

Pharmacological Inhibition of IKK to Tackle Latency and Hyperinflammation in Chronic HIV-1 Infection

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
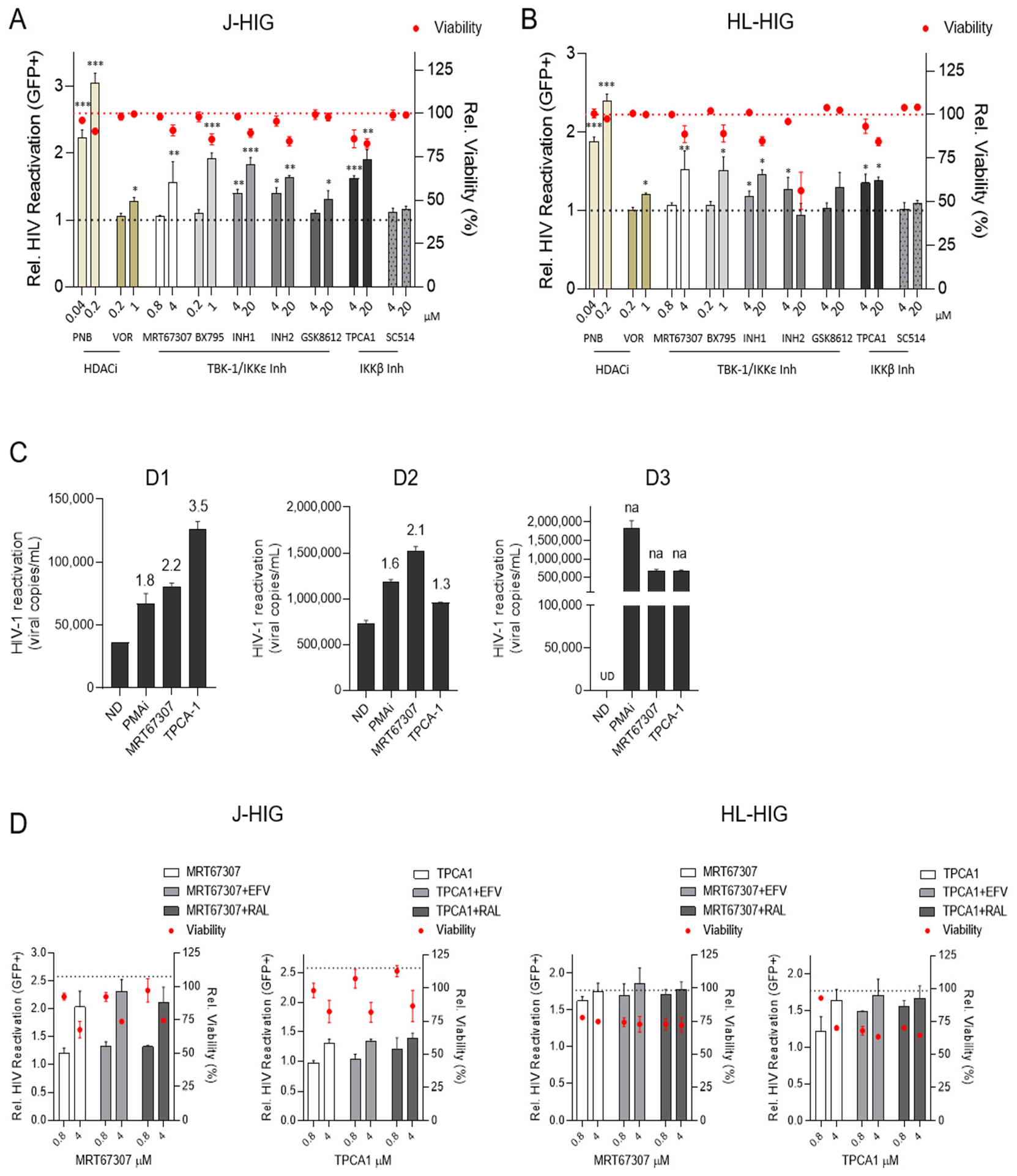
2.1. IKKis Induce HIV Reactivation in Distinct Models of HIV Latency In Vitro
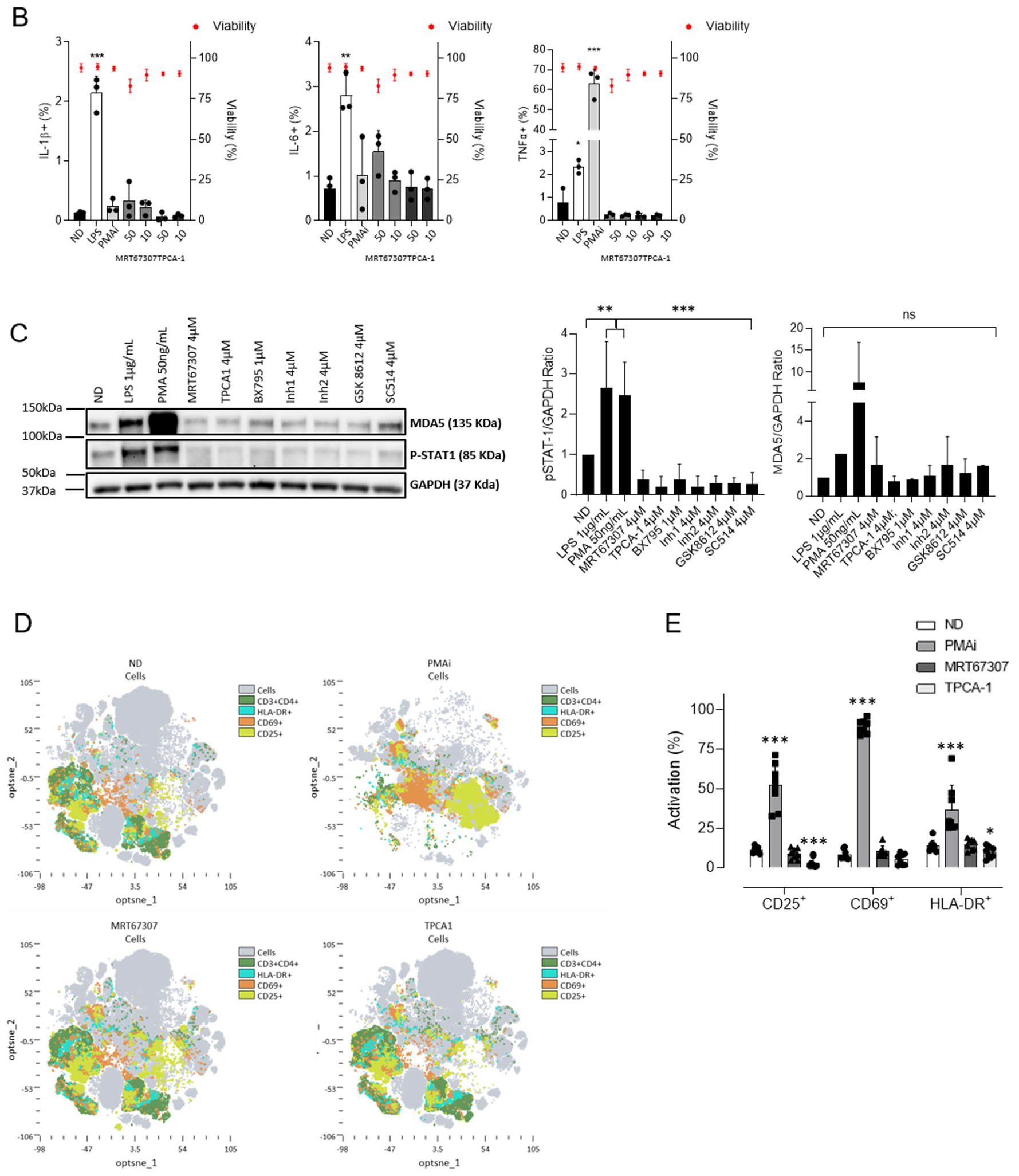
2.2. TBK1/IKKε and IKKβ Inhibitors Induce HIV Reactivation Ex Vivo in CD4+ T Cells from HIV+ Individuals
2.3. Antiretroviral Drugs Do Not Interfere with HIV Reactivation Activity Induced by IKKis
2.4. IKKis Do Not Modify Cell Activation in Ex Vivo Treated Primary CD4+ T Cells
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Compounds
4.3. Virus
4.4. Generation of Latently Infected Cells
4.5. HIV Reactivation In Vitro in Latently Infected Cells
4.6. Measurement of HIV Production in CD4+ T Cells from HIV+ Individuals
4.7. Quantitative RT-Polymerase Chain Reaction (qRT-PCR)
4.8. Western Blot
4.9. Flow Cytometry
4.10. Evaluation of Cytotoxicity
4.11. Anti-HIV Assays
4.12. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- García, M.; Buzón, M.J.; Benito, J.M.; Rallón, N. Peering into the HIV reservoir. Rev. Med. Virol. 2018, 28, e1981. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Archin, N.; Cannon, P.; Collins, S.; Jones, R.B.; de Jong, M.A.W.P.; Lambotte, O.; Lamplough, R.; Ndung’u, T.; Sugarman, J.; et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nat. Med. 2021, 27, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.M.; Jiang, G. NF-ΚB Sub-Pathways and HIV Cure: A Revisit. eBioMedicine 2021, 63, 103159. [Google Scholar] [CrossRef] [PubMed]
- Grau-Expósito, J.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badía, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLOS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, B.I.; Laws, E.I.; Ndhlovu, L.C. Impact of Myeloid Reservoirs in HIV Cure Trials. Curr. HIV/AIDS Rep. 2019, 16, 129–140. [Google Scholar] [CrossRef]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV Reservoir in Monocytes and Macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Olson, R.M.; Gornalusse, G.; Whitmore, L.S.; Newhouse, D.; Tisoncik-Go, J.; Smith, E.; Ochsenbauer, C.; Hladik, F.; Gale, M. Innate immune regulation in HIV latency models. Retrovirology 2022, 19, 15. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Saifuddin, M. Regulation of HIV-1 Transcription. Gene Expr. 1999, 8, 67–84. [Google Scholar]
