
Congenital Stationary Night Blindness: Clinical and Genetic Features

 , ,
, ,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
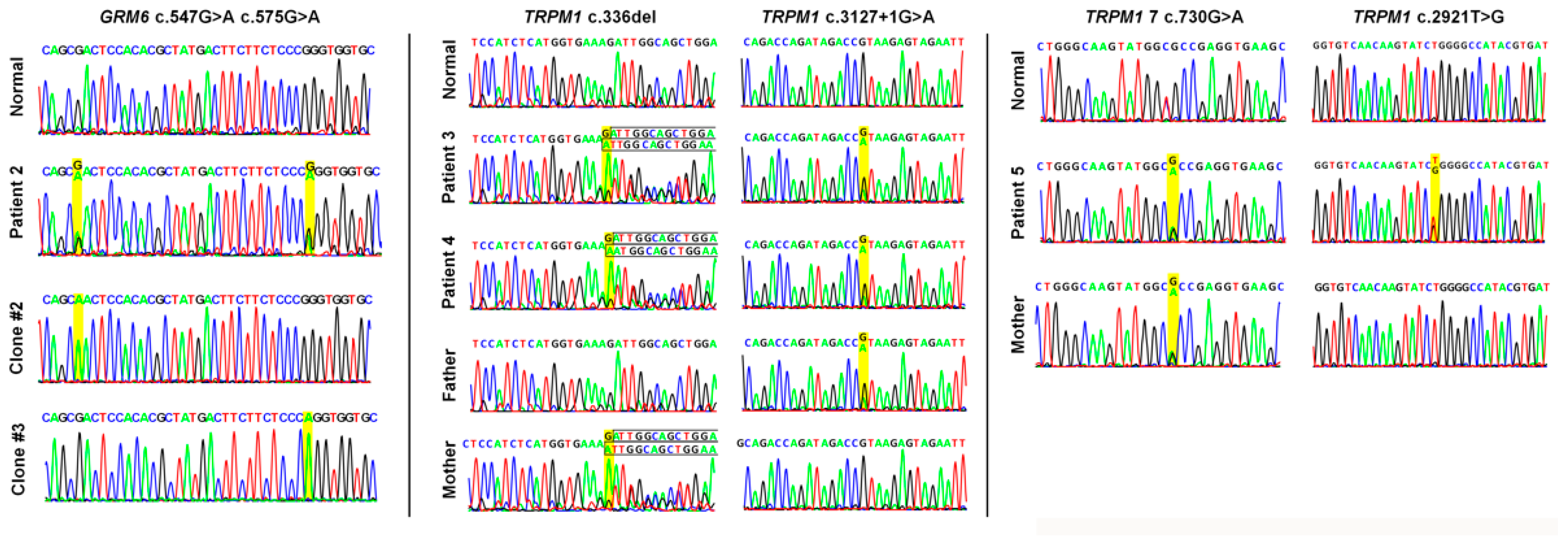
2.1. Demographics and Genetic Results
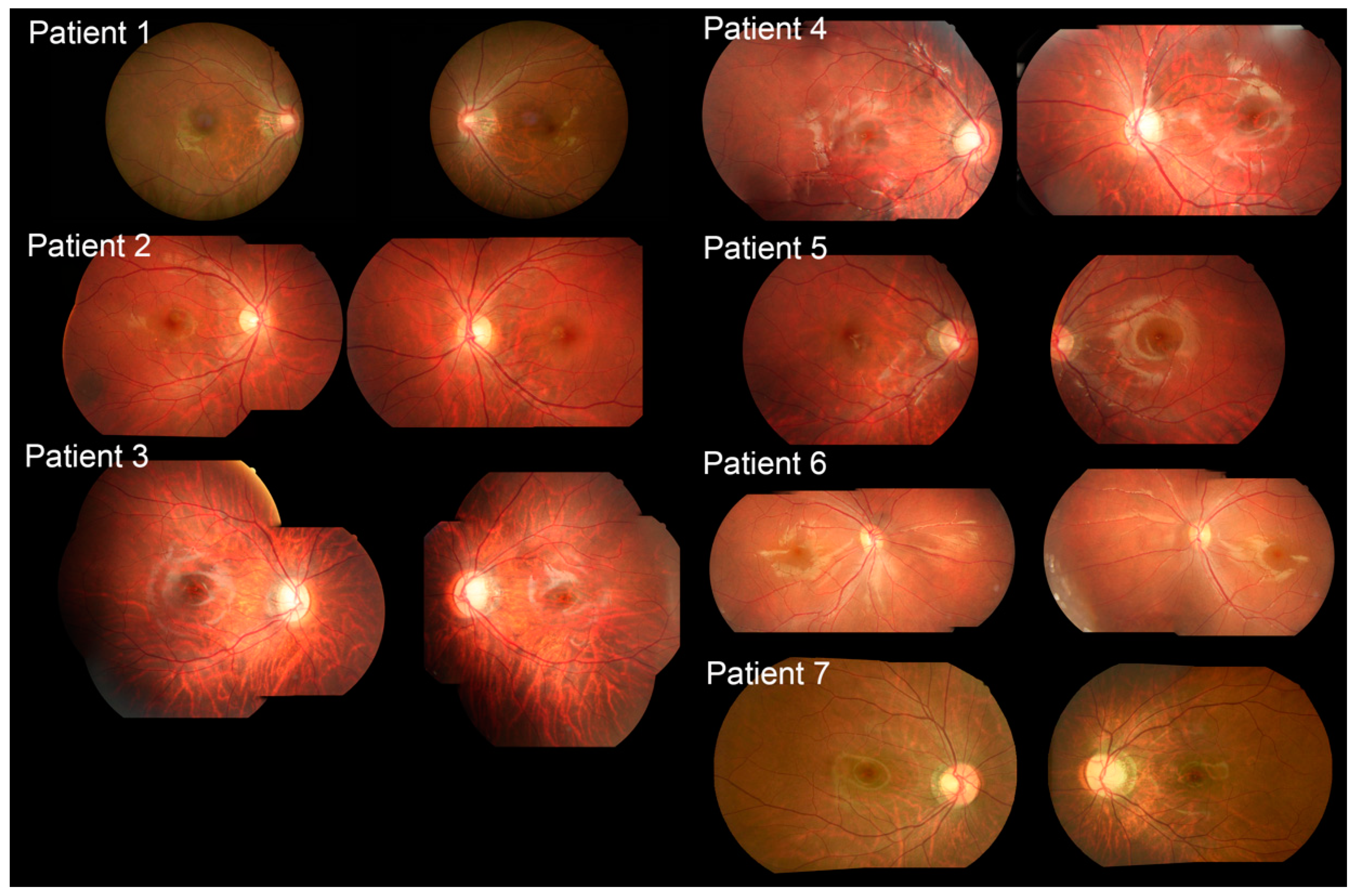
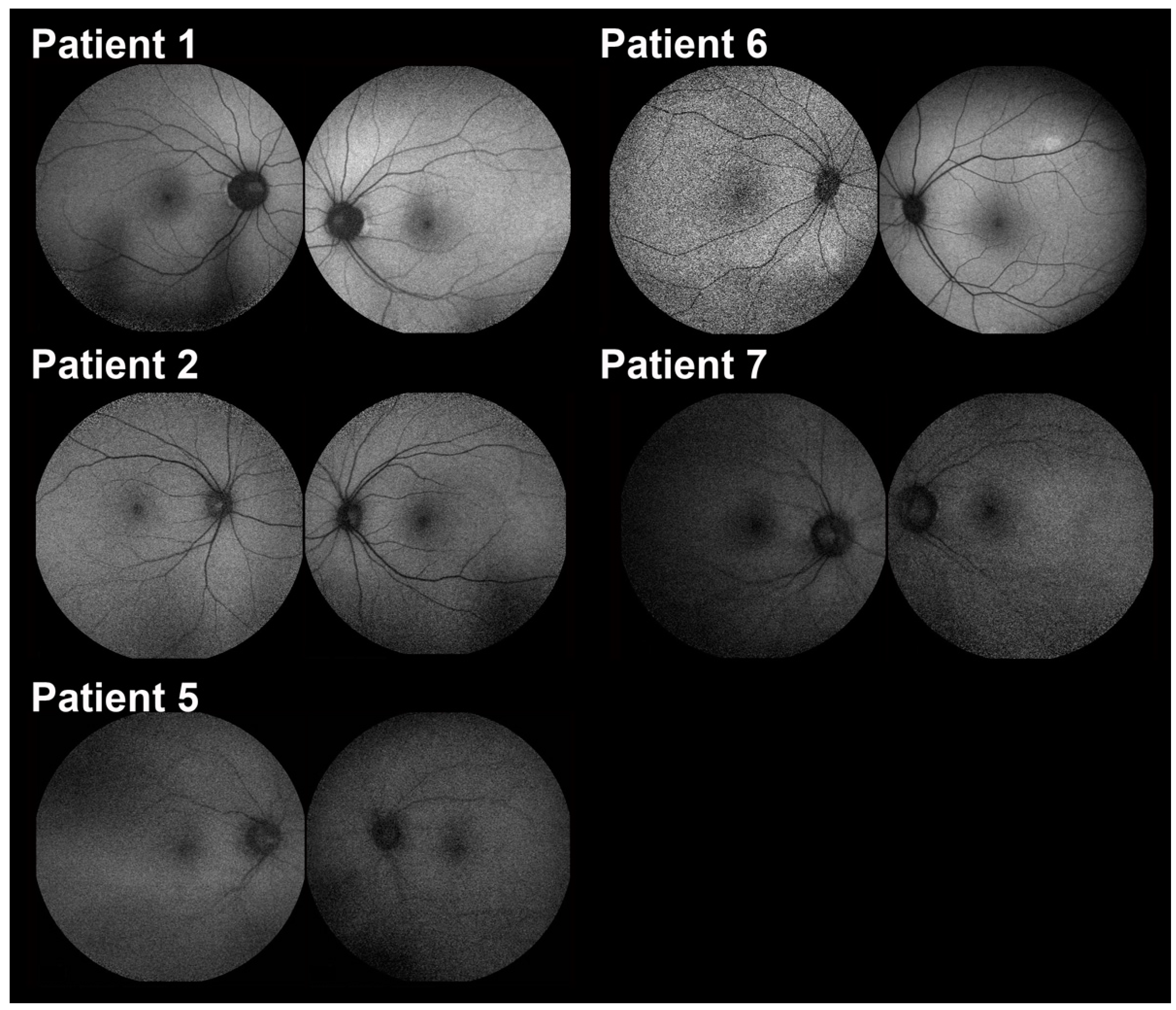
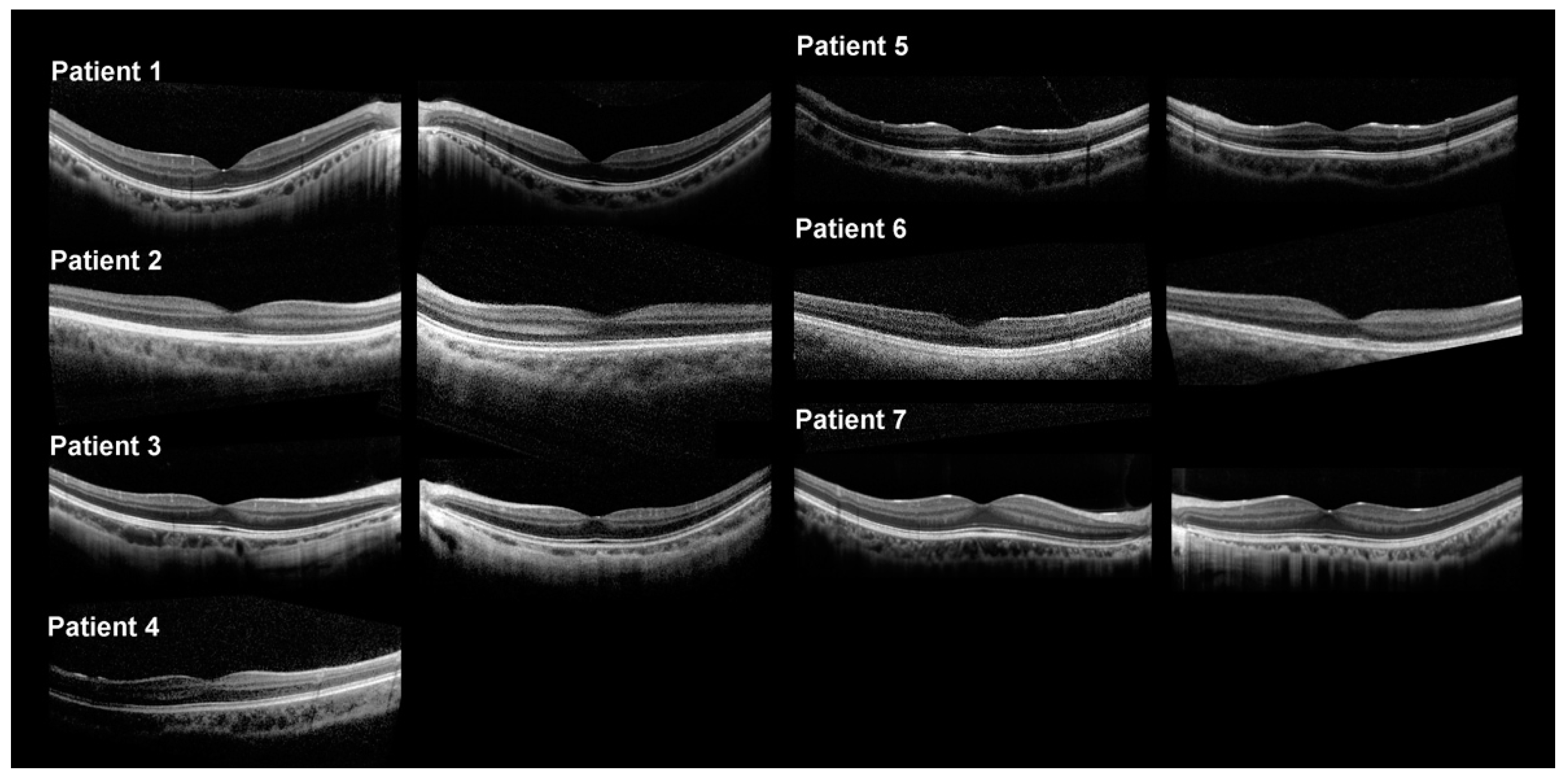
2.2. Clinical Features
3. Discussion
3.1. Pathophysiology and Clinical Presentations
3.2. Variant Interpretation and Clinical Significance Evaluation
3.3. Clinical Therapeutics
4. Methods and Materials
4.1. Patients
4.2. Ophthalmologic Examination
4.3. Genetic Analysis
4.4. Functional Impact Prediction of Missense Variants
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cunier, F. Histoire d’une héméralogie héréditaire depuis II siecles dans une famille de la commune de Vendérnian, près Montpellier. Ann. Soc. Méd. Gand. 1838, 4, 383–395. [Google Scholar]
- Tsang, S.H.; Sharma, T. Congenital Stationary Night Blindness. Adv. Exp. Med. Biol. 2018, 1085, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Pieh, C.; Simonsz-Toth, B.; Gottlob, I. Nystagmus characteristics in congenital stationary night blindness (CSNB). Br. J. Ophthalmol. 2008, 92, 236–240. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef]
- Marmor, M.F.; Zeitz, C. Riggs-type dominant congenital stationary night blindness: ERG findings, a new GNAT1 mutation and a systemic association. Doc. Ophthalmol. 2018, 137, 57–62. [Google Scholar] [CrossRef]
- Kabanarou, S.A.; Holder, G.E.; Fitzke, F.W.; Bird, A.C.; Webster, A.R. Congenital stationary night blindness and a "Schubert-Bornschein" type electrophysiology in a family with dominant inheritance. Br. J. Ophthalmol. 2004, 88, 1018–1022. [Google Scholar] [CrossRef]
- Miyake, Y.; Yagasaki, K.; Horiguchi, M.; Kawase, Y. On- and off-responses in photopic electroretinogram in complete and incomplete types of congenital stationary night blindness. Jpn. J. Ophthalmol. 1987, 31, 81–87. [Google Scholar]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef]
- Kobal, N.; Krasovec, T.; Sustar, M.; Volk, M.; Peterlin, B.; Hawlina, M.; Fakin, A. Stationary and Progressive Phenotypes Caused by the p.G90D Mutation in Rhodopsin Gene. Int. J. Mol. Sci. 2021, 22, 2133. [Google Scholar] [CrossRef]
- Bijveld, M.M.; Florijn, R.J.; Bergen, A.A.; van den Born, L.I.; Kamermans, M.; Prick, L.; Riemslag, F.C.; van Schooneveld, M.J.; Kappers, A.M.; van Genderen, M.M. Genotype and phenotype of 101 dutch patients with congenital stationary night blindness. Ophthalmology 2013, 120, 2072–2081. [Google Scholar] [CrossRef]
