
Cholesterol Stationary Phase in the Separation and Identification of siRNA Impurities by Two-Dimensional Liquid Chromatography-Mass Spectrometry

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
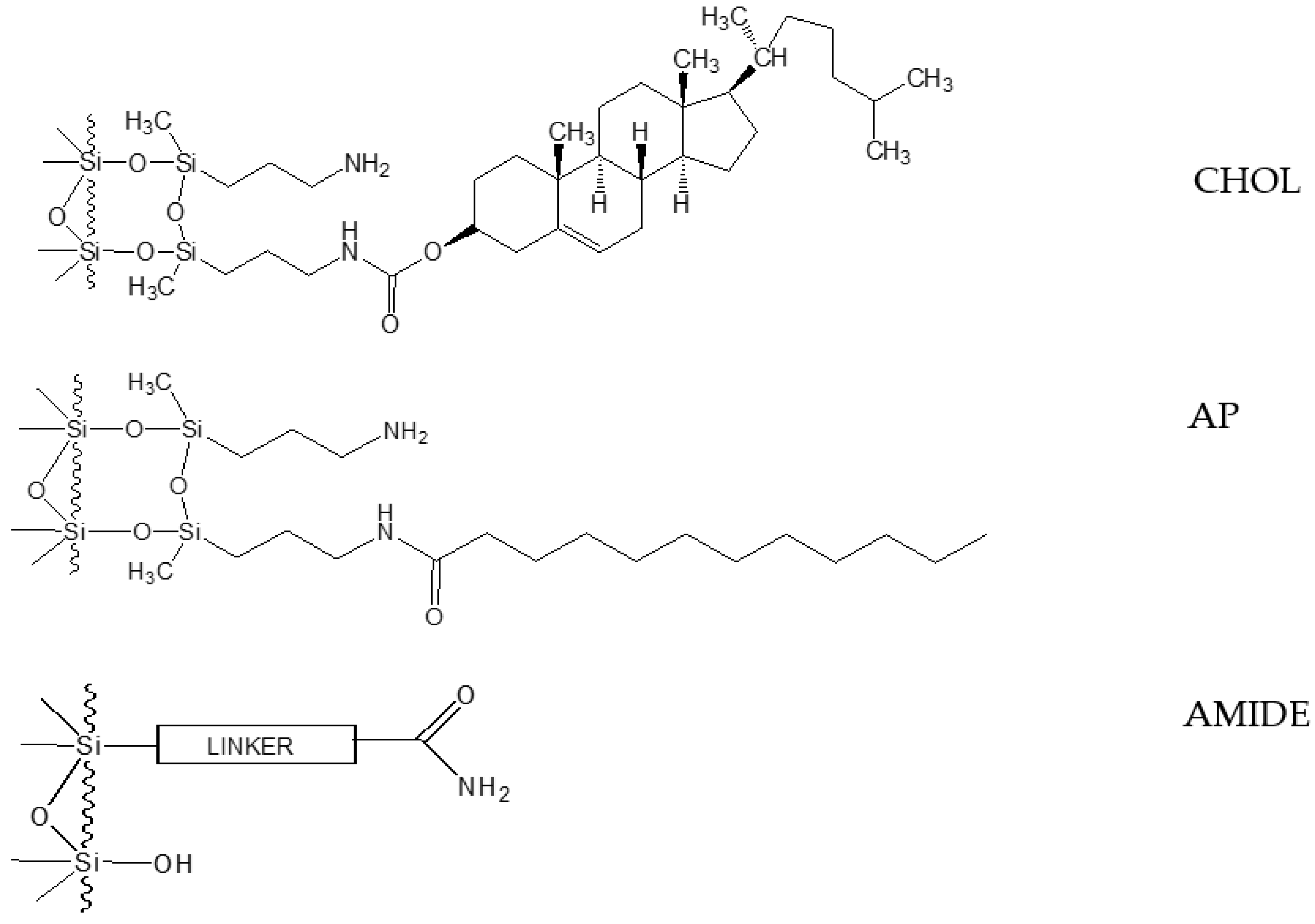
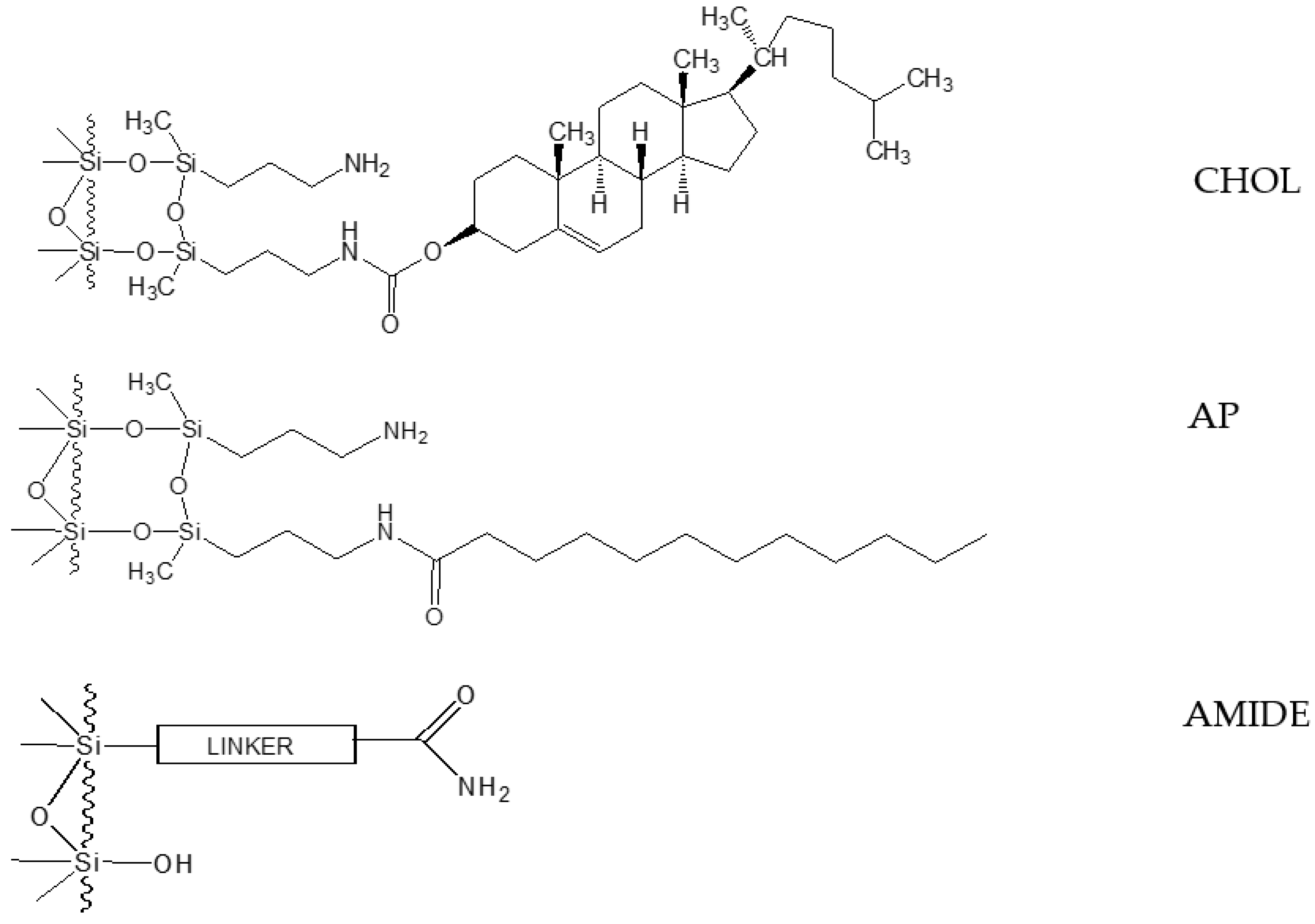
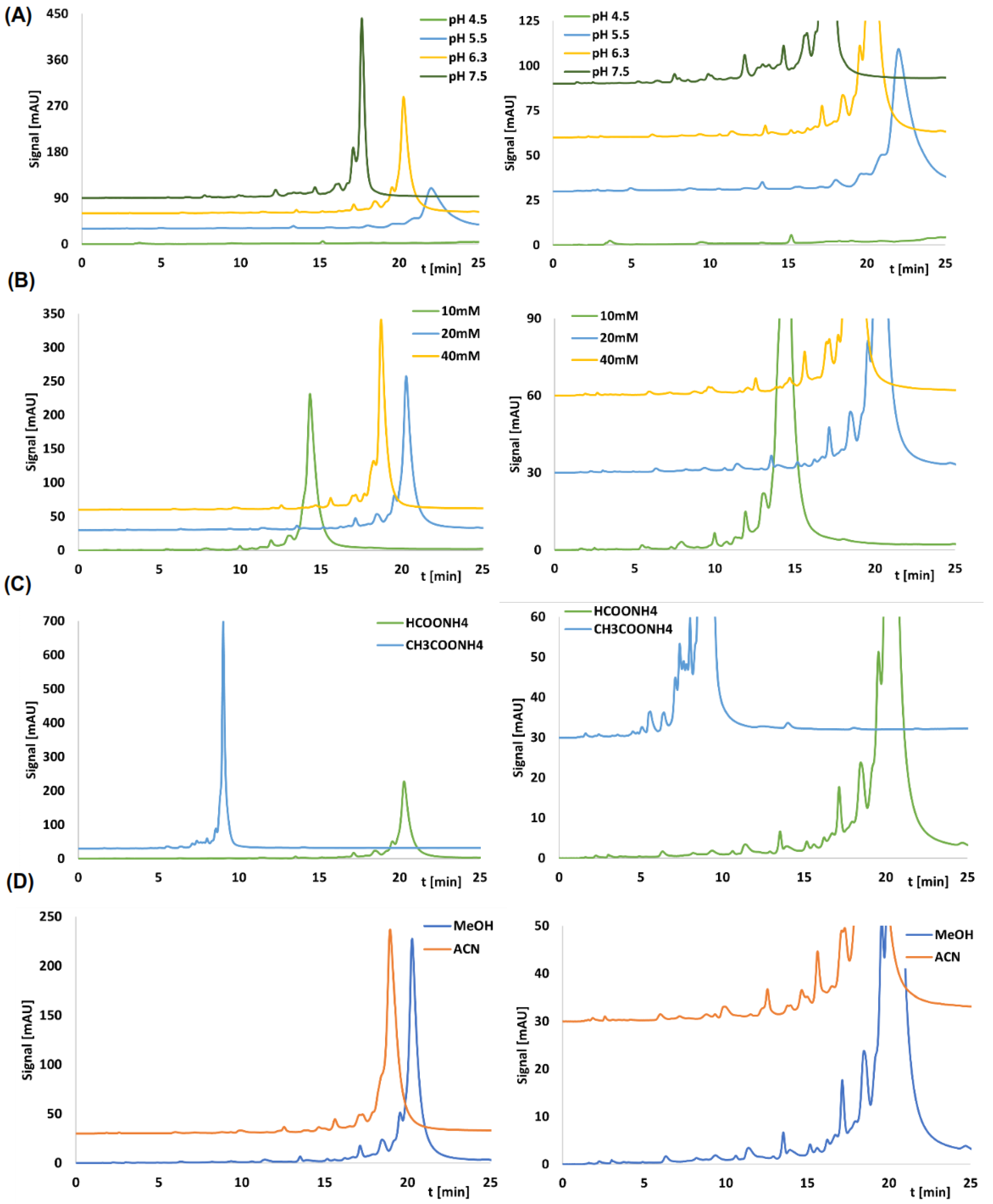
2.1. Retention and Separation Studies for CHOL and AP Columns
2.2. Impurities Analysis with the Use of Q-TOF-MS
2.2.1. The Chemistry behind the Origin of Impurities
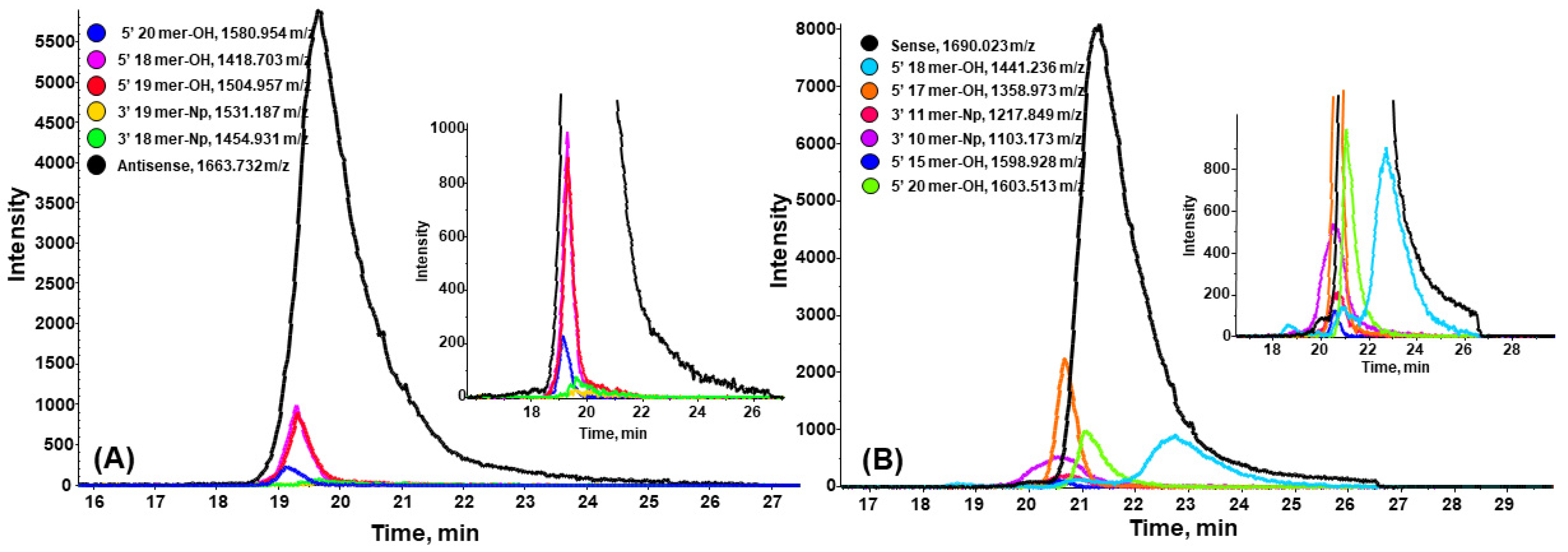
2.2.2. Antisense Strand of Patisiran
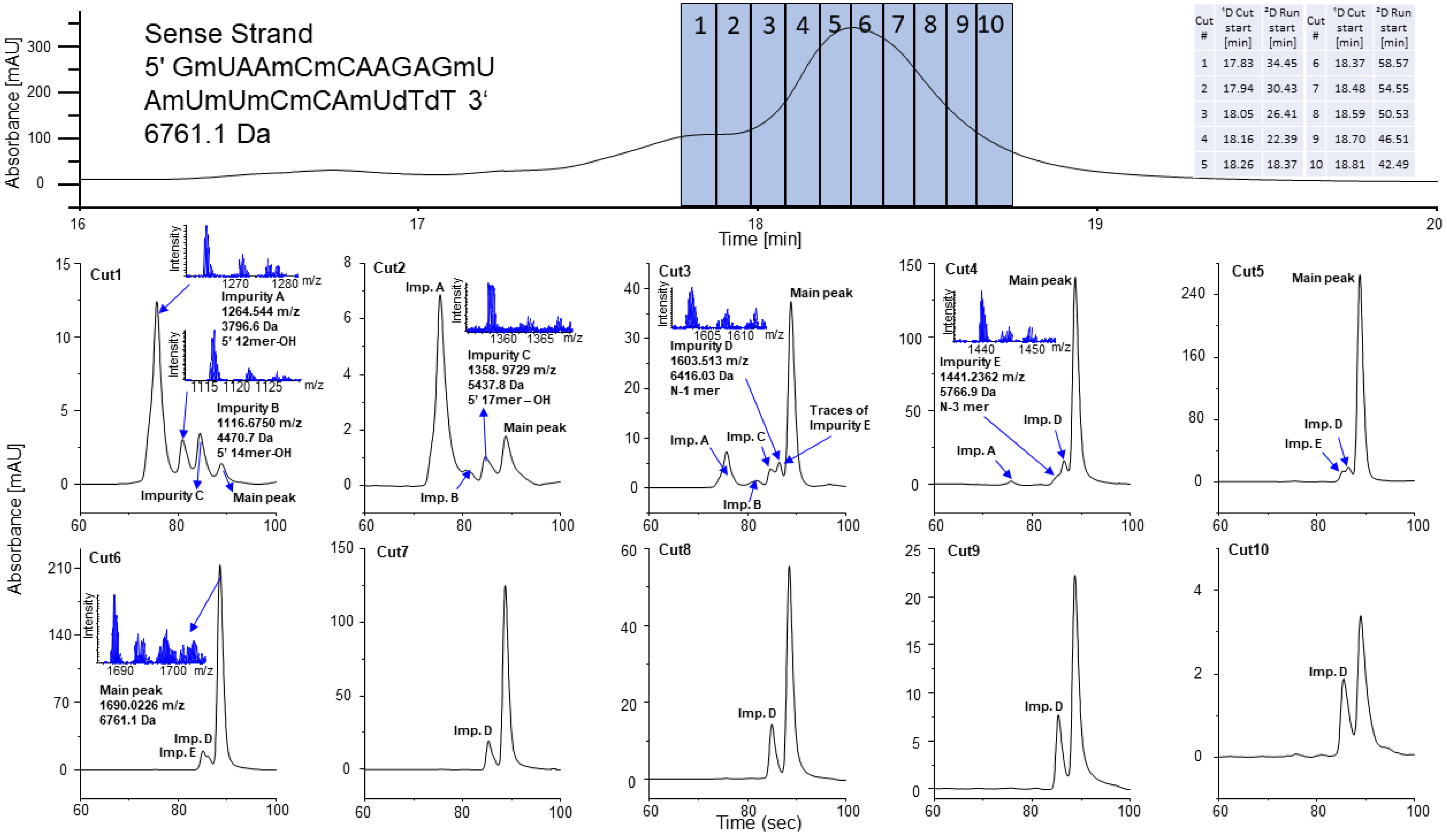
2.2.3. Sense Strand of Patisiran
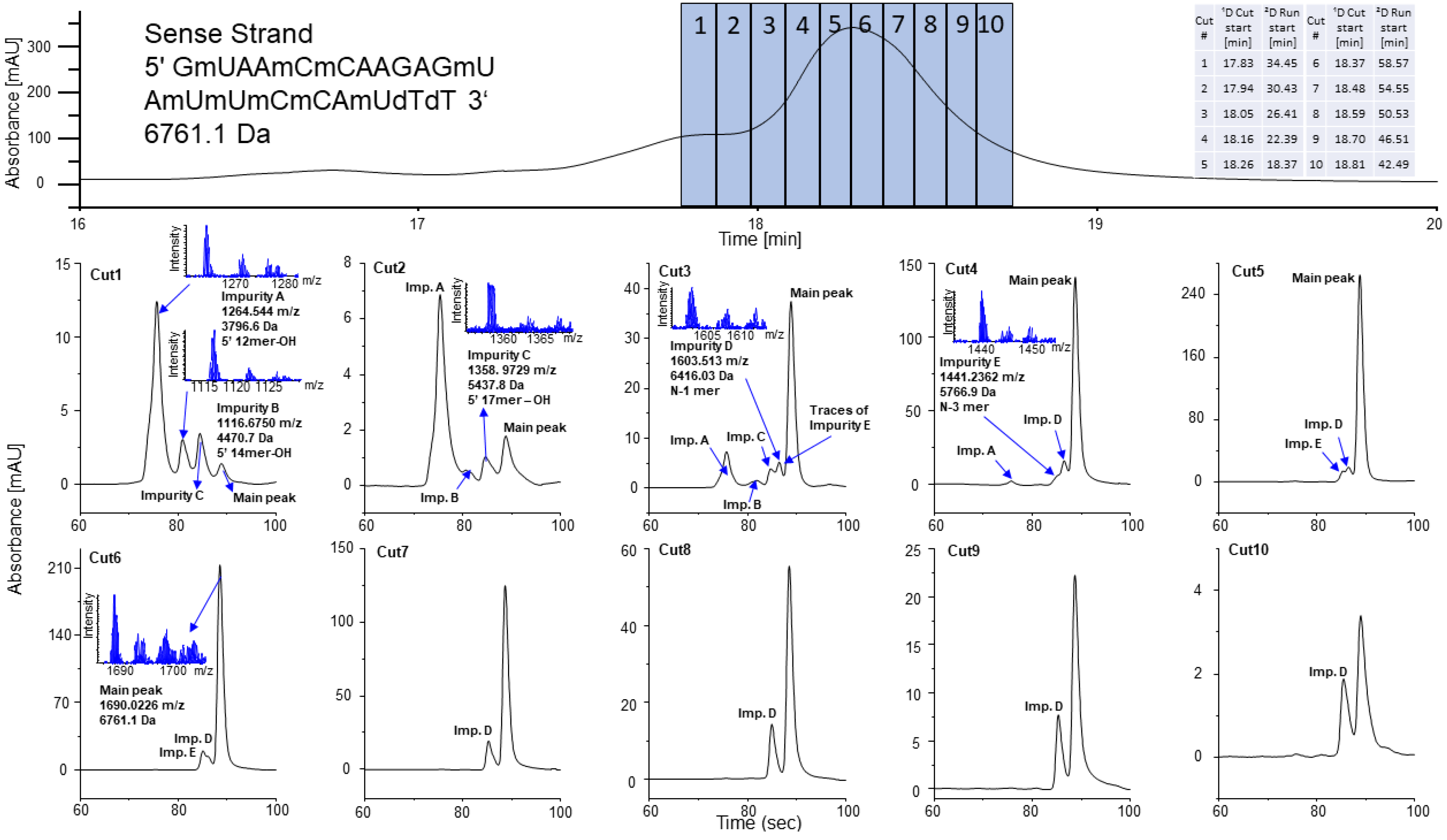
2.3. Closely Related Impurities in the Main Peak of Sense and Antisense Strands
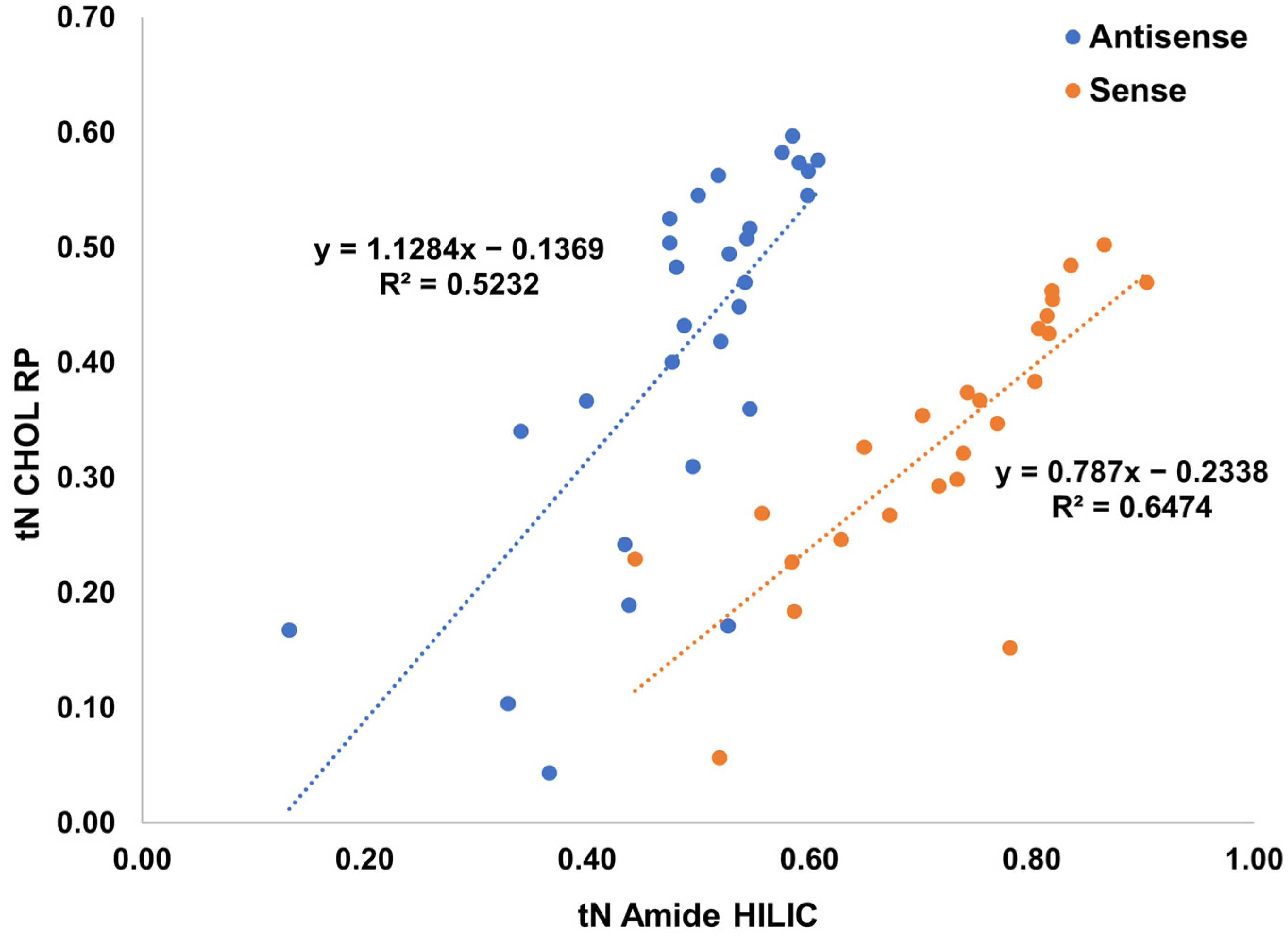
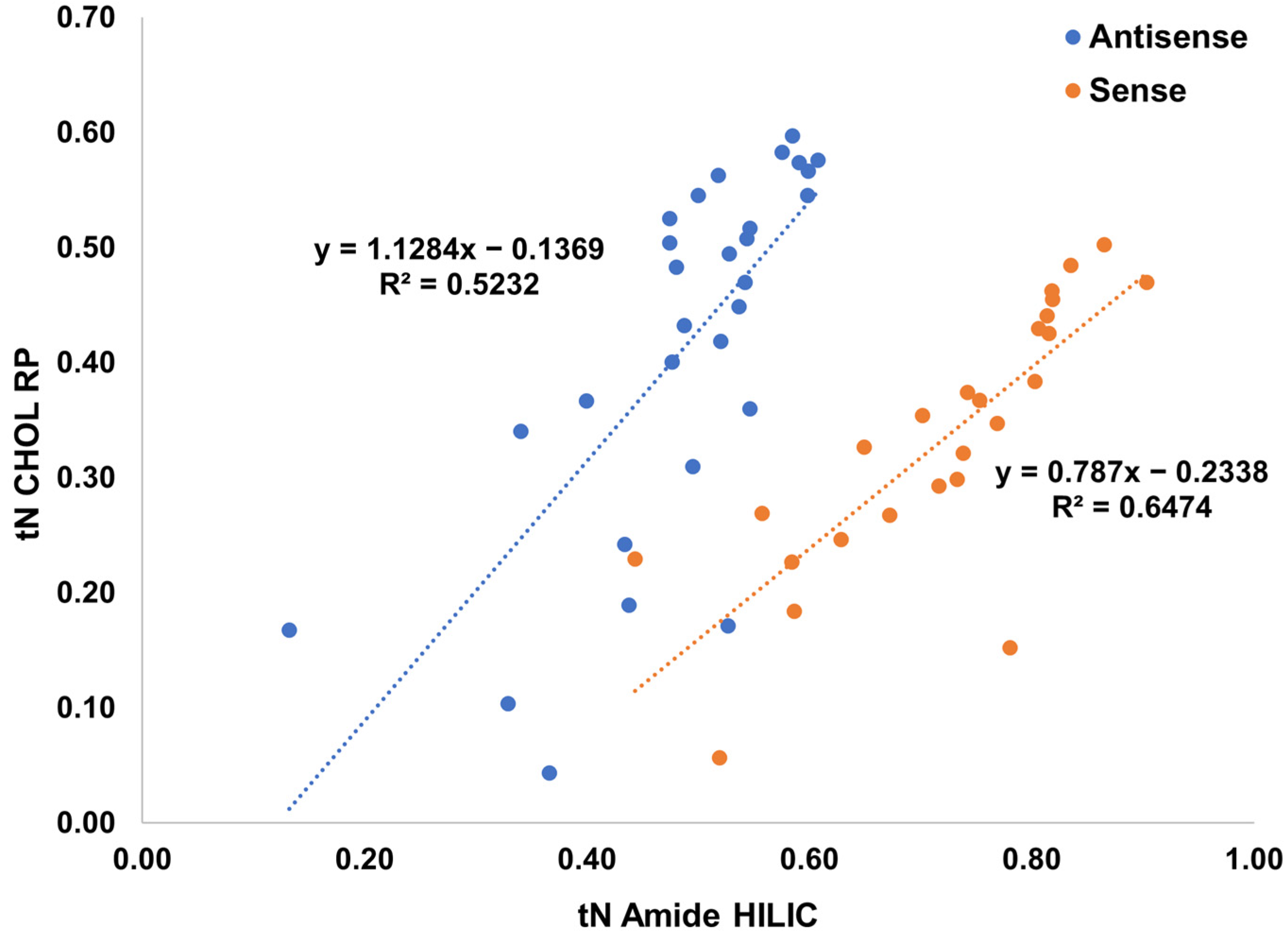
2.4. Selective Comprehensive (High-Resolution Sampling) sRP × HILIC-2DLC for Patisiran
3. Materials and Methods
3.1. Materials
3.2. Oligonucleotides
3.3. Instrumentation
