
Genome-Wide DNA Methylation Profile Indicates Potential Epigenetic Regulation of Aging in the Rhesus Macaque Thymus



Abstract
1. Introduction
2. Results
2.1. WGBS Data Processing and Alignment
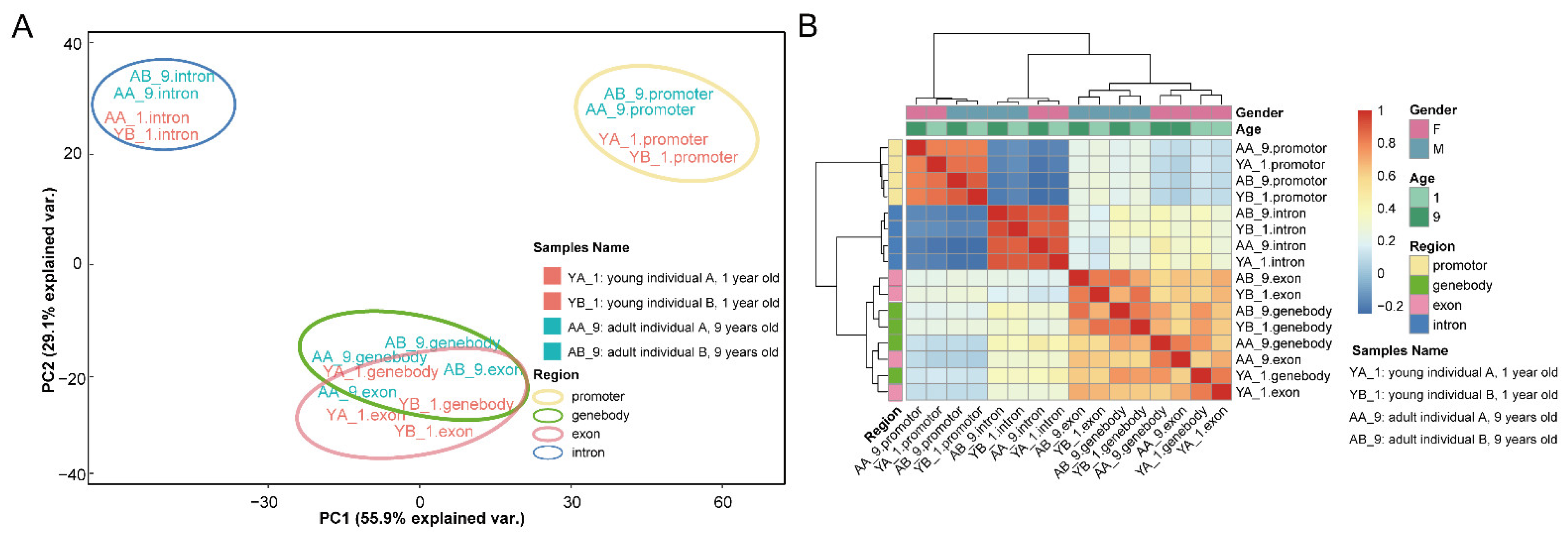
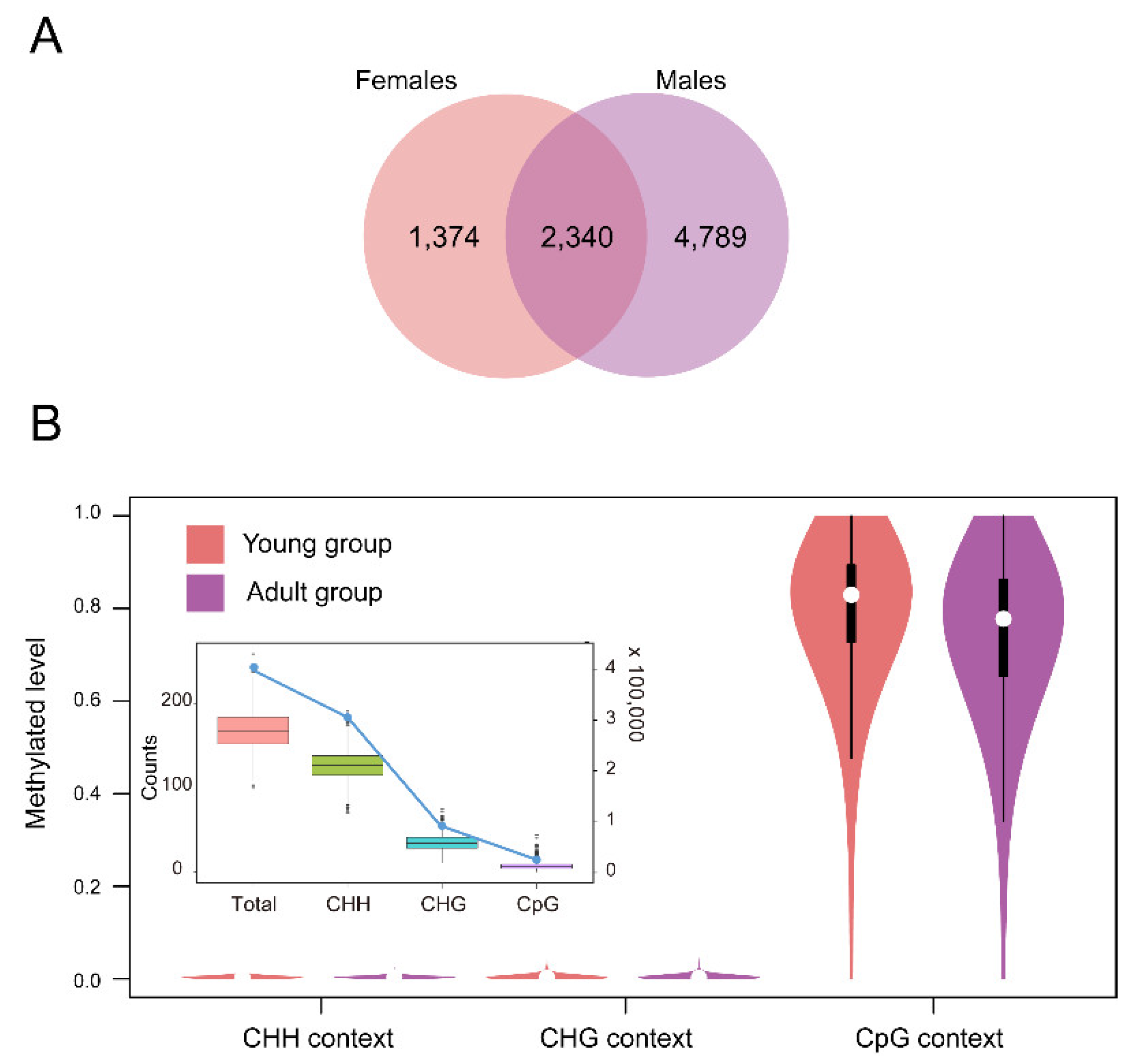
2.2. DNA Methylation Profiles of the Thymus in Rhesus Macaques
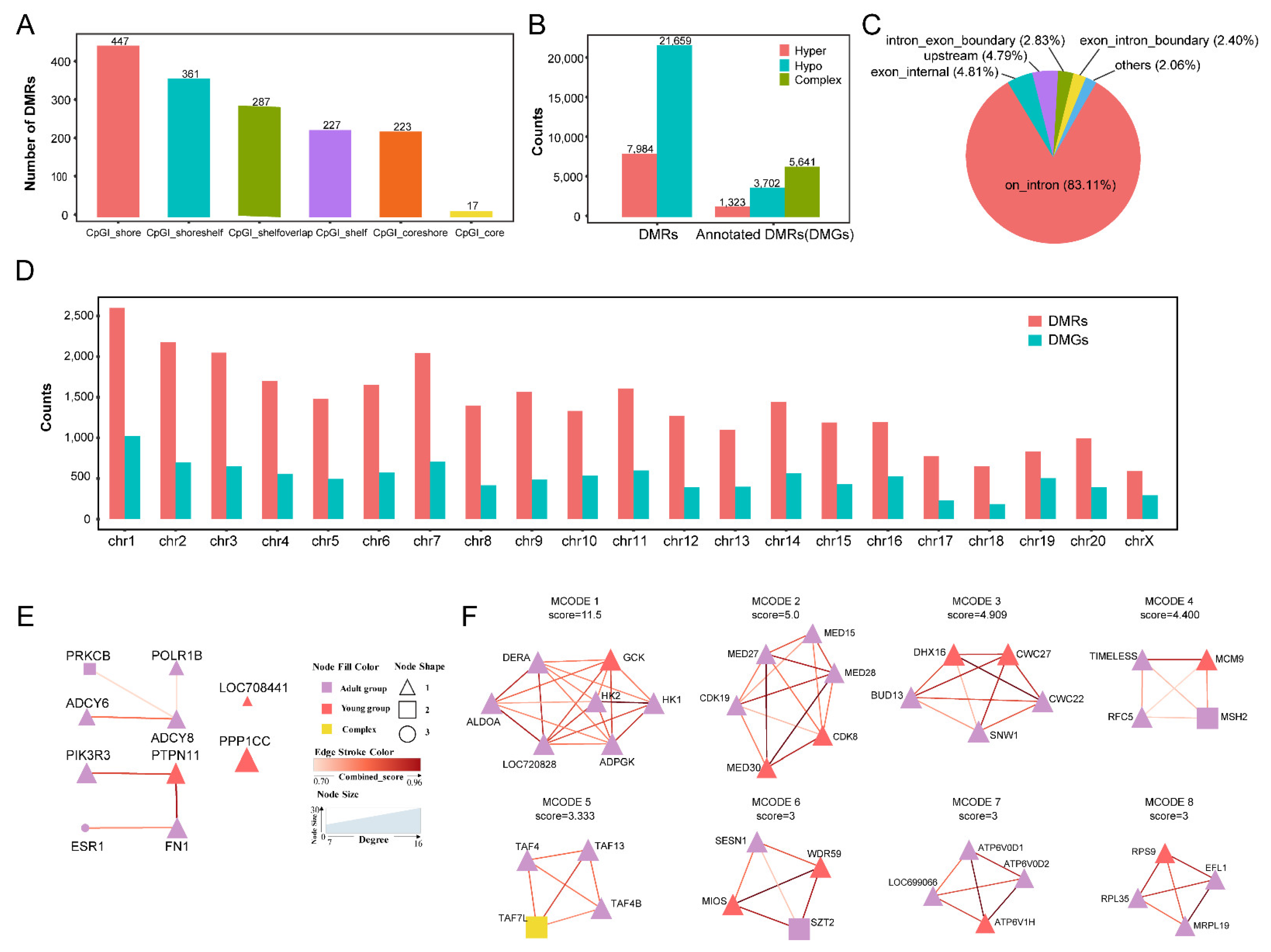
2.3. DMGs and ASMs from the Thymus between the Two Age Groups
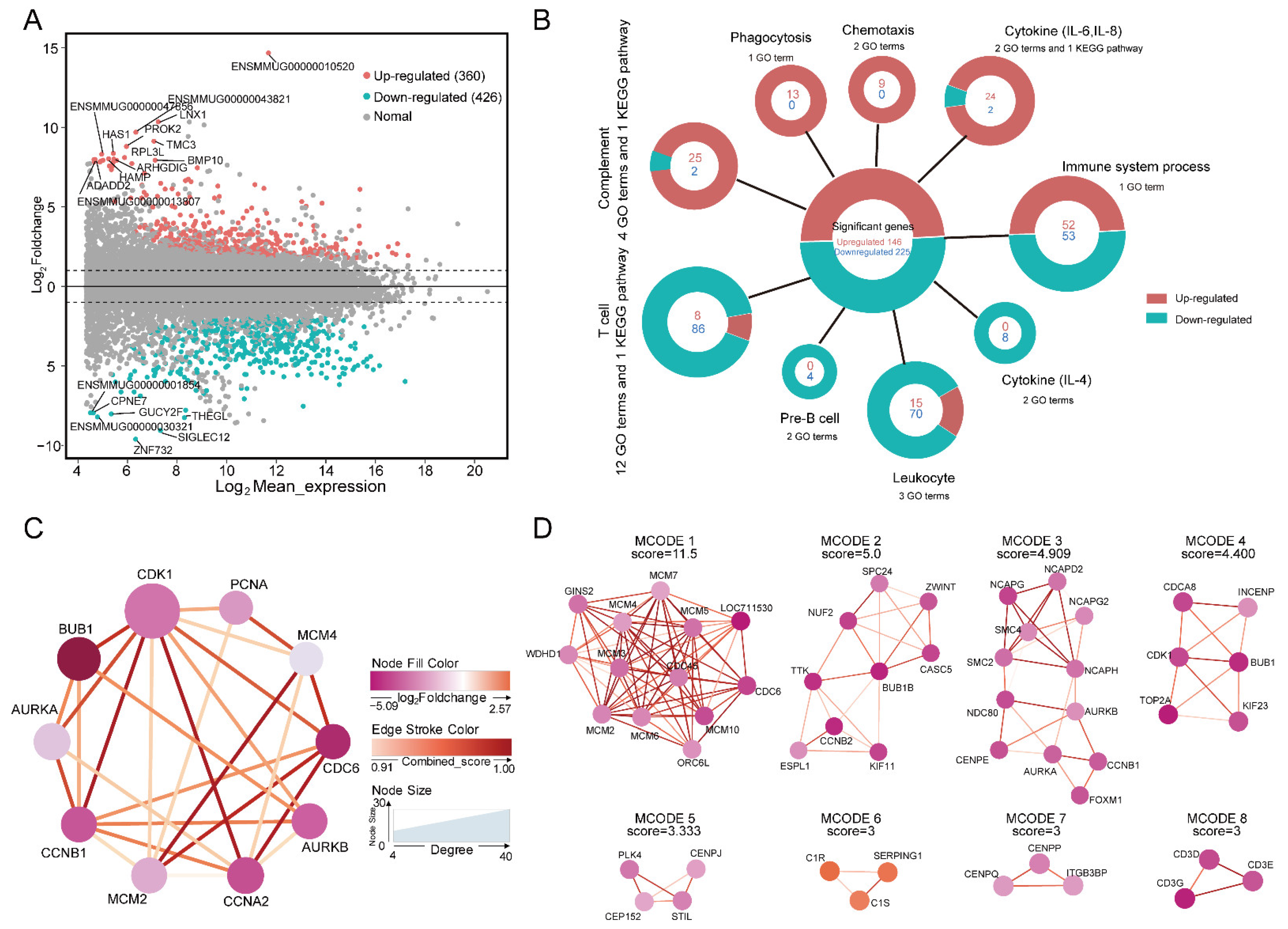
2.4. DEGs of the Thymus between the Two Age Groups in Macaques
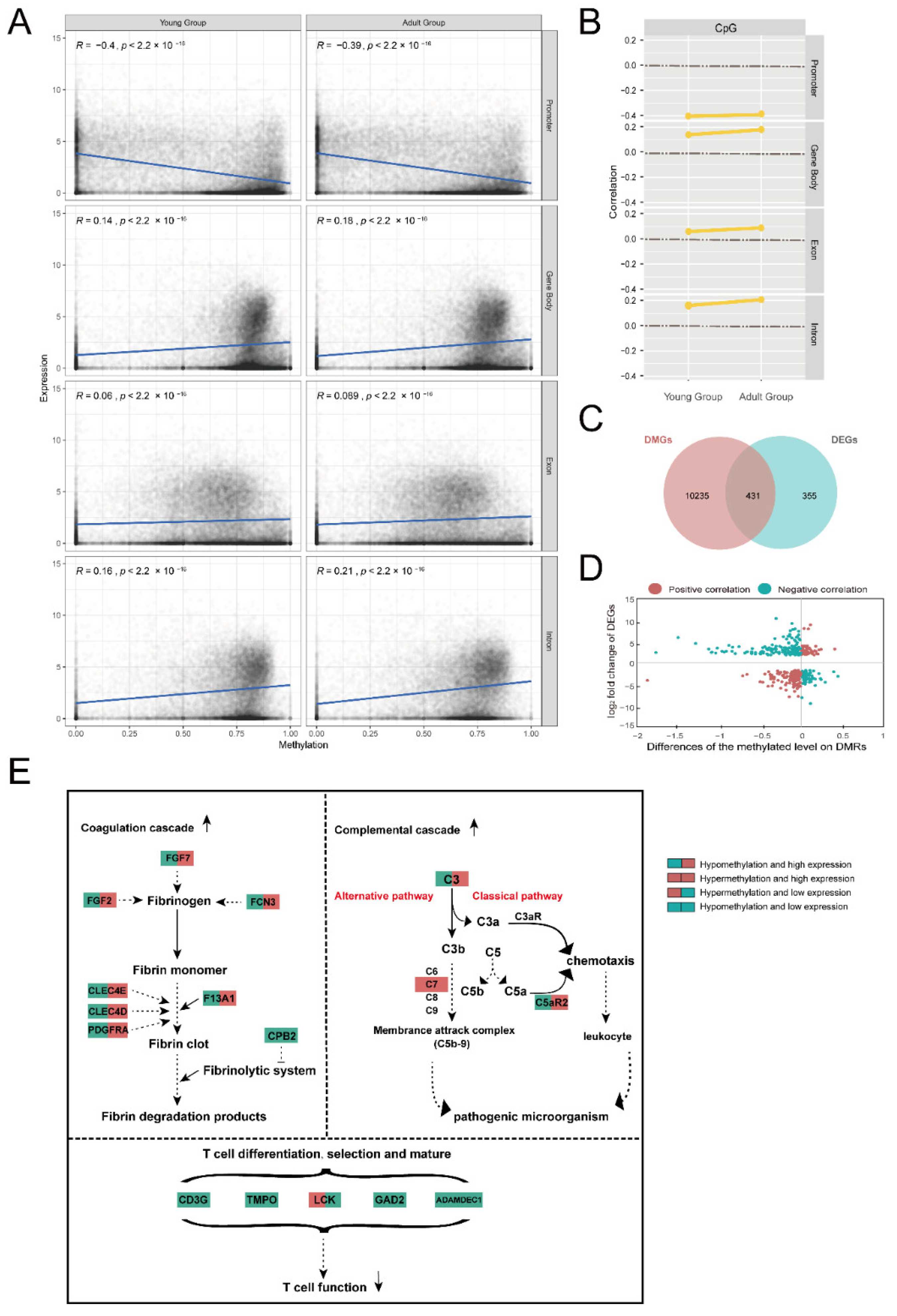
2.5. Associations of Methylation and Expression
2.6. Promoter Methylation
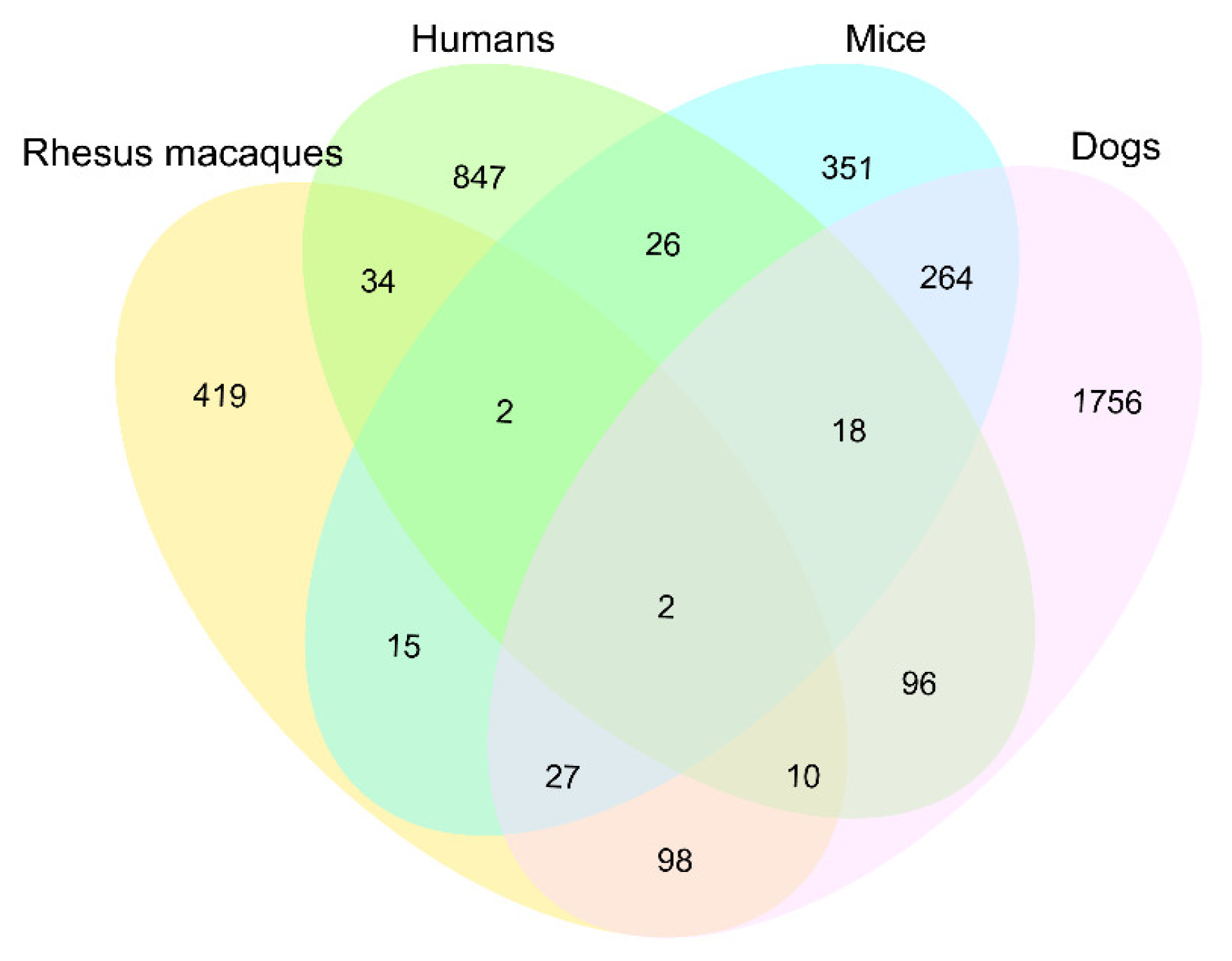
2.7. Comparisons of the Age-Related Methylated Genes in Four Mammals
3. Discussion
3.1. DNA Methylation Profiles of the Thymus in the Rhesus Macaque
3.2. Age-Related Alterations of DNA Methylation and Gene Expression
3.3. Changes in Immune Function of the Thymus with Age
3.4. Comparisons of Age-Related Methylated Genes in Four Mammals
4. Materials and Methods
4.1. Sample Collection
4.2. Whole-Genome Bisulfite Library and RNA Library Preparation and Sequencing
4.3. Data Quality Control and Alignment
4.4. Differentially Methylated Regions Analysis and Annotation
4.5. Promoter Methylation Analysis
4.6. Associations of Methylation and Expression
4.7. Protein–Protein Interaction Network Analysis
4.8. Gene Annotation and Multi-Species Synteny
4.9. Identification of ASM Sites and aDMRs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Owen, D.L.; Sjaastad, L.E.; Farrar, M.A. Regulatory T Cell Development in the Thymus. J. Immunol. 2019, 203, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.P.; Dabrowska-Bernstein, B. Immunoregulatory Role of Thymus; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Aspinall, R.; Pitts, D.; Lapenna, A.; Mitchell, W. Immunity in the elderly: The role of the thymus. J. Comp. Pathol. 2010, 142 (Suppl. 1), S111–S115. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.M.; Keown, A.; Policheni, A.; Roesley, S.N.A.; Rivlin, N.; Kadouri, N.; Ritchie, M.E.; Jain, R.; Abramson, J.; Heng, T.S.P.; et al. Thymospheres Are Formed by Mesenchymal Cells with the Potential to Generate Adipocytes, but Not Epithelial Cells. Cell Rep. 2017, 21, 934–942. [Google Scholar] [CrossRef]
- Palmer, D.B. The effect of age on thymic function. Front. Immunol. 2013, 4, 316. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun. Ageing 2020, 17, 2. [Google Scholar] [CrossRef]