- Liu, X.; Zhuang, S.; Hu, S.; Zhang, F.; Lin, B.; Li, X.; Xu, D.; Chen, S.H. A dominant form of congenital stationary night blindness (adCSNB) in a large Chinese family. Ann. Hum. Genet. 2005, 69, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Gross, A.K.; Leifert, D.; Kloeckener-Gruissem, B.; McAlear, S.D.; Lemke, J.; Neidhardt, J.; Berger, W. Identification and functional characterization of a novel rhodopsin mutation associated with autosomal dominant CSNB. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4105–4114. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; Lo, C.T.; Sheu, S.J.; Yin, L.T. Risk factors for and progression of myopia in young Taiwanese men. Ophthalmic Epidemiol. 2015, 22, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.; Ostermaier, M.K.; Vishnivetskiy, S.A.; Panneels, V.; Homan, K.T.; Tesmer, J.J.; Veprintsev, D.; Deupi, X.; Gurevich, V.V.; Schertler, G.F.; et al. Insights into congenital stationary night blindness based on the structure of G90D rhodopsin. EMBO Rep. 2013, 14, 520–526. [Google Scholar] [CrossRef]
- Papageorgiou, E.; McLean, R.J.; Gottlob, I. Nystagmus in childhood. Pediatr. Neonatol. 2014, 55, 341–351. [Google Scholar] [CrossRef]
- Morgans, C.W.; Brown, R.L.; Duvoisin, R.M. TRPM1: The endpoint of the mGluR6 signal transduction cascade in retinal ON-bipolar cells. Bioessays 2010, 32, 609–614. [Google Scholar] [CrossRef]
- Morgans, C.W.; Zhang, J.; Jeffrey, B.G.; Nelson, S.M.; Burke, N.S.; Duvoisin, R.M.; Brown, R.L. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19174–19178. [Google Scholar] [CrossRef]
- Pearring, J.N.; Bojang, P., Jr.; Shen, Y.; Koike, C.; Furukawa, T.; Nawy, S.; Gregg, R.G. A role for nyctalopin, a small leucine-rich repeat protein, in localizing the TRP melastatin 1 channel to retinal depolarizing bipolar cell dendrites. J. Neurosci. 2011, 31, 10060–10066. [Google Scholar] [CrossRef]
- Kim, H.M.; Joo, K.; Han, J.; Woo, S.J. Clinical and Genetic Characteristics of Korean Congenital Stationary Night Blindness Patients. Genes 2021, 12, 789. [Google Scholar] [CrossRef]
- Sergouniotis, P.I.; Robson, A.G.; Li, Z.; Devery, S.; Holder, G.E.; Moore, A.T.; Webster, A.R. A phenotypic study of congenital stationary night blindness (CSNB) associated with mutations in the GRM6 gene. Acta Ophthalmol. 2012, 90, e192–e197. [Google Scholar] [CrossRef]