3.4. LC-MS Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Ann. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- El Zahar, N.M.; Magdy, N.; El-Kosasy, A.M.; Bartlett, M.G. Chromatographic approaches for the characterization and quality control of therapeutic oligonucleotide impurities. Biomed. Chromatogr. 2017, 32, e4088. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-targeted therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [Green Version]
- Deleavey, G.F.; Damha, M.J. Designing chemically modified oligonucleotides for targeted gene silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Pourshahian, S. Therapeutic oligonucleotides, impurities, degradants, and their characterization by mass spectrometry. Mass Spectrom. Rev. 2021, 40, 75–109. [Google Scholar] [CrossRef]
- Capaldi, D.; Teasdale, A.; Henry, S.; Akhtar, N.; den Besten, C.; Gao-Sheridan, S.; Kretschmer, M.; Sharpe, B.; Andrews, B.; Burm, B.; et al. Impurities in Oligonucleotide Drug Substances and Drug Products. Nucleic Acid Ther. 2017, 27, 1–14. [Google Scholar] [CrossRef]
- Goyon, A.; Yehl, P.; Zhang, K. Characterization of therapeutic oligonucleotides by liquid chromatography. J. Pharm. Biomed. Anal. 2000, 182, 113105. [Google Scholar] [CrossRef]
- Szekely, L.; Kiessig, S.; Schwarz, M.A.; Kalman, F. Capillary gel electrophoresis of therapeutic oligonucleotides–Analysis of single-and double-stranded forms. Electrophoresis 2009, 30, 1579–1586. [Google Scholar] [CrossRef]
- Stilianos, G.; Roussis, A. Novel and Intuitive Method of Displaying and Interacting with Mass Difference Information: Application to Oligonucleotide Drug Impurities. J. Am. Soc. Mass Spectrom. 2015, 26, 1150–1164. [Google Scholar]
- Elzahar, N.M.; Magdy, N.; El-Kosasy, A.M.; Bartlett, M.G. Degradation product characterization of therapeutic oligonucleotides using liquid chromatography mass spectrometry. Anal. Bioanal. Chem. 2018, 410, 3375–3384. [Google Scholar] [CrossRef]
- Roussis, S.G.; Koch, C.; Capaldi, D.; Rentel, C. Rapid oligonucleotide drug impurity determination by direct spectral comparison of ion-pair reversed-phase high-performance liquid chromatography electrospray ionization mass spectrometry data. Rapid Commun. Mass Spectrom. 