- Kilareski, E.M.; Shah, S.; Nonnemacher, M.R.; Wigdahl, B. Regulation of HIV-1 transcription in cells of the monocyte-macrophage lineage. Retrovirology 2009, 6, 118. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, G.; Zaikos, T.D.; Khan, S.Z.; Jacobi, A.M.; Behlke, M.A.; Zeichner, S.L. Targeting IκB Proteins for HIV Latency Activation: The Role of Individual IκB and NF-κB Proteins. J. Virol. 2013, 87, 3966–3978. [Google Scholar] [CrossRef] [Green Version]
- Amaya, M.; Keck, F.; Bailey, C.; Narayanan, A. The role of the IKK complex in viral infections. Pathog. Dis. 2014, 72, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Peggie, M.; Plater, L.; Sorcek, R.J.; Young, E.R.R.; Madwed, J.B.; Hough, J.; McIver, E.G.; Cohen, P. Novel cross-talk within the IKK family controls innate immunity. Biochem. J. 2011, 434, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, R.J.; Hagan, R.S.; Baldwin, A.S. Expanding the View of IKK: New Substrates and New Biology. Trends Cell Biol. 2021, 31, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-ΚB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Liu, R.; Tan, J.; Lin, Y.; Jia, R.; Yang, W.; Liang, C.; Geng, Y.; Qiao, W. HIV-1 Vpr activates both canonical and noncanonical NF-κB pathway by enhancing the phosphorylation of IKKα/β. Virology 2013, 439, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Prescott, J.; Cook, S. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.; Ansari, M.M.; Noor, S.; Mohammad, T.; Hasan, G.M.; Kazim, S.N.; Hassan, M.I. Therapeutic targeting of TANK-binding kinase signaling towards anticancer drug development: Challenges and opportunities. Int. J. Biol. Macromol. 2022, 207, 1022–1037. [Google Scholar] [CrossRef]
- Runde, A.P.; Mack, R.; Peter Breslin, S.J.; Zhang, J. The role of TBK1 in cancer pathogenesis and anticancer immunity. J. Exp. Clin. Cancer Res. 2022, 41, 135. [Google Scholar] [CrossRef]
- Gutierrez-Chamorro, L.; Felip, E.; Ezeonwumelu, I.J.; Margelí, M.; Ballana, E. Cyclin-dependent Kinases as Emerging Targets for Developing Novel Antiviral Therapeutics. Trends Microbiol. 2021, 29, 836–848. [Google Scholar] [CrossRef]
- Ezeonwumelu, I.J.; Garcia-Vidal, E.; Ballana, E. JAK-STAT Pathway: A Novel Target to Tackle Viral Infections. Viruses 2021, 13, 2379. [Google Scholar] [CrossRef]
- Ezeonwumelu, I.J.; García-Vidal, E.; Felip, E.; Puertas, M.C.; Oriol-Tordera, B.; Gutiérrez-Chamorro, L.; Gohr, A.; Ruiz-Riol, M.; Massanella, M.; Clotet, B.; et al. IRF7 expression correlates with HIV latency reversal upon specific blockade of immune activation. Front. Immunol. 2022, 13, 5026. [Google Scholar] [CrossRef] [PubMed]