- Zaina, S.; Perez-Luque, E.L.; Lund, G. Genetics talks to epigenetics? The interplay between sequence variants and chromatin structure. Curr. Genom. 2010, 11, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K.; et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef]
- Jeziorska, D.M.; Murray, R.J.S.; De Gobbi, M.; Gaentzsch, R.; Garrick, D.; Ayyub, H.; Chen, T.; Li, E.; Telenius, J.; Lynch, M.; et al. DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E7526–E7535. [Google Scholar] [CrossRef] [PubMed]
- Pai, A.A.; Bell, J.T.; Marioni, J.C.; Pritchard, J.K.; Gilad, Y. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 2011, 7, e1001316. [Google Scholar] [CrossRef]
- Michaelsen, S.R.; Aslan, D.; Urup, T.; Poulsen, H.S.; Gronbaek, K.; Broholm, H.; Kristensen, L.S. DNA Methylation Levels of the ELMO Gene Promoter CpG Islands in Human Glioblastomas. Int. J. Mol. Sci. 2018, 19, 679. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, V.; Lurie, E.; Carrero, I.; Pawliczek, P.; Patel, R.Y.; Rozowsky, J.; Galeev, T.; Huang, Z.; Altshuler, R.C.; Zhang, Z.; et al. Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci. Science 2018, 361, eaar3146. [Google Scholar] [CrossRef] [PubMed]
- Gertz, J.; Varley, K.E.; Reddy, T.E.; Bowling, K.M.; Pauli, F.; Parker, S.L.; Kucera, K.S.; Willard, H.F.; Myers, R.M. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011, 7, e1002228. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Sun, M.A.; Wang, Z.; Wang, Q.; Li, Q.; Xie, H. Characterization and machine learning prediction of allele-specific DNA methylation. Genomics 2015, 106, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.P.; Bartolomei, M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef] [PubMed]
- Kerkel, K.; Spadola, A.; Yuan, E.; Kosek, J.; Jiang, L.; Hod, E.; Li, K.; Murty, V.V.; Schupf, N.; Vilain, E.; et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 2008, 40, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F.; et al. Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 2007, 357, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Andrew, M.; Paes, B.; Milner, R.; Johnston, M.; Mitchell, L.; Tollefsen, D.M.; Powers, P. Development of the human coagulation system in the full-term infant. Blood 1987, 70, 165–172. [Google Scholar] [CrossRef]
- Maegawa, S.; Lu, Y.; Tahara, T.; Lee, J.T.; Madzo, J.; Liang, S.; Jelinek, J.; Colman, R.J.; Issa, J.J. Caloric restriction delays age-related methylation drift. Nat. Commun. 2017, 8, 539. [Google Scholar] [CrossRef]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef]