- Bijveld, M.M.; van Genderen, M.M.; Hoeben, F.P.; Katzin, A.A.; van Nispen, R.M.; Riemslag, F.C.; Kappers, A.M. Assessment of night vision problems in patients with congenital stationary night blindness. PLoS ONE 2013, 8, e62927. [Google Scholar] [CrossRef] [PubMed]
- Miraldi Utz, V.; Pfeifer, W.; Longmuir, S.Q.; Olson, R.J.; Wang, K.; Drack, A.V. Presentation of TRPM1-Associated Congenital Stationary Night Blindness in Children. JAMA Ophthalmol. 2018, 136, 389–398. [Google Scholar] [CrossRef]
- Nakamura, M.; Sanuki, R.; Yasuma, T.R.; Onishi, A.; Nishiguchi, K.M.; Koike, C.; Kadowaki, M.; Kondo, M.; Miyake, Y.; Furukawa, T. TRPM1 mutations are associated with the complete form of congenital stationary night blindness. Mol. Vis. 2010, 16, 425–437. [Google Scholar] [PubMed]
- van Genderen, M.M.; Bijveld, M.M.; Claassen, Y.B.; Florijn, R.J.; Pearring, J.N.; Meire, F.M.; McCall, M.A.; Riemslag, F.C.; Gregg, R.G.; Bergen, A.A.; et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am. J. Hum. Genet. 2009, 85, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gao, Y.; Li, S.; Guo, X.; Zhang, Q. Mutation screening of TRPM1, GRM6, NYX and CACNA1F genes in patients with congenital stationary night blindness. Int. J. Mol. Med. 2012, 30, 521–526. [Google Scholar] [CrossRef]
- al-Jandal, N.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Bannon, N.; Findlay, J.B.; Humphries, P.; Kenna, P.F. A novel mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal dominant congenital stationary night blindness. Hum. Mutat. 1999, 13, 75–81. [Google Scholar] [CrossRef]
- Hasan, N.; Pangeni, G.; Cobb, C.A.; Ray, T.A.; Nettesheim, E.R.; Ertel, K.J.; Lipinski, D.M.; McCall, M.A.; Gregg, R.G. Presynaptic Expression of LRIT3 Transsynaptically Organizes the Postsynaptic Glutamate Signaling Complex Containing TRPM1. Cell Rep. 2019, 27, 3107–3116.e3103. [Google Scholar] [CrossRef]
- Scalabrino, M.L.; Boye, S.L.; Fransen, K.M.; Noel, J.M.; Dyka, F.M.; Min, S.H.; Ruan, Q.; De Leeuw, C.N.; Simpson, E.M.; Gregg, R.G.; et al. Intravitreal delivery of a novel AAV vector targets ON bipolar cells and restores visual function in a mouse model of complete congenital stationary night blindness. Hum. Mol. Genet. 2015, 24, 6229–6239. [Google Scholar] [CrossRef]
- Blair, K.; Cibis, G.; Gulani, A.C. Amblyopia; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Holmes, J.M.; Lazar, E.L.; Melia, B.M.; Astle, W.F.; Dagi, L.R.; Donahue, S.P.; Frazier, M.G.; Hertle, R.W.; Repka, M.X.; Quinn, G.E.; et al. Effect of age on response to amblyopia treatment in children. Arch. Ophthalmol. 2011, 129, 1451–1457. [Google Scholar] [CrossRef]
