCT04674956 | PFS | |
| Apatinib | PD-1 | II | 48 | NCT04415385 | ORR | |
| Capecitabine | PD-1 | I | 20 | NCT04932187 | AEs | |
| Nivolumab | Tadalafil + oral vancomycin | PD-1 | II | 22 | NCT03785210 | BOR |
| Recombinant Human IL12/15-PDL1B Oncolytic HSV-1 Injection (Vero Cell) | PD-1 | I/II | 51 | NCT05162118 | DCR (phase 2), MTD, AEs, DLT (phase 1), RP2D (phase 1) | |
| Cyclophosphamide + Ipilimumab + GVAX Pancreas Vaccine + CRS-207 | PD-1 | II | 63 | NCT03190265 | ORR | |
| Ipilimumab + Stereotactic body radiation therapy + Low dose irradiation | PD-1 | I | 10 | NCT05088889 | ORR | |
| Nivolumab | Anetumab Ravtansine + Gemcitabine Hydrochloride + Ipilimumab | PD-1 | I/II | 74 | NCT03816358 | MTD |
| Irreversible Electroporation (IRE) + Toll-Like Receptor 9 | PD-1 | I | 18 | NCT04612530 | Safety | |
| Irreversible electroporation (IRE) | PD-1 | II | 12 | NCT05435053 | Safety, tolerability | |
| SX-682 | PD-1 | I | 20 | NCT04477343 | MTD | |
| Cyclophosphamide + GVAX Pancreas Vaccine + Stereotactic Body Radiation (SBRT) | PD-1 | II | 30 | NCT03161379 | CD8 count in the TME | |
| Cabiralizumab | PD-1 | I | 313 | NCT02526017 | ORR, RD, Safety | |
| AK105 | Anlotinib | PD-1 | I/II | 29 | NCT04803851 | DCR |
| Camrelizumab | Paclitaxel (Albumin Bound) + Gemcitabine | PD-1 | III | 401 | NCT04674956 | PFS |
| Anti-PD-1 mAb | PD-1 | III | 830 | NCT03983057 | PFS | |
| Anti-PD-1 antibody | Chemotherapy | PD-1 | III | 110 | NCT03977272 | OS |
| Radiation | PD-1 | II | 21 | NCT03374293 | Local control | |
| SBRT | PD-1 | I | 36 | NCT03716596 | OS | |
| Radiotherapy + Gemcitabine + cisplatin + Apatinib | PD-1 | 150 | NCT04365049 | PFS | ||
| Durvalumab | Gemcitabine | PD-L1 | II | 71 | NCT03572400 | DFS |
| Tremelimumab + Gemcitabine + Minimally Invasive Surgical Microwave Ablation | PD-L1 | II | 20 | NCT04156087 | PFS | |
| Olaparib + Radiation Therapy | PD-L1 | I | 18 | NCT05411094 | DLTs | |
| Radiotherapy + Tremelimumab | PD-L1 | I | 30 | NCT02639026 | Number of AEs | |
| Gemcitabine + Nab-paclitaxel + Oleclumab | PD-L1 | II | 30 | NCT04940286 | Incidence of AEs, RR | |
| Romidepsin + Azacitidine + nab-Paclitaxel + Gemcitabine + Lenalidomide capsule | PD-L1 | I/II | 75 | NCT04257448 | RDE, safety and tolerability | |
| Capecitabine + Bevacizumab + CV301 | PD-L1 | I/II | 8 | NCT03376659 | PFS(4 and 8.5 month) RP2D | |
| Oleclumab+gemcitabine + nab-paclitaxel + oxaliplatin + leucovorin + 5-FU | PD-L1 | I/II | 212 | NCT03611556 | Incidence of AEs, clinically significant ECG abnormalities and laboratory values, ORR | |
| Tazemetostat | PD-L1 | II | 173 | NCT04705818 | Assessment of antitumor activity | |
| Galunisertib | PD-L1 | I | 37 | NCT02734160 | DLTs | |