- Badia, R.; Grau, J.; Riveira-Muñoz, E.; Ballana, E.; Giannini, G.; Esté, J.A. The thioacetate-ω(γ-lactam carboxamide) HDAC inhibitor ST7612AA1 as HIV-1 latency reactivation agent. Antivir. Res. 2015, 123, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Nalli, M.; Armijos Rivera, J.I.; Masci, D.; Coluccia, A.; Badia, R.; Riveira-Muñoz, E.; Brambilla, A.; Cinquina, E.; Turriziani, O.; Falasca, F.; et al. New indolylarylsulfone non-nucleoside reverse transcriptase inhibitors show low nanomolar inhibition of single and double HIV-1 mutant strains. Eur. J. Med. Chem. 2020, 208, 112696. [Google Scholar] [CrossRef]
- Board, N.L.; Moskovljevic, M.; Wu, F.; Siliciano, R.F.; Siliciano, J.D. Engaging innate immunity in HIV-1 cure strategies. Nat. Rev. Immunol. 2022, 22, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Z.; Gasperino, S.; Zeichner, S.L. Nuclear Transit and HIV LTR Binding of NF-κB Subunits Held by IκB Proteins: Implications for HIV-1 Activation. Viruses 2019, 11, 1162. [Google Scholar] [CrossRef] [Green Version]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.S.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef]
- Acchioni, C.; Remoli, A.L.; Marsili, G.; Acchioni, M.; Nardolillo, I.; Orsatti, R.; Farcomeni, S.; Palermo, E.; Perrotti, E.; Barreca, M.L.; et al. Alternate NF-κB-Independent Signaling Reactivation of Latent HIV-1 Provirus. J. Virol. 2019, 93, e00495-19. [Google Scholar] [CrossRef] [Green Version]
- Acchioni, C.; Palermo, E.; Sandini, S.; Acchioni, M.; Hiscott, J.; Sgarbanti, M. Fighting HIV-1 Persistence: At the Crossroads of “Shoc-K and B-Lock”. Pathogens 2021, 10, 1517. [Google Scholar] [CrossRef]
- Mediouni, S.; Lyu, S.; Schader, S.M.; Valente, S.T. Forging a Functional Cure for HIV: Transcription Regulators and Inhibitors. Viruses 2022, 14, 1980. [Google Scholar] [CrossRef]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone Deacetylase Inhibitor Romidepsin Induces HIV Expression in CD4 T Cells from Patients on Suppressive Antiretroviral Therapy at Concentrations Achieved by Clinical Dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef] [Green Version]
- Novis, C.L.; Archin, N.M.; Buzon, M.J.; Verdin, E.; Round, J.L.; Lichterfeld, M.; Margolis, D.M.; Planelles, V.; Bosque, A. Reactivation of latent HIV-1 in central memory CD4+T cells through TLR-1/2 stimulation. Retrovirology 2013, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Brogdon, J.; Ziani, W.; Wang, X.; Veazey, R.S.; Xu, H. In vitro effects of the small-molecule protein kinase C agonists on HIV latency reactivation. Sci. Rep. 2016, 6, 39032. [Google Scholar] [CrossRef] [Green Version]