- Reichel, F.F.; Michalakis, S.; Wilhelm, B.; Zobor, D.; Muehlfriedel, R.; Kohl, S.; Weisschuh, N.; Sothilingam, V.; Kuehlewein, L.; Kahle, N.; et al. Three-year results of phase I retinal gene therapy trial for CNGA3-mutated achromatopsia: Results of a non randomised controlled trial. Br. J. Ophthalmol. 2022, 106, 1567–1572. [Google Scholar] [CrossRef]
- Fischer, M.D.; Michalakis, S.; Wilhelm, B.; Zobor, D.; Muehlfriedel, R.; Kohl, S.; Weisschuh, N.; Ochakovski, G.A.; Klein, R.; Schoen, C.; et al. Safety and Vision Outcomes of Subretinal Gene Therapy Targeting Cone Photoreceptors in Achromatopsia: A Nonrandomized Controlled Trial. JAMA Ophthalmol. 2020, 138, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; McGee, T.L.; Berson, E.L.; Fishman, G.A.; Sandberg, M.A.; Alexander, K.R.; Derlacki, D.J.; Rajagopalan, A.S. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc. Natl. Acad. Sci. USA 2005, 102, 4884–4889. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ito, S.; Piao, C.H.; Terasaki, H.; Miyake, Y. Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family. Arch. Ophthalmol. 2003, 121, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Scheiman, M.M.; Hertle, R.W.; Beck, R.W.; Edwards, A.R.; Birch, E.; Cotter, S.A.; Crouch, E.R., Jr.; Cruz, O.A.; Davitt, B.V.; Donahue, S.; et al. Randomized trial of treatment of amblyopia in children aged 7 to 17 years. Arch. Ophthalmol. 2005, 123, 437–447. [Google Scholar] [CrossRef]
- Wallace, D.K.; Repka, M.X.; Lee, K.A.; Melia, M.; Christiansen, S.P.; Morse, C.L.; Sprunger, D.T.; American Academy of Pediatric Ophthalmology/Strabismus Preferred Practice Pattern Pediatric Ophthalmology Panel. Amblyopia Preferred Practice Pattern(R). Ophthalmology 2018, 125, P105–P142. [Google Scholar] [CrossRef]
- Pierce, E.A.; Bennett, J. The Status of RPE65 Gene Therapy Trials: Safety and Efficacy. Cold Spring Harb. Perspect. Med. 2015, 5, a017285. [Google Scholar] [CrossRef]
- Asper, L.; Watt, K.; Khuu, S. Optical treatment of amblyopia: A systematic review and meta-analysis. Clin. Exp. Optom. 2018, 101, 431–442. [Google Scholar] [CrossRef]
- Pearson, R.A.; Barber, A.C.; Rizzi, M.; Hippert, C.; Xue, T.; West, E.L.; Duran, Y.; Smith, A.J.; Chuang, J.Z.; Azam, S.A.; et al. Restoration of vision after transplantation of photoreceptors. Nature 2012, 485, 99–103. [Google Scholar] [CrossRef]
- Varin, J.; Bouzidi, N.; Dias, M.M.S.; Pugliese, T.; Michiels, C.; Robert, C.; Desrosiers, M.; Sahel, J.A.; Audo, I.; Dalkara, D.; et al. Restoration of mGluR6 Localization Following AAV-Mediated Delivery in a Mouse Model of Congenital Stationary Night Blindness. Investig. Ophthalmol. Vis. Sci. 2021, 62, 24. [Google Scholar] [CrossRef]