2018, 32, 1099–1106. [Google Scholar] [CrossRef]
- Roussis, S.G.; Cedillo, I.; Rentel, C. Automated determination of early eluting oligonucleotide impurities using ion-pair reversed-phase liquid chromatography high resolution-mass spectrometry. Anal. Biochem. 2020, 595, 113623. [Google Scholar] [CrossRef] [PubMed]
- Roussis, S.G.; Pearce, M.; Rentel, C. Small Alkyl Amines as Ion-Pair Reagents for the Separation of Positional Isomers of Impurities in Phosphate Diester Oligonucleotides. J. Chromatogr. A 2019, 1594, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Roussis, S.G.; Rodriguez, A.A.; Rentel, C. Determination of individual liogonucleotide impurities by small amine ion pair-RP HPLC MS and MS/MS: N-1 impurities. J. Chromatogr. B 2021, 1169, 122611. [Google Scholar] [CrossRef]
- Rütters, H.; Möhring, T.; Rullkötter, J.; Griep-Raming, J.; Metzger, J.O. The persistent memory effect of triethylamine in the analysis of phospholipids by liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 122–123. [Google Scholar] [CrossRef]
- Zimmermann, A.; Horak, J.; Sánchez-Muñoz, O.L.; Lämmerhofer, M. Surface charge fine tuning of reversed-phase/weak anion-exchange type mixed-mode stationary phases for milder elution conditions. J. Chromatogr. A 2015, 1409, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Lobue, P.A.; Jora, M.; Addepalli, B.; Limbach, P.A. Oligonucleotide analysis by hydrophilic interaction liquid chromatography-mass spectrometry in the absence of ion-pair reagents. J. Chromatogr. A 2019, 1595, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Xu, X.; Qiu, H.; Li, N. Analytical characterization of DNA and RNA oligonucleotides by hydrophilic interaction liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2021, 1648, 462184. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarkiewicz, A.; Buszewski, B.; Studzińska, S. Application of hydrophilic interaction liquid chromatography coupled with tandem mass spectrometry for the retention and sensitivity studies of antisense oligonucleotides. J. Chromatogr. A 2020, 1622, 461100. [Google Scholar]