- Vemula, S.V.; Maxwell, J.W.; Nefedov, A.; Wan, B.-L.; Steve, J.; Newhard, W.; Sanchez, R.I.; Tellers, D.; Barnard, R.J.; Blair, W.; et al. Identification of proximal biomarkers of PKC agonism and evaluation of their role in HIV reactivation. Antivir. Res. 2017, 139, 161–170. [Google Scholar] [CrossRef]
- Badia, R.; Pujantell, M.; Riveira-Muñoz, E.; Puig, T.; Torres-Torronteras, J.; Martí, R.; Clotet, B.; Ampudia, R.M.; Vives-Pi, M.; Esté, J.A.; et al. The G1/S Specific Cyclin D2 Is a Regulator of HIV-1 Restriction in Non-proliferating Cells. PLOS Pathog. 2016, 12, e1005829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, A.; Pauls, E.; Badia, R.; Torres-Torronteras, J.; Riveira-Muñoz, E.; Clotet, B.; Martí, R.; Ballana, E.; Esté, J.A. Cyclin D3-dependent control of the dNTP pool and HIV-1 replication in human macrophages. Cell Cycle 2015, 14, 1657–1665. [Google Scholar] [CrossRef]
- Li, P.; Kaiser, P.; Lampiris, H.W.; Kim, P.; Yukl, S.A.; Havlir, D.V.; Greene, W.C.; Wong, J.K. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat. Med. 2016, 22, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Vidal, E.; Castellví, M.; Pujantell, M.; Badia, R.; Jou, A.; Gomez, L.; Puig, T.; Clotet, B.; Ballana, E.; Riveira-Muñoz, E.; et al. Evaluation of the Innate Immune Modulator Acitretin as a Strategy To Clear the HIV Reservoir. Antimicrob. Agents Chemother. 2017, 61, e01368-17. [Google Scholar] [CrossRef] [Green Version]
- Vandergeeten, C.; Fromentin, R.; DaFonseca, S.; Lawani, M.B.; Sereti, I.; Lederman, M.M.; Ramgopal, M.; Routy, J.P.; Sékaly, R.P.; Chomont, N. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 2013, 121, 4321–4329. [Google Scholar] [CrossRef]
- Pujantell, M.; Badia, R.; Ramirez, C.; Puig, T.; Clotet, B.; Ballana, E.; Esté, J.A.; Riveira-Muñoz, E. Long-term HIV-1 infection induces an antiviral state in primary macrophages. Antivir. Res. 2016, 133, 145–155. [Google Scholar] [CrossRef]