- Varin, J.; Bouzidi, N.; Gauvain, G.; Joffrois, C.; Desrosiers, M.; Robert, C.; De Sousa Dias, M.M.; Neuille, M.; Michiels, C.; Nassisi, M.; et al. Substantial restoration of night vision in adult mice with congenital stationary night blindness. Mol. Ther. Methods Clin. Dev. 2021, 22, 15–25. [Google Scholar] [CrossRef]
- Miyadera, K.; Santana, E.; Roszak, K.; Iffrig, S.; Visel, M.; Iwabe, S.; Boyd, R.F.; Bartoe, J.T.; Sato, Y.; Gray, A.; et al. Targeting ON-bipolar cells by AAV gene therapy stably reverses LRIT3-congenital stationary night blindness. Proc. Natl. Acad. Sci. USA 2022, 119, e2117038119. [Google Scholar] [CrossRef]
- Pearson, R.A. Advances in repairing the degenerate retina by rod photoreceptor transplantation. Biotechnol. Adv. 2014, 32, 485–491. [Google Scholar] [CrossRef]
- Liu, C.F.; Liu, L.; Lai, C.C.; Chou, J.C.; Yeh, L.K.; Chen, K.J.; Chen, Y.P.; Wu, W.C.; Chuang, L.H.; Sun, C.C.; et al. Multimodal imaging including spectral-domain optical coherence tomography and confocal near-infrared reflectance for characterization of lacquer cracks in highly myopic eyes. Eye 2014, 28, 1437–1445. [Google Scholar] [CrossRef]
- Wang, N.K.; Chuang, L.H.; Lai, C.C.; Chou, C.L.; Chu, H.Y.; Yeung, L.; Chen, Y.P.; Chen, K.J.; Wu, W.C.; Chen, T.L.; et al. Multimodal fundus imaging in fundus albipunctatus with RDH5 mutation: A newly identified compound heterozygous mutation and review of the literature. Doc. Ophthalmol. 2012, 125, 51–62. [Google Scholar] [CrossRef]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M.; International Society for Clinical Electrophysiology of Vision. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef]
- Seo, G.H.; Kim, T.; Choi, I.H.; Park, J.Y.; Lee, J.; Kim, S.; Won, D.G.; Oh, A.; Lee, Y.; Choi, J.; et al. Diagnostic yield and clinical utility of whole exome sequencing using an automated variant prioritization system, EVIDENCE. Clin. Genet. 2020, 98, 562–570. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inheritance Pattern | Type | Genes | ERG Findings |
|---|---|---|---|
| Autosomal dominant | Riggs | RHO GNAT1 PDE6B | DA 0.01: non-detectable DA 3.0 and 10.0: decreased with decreased b:a ratio (possibly electronegative) LA 30 Hz and 3.0: normal photopic ERG |