- Zhang, M.; Xi, Z.; Ghani, M.; Jia, P.; Pal, M.; Werynska, K.; Moreno, D.; Sato, C.; Liang, Y.; Robertson, J.; et al. Genetic and epigenetic study of ALS-discordant identical twins with double mutations in SOD1 and ARHGEF28. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Bimber, B.N.; Yan, M.Y.; Peterson, S.M.; Ferguson, B. mGAP: The macaque genotype and phenotype resource, a framework for accessing and interpreting macaque variant data, and identifying new models of human disease. BMC Genom. 2019, 20, 176. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Guan, P.; Zhu, Z.; Cheng, S.; Zhou, C.; Wang, H.; Xu, Q.; Sung, W.K.; Li, G. ASMdb: A comprehensive database for allele-specific DNA methylation in diverse organisms. Nucleic Acids Res. 2022, 50, D60–D71. [Google Scholar] [CrossRef] [PubMed]
- Shanley, D.P.; Aw, D.; Manley, N.R.; Palmer, D.B. An evolutionary perspective on the mechanisms of immunosenescence. Trends Immunol. 2009, 30, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Buse, E.; Habermann, G.; Vogel, F. Thymus development in Macaca fascicularis (Cynomolgus monkey): An approach for toxicology and embryology. J. Mol. Histol. 2006, 37, 161–170. [Google Scholar] [CrossRef]
- Rezzani, R.; Nardo, L.; Favero, G.; Peroni, M.; Rodella, L.F. Thymus and aging: Morphological, radiological, and functional overview. Age 2014, 36, 313–351. [Google Scholar] [CrossRef]
- Palmer, S.; Albergante, L.; Blackburn, C.C.; Newman, T.J. Thymic involution and rising disease incidence with age. Proc. Natl. Acad. Sci. USA 2018, 115, 1883–1888. [Google Scholar] [CrossRef]
- Polyakova, V.O.; Linkova, N.S.; Kvetnoy, I.M.; Khavinson, V. Functional unity of the thymus and pineal gland and study of the mechanisms of aging. Bull. Exp. Biol. Med. 2011, 151, 627–630. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, L.; Huang, Y.; Shen, F.; Wu, H.; Yang, Z.; Hou, R.; Song, Z.; Yue, B.; Zhang, X. Epigenomic profiling indicates a role for DNA methylation in the postnatal liver and pancreas development of giant pandas. Genomics 2022, 114, 110342. [Google Scholar] [CrossRef]