- Clark, K.; Plater, L.; Peggie, M.; Cohen, P. Use of the Pharmacological Inhibitor BX795 to Study the Regulation and Physiological Roles of TBK1 and IκB Kinase Epsilon: A Distinct Upstream Kinase Mediates Ser-172 Phosphorylation and Activation. J. Biol. Chem. 2009, 284, 14136–14146. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018, 8, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Muvaffak, A.; Pan, Q.; Yan, H.; Fernandez, R.; Lim, J.; Dolinski, B.; Nguyen, T.T.; Strack, P.; Wu, S.; Chung, R.; et al. Evaluating TBK1 as a Therapeutic Target in Cancers with Activated IRF3. Mol. Cancer Res. 2014, 12, 1055–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, D.W.; Poeckel, D.; Zinn, N.; Rau, C.; Strohmer, K.; Wagner, A.J.; Graves, A.P.; Perrin, J.; Bantscheff, M.; Duempelfeld, B.; et al. Discovery of GSK8612, a Highly Selective and Potent TBK1 Inhibitor. ACS Med. Chem. Lett. 2019, 10, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Kishore, N.; Sommers, C.; Mathialagan, S.; Guzova, J.; Yao, M.; Hauser, S.; Huynh, K.; Bonar, S.; Mielke, C.; Albee, L.; et al. A Selective IKK-2 Inhibitor Blocks NF-κ B-Dependent Gene Expression in Interleukin-1 Beta-Stimulated Synovial Fibroblasts. J. Biol. Chem. 2003, 278, 32861–32871. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezeonwumelu, I.J.; Garcia-Vidal, E.; Riveira-Muñoz, E.; Felip, E.; Gutiérrez-Chamorro, L.; Calba, I.; Massanella, M.; Sirera, G.; Clotet, B.; Ballana, E.; et al. Pharmacological Inhibition of IKK to Tackle Latency and Hyperinflammation in Chronic HIV-1 Infection. Int. J. Mol. Sci. 2022, 23, 15000. https://doi.org/10.3390/ijms232315000
Ezeonwumelu IJ, Garcia-Vidal E, Riveira-Muñoz E, Felip E, Gutiérrez-Chamorro L, Calba I, Massanella M, Sirera G, Clotet B, Ballana E, et al. Pharmacological Inhibition of IKK to Tackle Latency and Hyperinflammation in Chronic HIV-1 Infection. International Journal of Molecular Sciences. 2022; 23(23):15000. https://doi.org/10.3390/ijms232315000
Chicago/Turabian StyleEzeonwumelu, Ifeanyi Jude, Edurne Garcia-Vidal, Eva Riveira-Muñoz, Eudald Felip, Lucía Gutiérrez-Chamorro, Ignasi Calba, Marta Massanella, Guillem Sirera, Bonaventura Clotet, Ester Ballana, and et al. 2022. "Pharmacological Inhibition of IKK to Tackle Latency and Hyperinflammation in Chronic HIV-1 Infection" International Journal of Molecular Sciences 23, no. 23: 15000. https://doi.org/10.3390/ijms232315000
APA StyleEzeonwumelu, I. J., Garcia-Vidal, E., Riveira-Muñoz, E., Felip, E., Gutiérrez-Chamorro, L., Calba, I., Massanella, M., Sirera, G., Clotet, B., Ballana, E., & Badia, R. (2022). Pharmacological Inhibition of IKK to Tackle Latency and Hyperinflammation in Chronic HIV-1 Infection. International Journal of Molecular Sciences, 23(23), 15000. https://doi.org/10.3390/ijms232315000