| Autosomal recessive | Abnormal fundus | SAG GRK1 RDH5 RLBP1 RPE65 | DA 0.01: reduced DA 3.0 and 10.0: decreased with decreased b:a ratio (possibly electronegative); recovery of scotopic ERG after prolonged dark adaptation LA 30 Hz and 3.0: normal photopic ERG |
| Complete | GRM6 TRPM1 GPR179 LRIT3 | DA 0.01: absent DA 3.0 and 10.0: decreased with decreased b:a ratio (electronegative) LA 30 Hz: normal amplitude, possibly increased latency LA 3.0: normal a-wave with wide trough; sharply rising b-wave with no oscillatory potentials with decreased reduced b:a ratio | |
| Incomplete | CABP4 CACNA2D4 | DA 0.01: reduced amplitude DA 3.0: slightly decreased a-wave, decreased a-wave DA 10.0: normal a-wave bright flash, reduced b-wave (electronegative) bright flash scotopic LA 30 Hz: reduced with increased latency, bifid peaks LA 3.0: reduced amplitude, decreased b:a ratio | |
| Riggs | SLC24A1 GNAT1 | DA 0.01: non-detectable DA 10.0: decreased bright flash scotopic ERG with decreased b:a ratio (possibly electronegative) LA 30 Hz and 3.0: normal photopic ERG | |
| X-linked | Complete | NYX | DA 0.01: absent DA 10.0: decreased with decreased b:a ratio (electronegative) LA 30 Hz: normal amplitude, possibly increased latency LA 3.0: normal a-wave with wide trough; sharply rising b-wave with no oscillatory potentials with decreased reduced b:a ratio |
| Incomplete | CACNA1F | DA 0.01: reduced amplitude DA 10.0: normal a-wave bright flash, reduced b-wave (electronegative) bright flash scotopic LA 30 Hz: reduced with increased latency, bifid peaks LA 3.0: reduced amplitude, decreased b:a ratio |
| Patient No. | Age | Sex | BCVA OD OS | Refractive Error (D) | Clinical Diagnosis | Gene (OMIM No.) | Transcript | Variants | ACMG Classification | Zygosity |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 22 | M | 20/20 20/20 | −3.75 −3.25 | AD CSNB | RHO (180380) | NM_000539.3 | c.281C>T (p.Thr94Ile) | Likely pathogenic | Heterozygous |
| 2 | 28 | M | 20/200 20/200 | −1.75 −1.25 | AR cCSNB | GRM6 (604096) | NM_000843.4 | c.547G>A (p.Asp183Asn) c.575G>A (p.Arg192Gln) | VUS VUS | Compound Heterozygous |
| 3 § | 7 | M | 20/30 20/100 | −5.0 −5.0 | AR cCSNB | TRPM1 (603576) | NM_001252024.2 | c.336del (p.Asp113IlefsTer10) c.3127+1G>A | Pathogenic Pathogenic | Compound Heterozygous |
| 4 § | 5 | M | 20/200 20/200 | −5.0 −5.25 | AR cCSNB | TRPM1 (603576) | NM_001252024.2 | c.336del (p.Asp113IlefsTer10) c.3127+1G>A | Pathogenic Pathogenic | Compound Heterozygous |
| 5 | 15 | M | 20/200 20/200 | −8.5 −9.0 | AR cCSNB | TRPM1 (603576) | NM_001252024.2 | c.2921T>G (p.Leu974Arg) c.730G>A (p.Ala244Thr) | VUS VUS | Unknown * |