- Price, A.J.; Collado-Torres, L.; Ivanov, N.A.; Xia, W.; Burke, E.E.; Shin, J.H.; Tao, R.; Ma, L.; Jia, Y.; Hyde, T.M.; et al. Divergent neuronal DNA methylation patterns across human cortical development reveal critical periods and a unique role of CpH methylation. Genome Biol. 2019, 20, 196. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, L.; Yang, Q.; Yan, C.; Jiang, P.; Lan, Y.; Wang, J.; Tang, R.; He, M.; Lei, G.; et al. Age-related gene expression and DNA methylation changes in rhesus macaque. Genomics 2020, 112, 5147–5156. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lei, X.; Lu, H.; Guo, W.; Wu, S.; Yin, Z.; Sun, Q.; Yang, X. Age-Related Changes on CD40 Promotor Methylation and Immune Gene Expressions in Thymus of Chicken. Front. Immunol. 2018, 9, 2731. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, K.; Zhang, K.; Ameri, A.; Huo, J.; Atoda, H.; Kurachi, S. Genetic and molecular mechanisms of age regulation (homeostasis) of blood coagulation. IUBMB Life 2000, 49, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.A. The human complement system: Assembly of the classical pathway C3 convertase. Biochem. J. 1980, 189, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Maegawa, S.; Chung, W.; Garriga, J.; Jelinek, J.; Estecio, M.R.; Shibata, T.; Hirata, I.; Arisawa, T.; Issa, J.P. Examination of whole blood DNA methylation as a potential risk marker for gastric cancer. Cancer Prev. Res. 2013, 6, 1093–1100. [Google Scholar] [CrossRef]
- Jost, J.P.; Bruhat, A. The formation of DNA methylation patterns and the silencing of genes. Prog. Nucleic Acid Res. Mol. Biol. 1997, 57, 217–248. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zoller, J.A.; Haghani, A.; Jasinska, A.J.; Raj, K.; Breeze, C.E.; Ernst, J.; Vaughan, K.L.; Mattison, J.A. Epigenetic clock and methylation studies in the rhesus macaque. Geroscience 2021, 43, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, H.; Sun, J.; Zhou, Y.; Zhu, G.; Wu, S.; Bao, W. Age-associated changes in DNA methylation and expression of the TNFalpha gene in pigs. Genes Genet. Syst. 2018, 93, 191–198. [Google Scholar] [CrossRef]
- King, G.D.; Rosene, D.L.; Abraham, C.R. Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age 2012, 34, 1405–1419. [Google Scholar] [CrossRef]
- Gotze, O.; Muller-Eberhard, H.J. The c3-activator system: An alternate pathway of complement activation. J. Exp. Med. 1971, 134, 90–108. [Google Scholar] [CrossRef] [PubMed]
- Janatova, J. C3, C5 components and C3a, C4a, and C5a fragments of the complement system. Methods Enzymol. 1988, 162, 579–625. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, K.; Kurachi, S. Genetic mechanisms of age regulation of blood coagulation: Factor IX model. Arter. Thromb. Vasc. Biol. 2000, 20, 902–906. [Google Scholar] [CrossRef]