| Avelumab | Aldoxorubicin HCl + ALT-803 + ETBX-011 + GI-4000 etc. | PD-L1 | I/II | 173 | NCT03387098 | Incidence of AEs and SAEs, ORR |
| ALT-803 + ETBX-011 + GI-4000 + haNK etc. | PD-L1 | I/II | 80 | NCT03329248 | Incidence of AEs and SAEs, ORR | |
| Aldoxorubicin HCl + ALT-803 + ETBX-011 + ETBX-021 etc. | PD-L1 | I/II | 173 | NCT03586869 | Incidence of AEs and SAEs, ORR | |
| Cyclophosphamide + Oxaliplatin + Capecitabine + 5-Fluorouracil etc. | PD-L1 | I/II | 3 | NCT03136406 | Incidence of AEs and SAEs, ORR | |
| Atezolizumab | Cabozantinib | PD-L1 | II | 29 | NCT04820179 | ORR or stable disease |
| KY1044 | PD-L1 | I/II | 412 | NCT03829501 | Safety, tolerability, DLTs, ORR, survival rate | |
| FT500 + Nivolumab + Pembrolizuma + Cyclophosphamide etc. | PD-L1 | I | 37 | NCT03841110 | DLTs | |
| RO7198457 + mFOLFIRINOX | PD-L1 | I | 29 | NCT04161755 | Toxicity | |
| MEDI4736 | Tremelimumab | PD-L1 | II | 64 | NCT02527434 | OR |
| SHR-1701 (PD-L1/TGF-β bsAb) | PD-L1 | I/II | 56 | NCT04624217 | ORR, RP2D | |
| Pembrolizumab | Cabozantinib (RKTs Inhibitor) | PD-L2 | II | 21 | NCT05052723 | PFS |
| PEGPH20 | PD-L2 | II | 35 | NCT03634332 | PFS | |
| CD276 CAR-T cells | B7-H3 | I/II | 10 | NCT05143151 | ORR | |
| MGD009 (B7-H3/CD3 bsAb) | B7-H3 | I | 67 | NCT02628535 | AEs | |
| SGN-B7H4V (B7-H4 ADC) | B7-H4 | I | 375 | NCT05194072 | AEs, DLTs, laboratory abnormalities | |
| HS-20089 (B7-H4 ADC) | B7-H4 | I | 177 | NCT05263479 | MTD | |
| JNJ-61610588 (B7-H5 mab) | VISTA | I | 12 | NCT02671955 | Markers of monocyte activation, T-cell activation, immune infiltration; total blood cell counts; protein expression of VISTA; DLTs; AEs; SAEs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Li, J.; Chen, Y.; Que, Z.; Du, J.; Zhang, J. B7 Family Members in Pancreatic Ductal Adenocarcinoma: Attractive Targets for Cancer Immunotherapy. Int. J. Mol. Sci. 2022, 23, 15005. https://doi.org/10.3390/ijms232315005
Chen X, Li J, Chen Y, Que Z, Du J, Zhang J. B7 Family Members in Pancreatic Ductal Adenocarcinoma: Attractive Targets for Cancer Immunotherapy. International Journal of Molecular Sciences. 2022; 23(23):15005. https://doi.org/10.3390/ijms232315005
Chicago/Turabian StyleChen, Xin, Jie Li, Yue Chen, Ziting Que, Jiawei Du, and Jianqiong Zhang. 2022. "B7 Family Members in Pancreatic Ductal Adenocarcinoma: Attractive Targets for Cancer Immunotherapy" International Journal of Molecular Sciences 23, no. 23: 15005. https://doi.org/10.3390/ijms232315005
APA StyleChen, X., Li, J., Chen, Y., Que, Z., Du, J., & Zhang, J. (2022). B7 Family Members in Pancreatic Ductal Adenocarcinoma: Attractive Targets for Cancer Immunotherapy. International Journal of Molecular Sciences, 23(23), 15005. https://doi.org/10.3390/ijms232315005