| 6 | 26 | M | 20/500 20/500 | +4.0 +4.25 | X-linked iCSNB | CACNA1F (300110) | NM_001256789.3 | c.2231C>A (p.Ala744Asp) | Likely pathogenic | Hemizygous |
| 7 | 22 | M | 20/20 20/20 | −6.25 −8.25 | X-linked cCSNB | NYX (300278) | NM_001378477.3 | c.217_218insA (p.Gly73Glufs * 42) | Likely pathogenic | Hemizygous |
| Patient No. | Dark-Adapted 0.01 ERG | Dark-Adapted 3.0 ERG | Light-Adapted ERG | ||||||
|---|---|---|---|---|---|---|---|---|---|
| a Wave (μV) OD/OS | b Wave (μV) OD/OS | IT (ms) OD/OS | a Wave (μV) OD/OS | b Wave (μV) OD/OS | a Wave(μV) OD/OS | b Wave (μV) OD/OS | 30 Hz Flicker (μV) OD/OS | 30 Hz Flicker IT (ms) OD/OS | |
| Patient 1 | −8.6/−12.7 | 4.1/3.7 | 80/81 | −119.1/−144.7 | 40.5/48.7 | −59.2/−68.6 | 130.7/153.6 | 94.2/118.8 | 27/27 |
| Patient 2 | −2.6/−1.3 | 33.35/19.36 | 92/90 | −228.6/−180.1 | 120.0/106.4 | −19.1/−33.4 | 83.2/70.0 | 71.9/56.8 | 30/28 |
| Patient 3 | −5.2/−7.2 | 13.3/24.7 | 64.5/64 | −145.0/−211.7 | 81.8/96.9 | −40.0/−47.6 | 72.3/88.0 | 54.2/60.8 | 31.5/31 |
| Patient 4 * | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Patient 5 | −2.2/−11.9 | 8.3/19.0 | 90/92 | −139.9/−161.6 | 56.0/69.5 | −21.6/−45.4 | 69.5/101.5 | 74.8/87.6 | 31.5/31.5 |
| Patient 6 | −9.2/−14.2 | 99.4/75.7 | 84/89 | −123.1/−141.6 | 126.1/142.1 | −42.9/−24.5 | 35.3/45.3 | 22.1/17.9 | 34/32 |
| Patient 7 | −7.8/−3.6 | 17.5/6.8 | 74/56 | −190.0/−160.2 | 112.5/99.4 | −43.9/−33.2 | 67.5/53.1 | 57.0/48.7 | 32.5/32.5 |
| Normal Reference | - | 218.5 ± 148.3 | 85.9 ± 14.1 | −210.1± 172.1 | 347.0 ± 134.1 | −36.4 ± 25.8 | 109.8 ± 67.8 | 121.4 ± 65.5 | 26.3 ± 3.8 |
| Gene | cDNA | Protein | Clinical Significance (ClinVar) | ACMG/AMP Classification | Allele Frequency (Gnomad v2.1.1) | Phylo P100 Way † | SIFT | PolyPhen | CADD | REVEL | MetaLR | Mutation Assessor | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 2 | GRM6 | c.547G>A | p.Asp183Asn | VUS | PM2+PP3 | 0.00000798 | 7.798 | Deleterious | Possibly damaging | Likely benign | Damaging | Damaging | High impact |
| GRM6 | c.575G>A | p.Arg192Gln | VUS | PM2+PP3 | 0.0000837 | 7.798 | Deleterious | Damaging | Likely deleterious | Damaging | Damaging | High impact | |
| Patient 5 | TRPM1 | c.2921T>G | p.Leu974Arg | VUS | PM2+PP3 | 0.0000318 | 9.317 | Deleterious | Possibly damaging | Likely benign | Possibly damaging | Damaging | Medium impact |
| TRPM1 | c.730G>A | p.Ala244Thr | VUS | PM2 | 0.0000441 | 6.160 | Tolerated | Possibly damaging | Likely benign | Likely benign | Tolerated | Low impact |