- Rallapalli, P.M.; Orengo, C.A.; Studer, R.A.; Perkins, S.J. Positive selection during the evolution of the blood coagulation factors in the context of their disease-causing mutations. Mol. Biol. Evol. 2014, 31, 3040–3056. [Google Scholar] [CrossRef] [PubMed]
- Anwar, R.; Valleley, E.M.; Trinh, C.H. Novel human pathological mutations. Gene symbol: F13A1. Disease: Factor XIII Deficiency. Hum. Genet. 2010, 127, 115–116. [Google Scholar] [PubMed]
- Gemmati, D.; Vigliano, M.; Burini, F.; Mari, R.; El Mohsein, H.H.; Parmeggiani, F.; Serino, M.L. Coagulation Factor XIIIA (F13A1): Novel Perspectives in Treatment and Pharmacogenetics. Curr. Pharm. Des. 2016, 22, 1449–1459. [Google Scholar] [CrossRef]
- Aw, D.; Palmer, D.B. It’s not all equal: A multiphasic theory of thymic involution. Biogerontology 2012, 13, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Labouesse, M.A.; Dong, E.; Grayson, D.R.; Guidotti, A.; Meyer, U. Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 2015, 10, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, N.R.; Chew, T.S.; Dunne, J.; Marnane, R.; Nedjat-Shokouhi, B.; Smith, P.J.; Bloom, S.L.; Smith, A.M.; Segal, A.W. Critical Role of the Disintegrin Metalloprotease ADAM-like Decysin-1 [ADAMDEC1] for Intestinal Immunity and Inflammation. J. Crohn’s Colitis 2016, 10, 1417–1427. [Google Scholar] [CrossRef]
- Rezzani, R.; Franco, C.; Hardeland, R.; Rodella, L.F. Thymus-Pineal Gland Axis: Revisiting Its Role in Human Life and Ageing. Int. J. Mol. Sci. 2020, 21, 8806. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Jasinska, A.J.; Haghani, A.; Zoller, J.A.; Li, C.Z.; Arneson, A.; Ernst, J.; Kavanagh, K.; Jorgensen, M.J.; Mattison, J.A.; Wojta, K.; et al. Epigenetic clock and methylation studies in vervet monkeys. Geroscience 2022, 44, 699–717. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Mozhui, K.; Lu, A.T.; Li, C.Z.; Haghani, A.; Sandoval-Sierra, J.V.; Wu, Y.; Williams, R.W.; Horvath, S. Genetic loci and metabolic states associated with murine epigenetic aging. Elife 2022, 11, e75244. [Google Scholar] [CrossRef] [PubMed]
- Petkovich, D.A.; Podolskiy, D.I.; Lobanov, A.V.; Lee, S.G.; Miller, R.A.; Gladyshev, V.N. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell Metab. 2017, 25, 954. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; Szladovits, B.; Haghani, A.; Zoller, J.A.; Li, C.Z.; Black, P.; Maddox, D.; Robeck, T.R.; Horvath, S. Epigenetic clock and methylation studies in cats. Geroscience 2021, 43, 2363–2378. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, T.M.; Bonder, M.J.; Stark, A.K.; Krueger, F.; Team, B.I.A.C.; von Meyenn, F.; Stegle, O.; Reik, W. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017, 18, 68. [Google Scholar] [CrossRef]
- Thompson, M.J.; vonHoldt, B.; Horvath, S.; Pellegrini, M. An epigenetic aging clock for dogs and wolves. Aging 2017, 9, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tsui, B.; Kreisberg, J.F.; Robertson, N.A.; Gross, A.M.; Yu, M.K.; Carter, H.; Brown-Borg, H.M.; Adams, P.D.; Ideker, T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017, 18, 57. [Google Scholar] [CrossRef] [PubMed]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb-prot5439. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Frazee, A.C.; Pertea, G.; Jaffe, A.E.; Langmead, B.; Salzberg, S.L.; Leek, J.T. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015, 33, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, P.A.; Chatterjee, A.; Rodger, E.J.; Morison, I.M. DMAP: Differential methylation analysis package for RRBS and WGBS data. Bioinformatics 2014, 30, 1814–1822. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Xia, L.; Tu, K.; Duan, M.; Kukurba, K.; Li-Pook-Than, J.; Xie, D.; Snyder, M. Longitudinal personal DNA methylome dynamics in a human with a chronic condition. Nat. Med. 2018, 24, 1930–1939. [Google Scholar] [CrossRef]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.V.; Davies, R.; Down, T.A.; et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer International Publishing: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jockel, K.H.; Erbel, R.; Muhleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef]
- Guevara, E.E.; Hopkins, W.D.; Hof, P.R.; Ely, J.J.; Bradley, B.J.; Sherwood, C.C. Comparative analysis reveals distinctive epigenetic features of the human cerebellum. PLoS Genet. 2021, 17, e1009506. [Google Scholar] [CrossRef] [PubMed]
- Kasprzyk, A. BioMart: Driving a paradigm change in biological data management. Database 2011, 2011, bar049. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Durinck, S.; Pandini, L.; Provero, P.; Allen, J.; Arnaiz, O.; Awedh, M.H.; Baldock, R.; Barbiera, G.; et al. The BioMart community portal: An innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015, 43, W589–W598. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhu, P.; Pellegrini, M.; Zhang, M.Q.; Wang, X.; Ni, Z. CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics 2018, 34, 381–387. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway and Term | Pathway and Term ID | Gene Name | Ensembl ID | DNA Methylation Difference | Expression Change |