| Population Cohort | Type | Gene | Average Age | Sample Size | Visual Acuity * (Snellen) | logMAR (Mean ± SD) | Spherical Equivalent (D) | Nystagmus (%) | Strabismus (%) |
|---|---|---|---|---|---|---|---|---|---|
| Korean [19] | Complete | TRPM1 | 4 | 1 | 20/50 | 0.1 ± 0 | −7 | 0 | 0 |
| Complete | NYX | 2.5 | 2 | 20/100 | 0.65 ± 0 | −7.8 | 50 | 50 | |
| Incomplete | CACNA1F | 2.86 | 14 | 20/123 | 0.74 ± 0.22 | −2.34 | 71 | 36 | |
| British [20] | Complete | GRM6 | 23.9 | 9 | 20/64 | 0.51 ± 0.57 | −5.375 | 55.56 | 22.22 |
| Dutch [21] | Complete | TRPM1 | 16 | 2 | 20/51 | 0.41 ± 0.16 | −3.75 | 100 | n/a |
| Complete | NYX | 24.8 | 6 | 20/31 | 0.21 ± 0.21 | −9.95 | 66.7 | n/a | |
| Incomplete | CACNA1F | 22.6 | 13 | 20/55 | 0.44 ± 0.29 | −5.56 | 61.5 | n/a | |
| Slovenian [9] | Autosomal dominant | RHO | 24.3 | 3 | 20/20 | 0 ± 0 | n/a | 0 | 0 |
| Taiwanese This paper | Autosomal dominant | RHO | 22 | 1 | 20/20 | 0 ± 0 | −3.5 | 0 | 0 |
| Complete | GRM6 | 28 | 1 | 20/200 | 1.0 ± 0 | −1.5 | 100 | 100 | |
| Complete | TRPM1 | 9 | 3 | 20/200 | 0.813 ± 0.32 | −5.125 | 100 | 100 | |
| Incomplete | CACNA1F | 26 | 1 | 20/500 | 1.40 ± 0 | +4.125 | 100 | 100 | |
| Complete | NYX | 22 | 1 | 20/20 | 0 ± 0 | −7.25 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, A.H.; Liu, P.-K.; Chang, Y.-H.; Kang, E.Y.-C.; Wang, H.-H.; Chen, N.; Tseng, Y.-J.; Seo, G.H.; Lee, H.; Liu, L.; et al. Congenital Stationary Night Blindness: Clinical and Genetic Features. Int. J. Mol. Sci. 2022, 23, 14965. https://doi.org/10.3390/ijms232314965
Kim AH, Liu P-K, Chang Y-H, Kang EY-C, Wang H-H, Chen N, Tseng Y-J, Seo GH, Lee H, Liu L, et al. Congenital Stationary Night Blindness: Clinical and Genetic Features. International Journal of Molecular Sciences. 2022; 23(23):14965. https://doi.org/10.3390/ijms232314965
Chicago/Turabian StyleKim, Angela H., Pei-Kang Liu, Yin-Hsi Chang, Eugene Yu-Chuan Kang, Hung-Hsuan Wang, Nelson Chen, Yun-Ju Tseng, Go Hun Seo, Hane Lee, Laura Liu, and et al. 2022. "Congenital Stationary Night Blindness: Clinical and Genetic Features" International Journal of Molecular Sciences 23, no. 23: 14965. https://doi.org/10.3390/ijms232314965
APA StyleKim, A. H., Liu, P.-K., Chang, Y.-H., Kang, E. Y.-C., Wang, H.-H., Chen, N., Tseng, Y.-J., Seo, G. H., Lee, H., Liu, L., Chao, A.-N., Chen, K.-J., Hwang, Y.-S., Wu, W.-C., Lai, C.-C., Tsang, S. H., Hsiao, M.-C., & Wang, N.-K. (2022). Congenital Stationary Night Blindness: Clinical and Genetic Features. International Journal of Molecular Sciences, 23(23), 14965. https://doi.org/10.3390/ijms232314965