|---|---|---|---|---|---|
| Complement and coagulation cascades | KEGG:04610 | C3 | ENSMMUG00000008693 | − | + |
| Complement and coagulation cascades | KEGG:04610 | F13A1 | ENSMMUG00000001248 | − | + |
| Complement and coagulation cascades | KEGG:04610 | CPB2 | ENSMMUG00000017029 | − | − |
| Complement and coagulation cascades; immune system process | KEGG:04610; GO:0002376 | C7 | ENSMMUG00000014171 | + | + |
| Immune system process | GO:0002376 | C5AR2 | ENSMMUG00000009277 | − | + |
| Immune system process | GO:0002376 | CLEC4D | ENSMMUG00000013703 | − | + |
| Immune system process | GO:0002376 | CLEC4E | ENSMMUG00000013706 | − | + |
| Immune system process | GO:0002376 | DNASE1L3 | ENSMMUG00000011235 | − | − |
| Immune system process | GO:0002376 | FCN3 | ENSMMUG00000018322 | − | + |
| Immune system process | GO:0002376 | HELLS | ENSMMUG00000017255 | + | − |
| Immune system process | GO:0002376 | LCK | ENSMMUG00000040694 | + | − |
| Immune system process; system development | GO:0002376; GO:0048733 | FANCD2 | ENSMMUG00000008966 | − | − |
| Immune system process; system development | GO:0002376; GO:0048732 | LILRB4 | ENSMMUG00000047009 | + | − |
| Immune system process; system development | GO:0002376; GO:0048734 | PDGFRA | ENSMMUG00000017395 | − | + |
| Immune system process; system development | GO:0002376; GO:0048731 | RARA | ENSMMUG00000012486 | − | + |
| System development | GO:0048731 | ADGRG3 | ENSMMUG00000015690 | − | + |
| System development | GO:0048731 | CD3G | ENSMMUG00000017600 | − | − |
| System development | GO:0048731 | NOX4 | ENSMMUG00000011116 | − | + |
| System development | GO:0048731 | PROX1 | ENSMMUG00000011914 | − | + |
| Carboxy-lyase activity | GO:0016831 | GAD2 | ENSMMUG00000012233 | − | − |
| Cation binding | GO:0043169 | TRAIP | ENSMMUG00000016476 | + | − |
| Cell division | GO:0051301 | MIS18BP1 | ENSMMUG00000016515 | + | − |
| Cell–cell junction | GO:0005911 | CLDN11 | ENSMMUG00000009274 | − | + |
| Cellular response to stress | GO:0033554 | UBE2T | ENSMMUG00000013795 | − | − |
| Cytokinesis | GO:0000910 | KIF23 | ENSMMUG00000014887 | + | − |
| DNA polymerase binding | GO:0070182 | FANCI | ENSMMUG00000011155 | − | − |
| Hydrolase activity, hydrolyzing N-glycosyl compounds | GO:0016799 | NEIL3 | ENSMMUG00000007394 | + | − |
| Hydrolase activity, hydrolyzing O-glycosyl compounds | GO:0004553 | MGAM | ENSMMUG00000016273 | − | + |
| Immune response | GO:0006955 | CTSV | ENSMMUG00000022971 | − | − |
| Intrinsic component of membrane | GO:0031224 | TMPO | ENSMMUG00000023719 | − | − |
| Metallopeptidase activity | GO:0008237 | ADAMDEC1 | ENSMMUG00000005318 | − | − |
| Metallopeptidase activity | GO:0008237 | ADAM28 | ENSMMUG00000005317 | − | − |
| Microfilament motor activity | GO:0000146 | MYH8 | ENSMMUG00000009763 | − | − |
| Molecular function | GO:0003674 | DLEU7 | ENSMMUG00000021744 | − | − |
| Nucleic acid metabolic process | GO:0090304 | MCM4 | ENSMMUG00000015360 | + | − |
| Nucleobase-containing compound metabolic process | GO:0006139 | POLE2 | ENSMMUG00000003913 | − | − |
| Phosphoric ester hydrolase activity | GO:0042578 | PTPN7 | ENSMMUG00000013789 | − | − |
| Plus-end-directed microtubule motor activity | GO:0008574 | KIF11 | ENSMMUG00000023266 | + | − |
| Positive regulation of epithelial cell proliferation | GO:0050679 | FGF7 | ENSMMUG00000009842 | − | + |
| Protein catabolic process | GO:0030163 | LRR1 | ENSMMUG00000037526 | + | − |
| Protein catabolic process | GO:0030163 | LNX1 | ENSMMUG00000041981 | − | + |
| Protein modification process | GO:0036211 | FBXO32 | ENSMMUG00000023778 | − | + |
| Protein-containing complex | GO:0032991 | POLE | ENSMMUG00000015463 | − | − |
| Regulation of gene expression | GO:0010468 | FGF2 | ENSMMUG00000007419 | − | + |
| Response to stress | GO:0006950 | RAD51AP1 | ENSMMUG00000015189 | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, H.; Li, H.; Fan, R.; Song, Y.; Pan, X.; Zhang, C.; Li, J. Genome-Wide DNA Methylation Profile Indicates Potential Epigenetic Regulation of Aging in the Rhesus Macaque Thymus. Int. J. Mol. Sci. 2022, 23, 14984. https://doi.org/10.3390/ijms232314984
Qiu H, Li H, Fan R, Song Y, Pan X, Zhang C, Li J. Genome-Wide DNA Methylation Profile Indicates Potential Epigenetic Regulation of Aging in the Rhesus Macaque Thymus. International Journal of Molecular Sciences. 2022; 23(23):14984. https://doi.org/10.3390/ijms232314984
Chicago/Turabian StyleQiu, Hong, Haobo Li, Ruiwen Fan, Yang Song, Xuan Pan, Chunhui Zhang, and Jing Li. 2022. "Genome-Wide DNA Methylation Profile Indicates Potential Epigenetic Regulation of Aging in the Rhesus Macaque Thymus" International Journal of Molecular Sciences 23, no. 23: 14984. https://doi.org/10.3390/ijms232314984
APA StyleQiu, H., Li, H., Fan, R., Song, Y., Pan, X., Zhang, C., & Li, J. (2022). Genome-Wide DNA Methylation Profile Indicates Potential Epigenetic Regulation of Aging in the Rhesus Macaque Thymus. International Journal of Molecular Sciences, 23(23), 14984. https://doi.org/10.3390/ijms232314984