- Li, F.; Su, X.; Bäurer, S.; Lämmerhofer, M. Multiple heart-cutting mixed-mode chromatography-reversed-phase 2D-liquid chromatography method for separation and mass spectrometric characterization of synthetic oligonucleotides. J. Chromatogr. A 2020, 1625, 461338. [Google Scholar] [CrossRef]
- Li, F.; Lämmerhofer, M. Impurity profiling of siRNA by two-dimensional liquid chromatography-mass spectrometry with quinine carbamate anion-exchanger and ion-pair reversed-phase chromatography. J. Chromatogr. A 2021, 1643, 462065. [Google Scholar] [CrossRef] [PubMed]
- Roussis, S.G.; Cedillo, I.; Rentel, C. Two-dimensional liquid chromatography-mass spectrometry for the characterization of modified oligonucleotide impurities. Anal. Biochem. 2018, 556, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lynen, F.; Wang, J.; Li, H.; Xu, G.; Sandra, P. Comprehensive hydrophilic interaction and ion-pair reversed-phase liquid chromatography for analysis of di-to deca-oligonucleotides. J. Chromatogr. A 2012, 1255, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Goyon, A.; Zhang, K. Characterization of antisense oligonucleotide impurities by ion pairing reversed-phase and anion exchange chromatography coupled to hydrophilic interaction liquid chromatography/mass spectrometry using a versatile two-dimensional liquid chromatography setup. Anal. Chem. 2020, 92, 5944–5951. [Google Scholar]
- Studzińska, S.; Pietrzak, L.; Buszewski, B. The effects of stationary phases on retention and selectivity of oligonucleotides in IP-RP-HPLC. Chromatographia 2014, 77, 1589–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studzińska, S.; Krzemińska, K.; Szumski, M.; Buszewski, B. Application of a cholesterol stationary phase in the analysis of phosphorothioate oligonucleotides by means of ion pair chromatography coupled with tandem mass spectrometry. Talanta 2016, 154, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Buszewski, B.; Jezierska-Świtała, M.; Kaliszan, R.; Wojtczak, A.; Albert, K.; Bochmann, S.; Matyska, M.T.; Pesek, J.J. Selectivity tuning and molecular modeling of new generation packings for RP HPLC. Chromatographia 2001, 53, 204–212. [Google Scholar] [CrossRef]
- Gadzała-Kopciuch, R.M.; Buszewski, B. A comparative study of hydrophobicity of octadecyl and alkylamide bonded phases based on methylene selectivity. J. Sep. Sci. 2003, 26, 1273–1283. [Google Scholar] [CrossRef]
- Guimaraes, G.J.; Bartlet, M.G. The critical role of mobile phase pH in the performance of oligonucleotide ion-pair liquid chromatography–mass spectrometry methods. Future Sci. OA 2021, 7, FSO753. [Google Scholar] [CrossRef] [PubMed]
- Sutton, J.M.; Guimaraes, G.J.; Annavarapu, V.; van Dongen, W.D.; Bartlett, M.G. Current state of oligonucleotide characterization using liquid chromatography-mass spectrometry: Insight into critical issues. J. Am. Soc. Mass Spectrom. 2020, 31, 1775–1782. [Google Scholar] [CrossRef] [PubMed]
- Sample, P.J.; Gaston, W.K.; Alfonzo, D.; Limbach, P.A. RoboOligo: Software for mass spectrometry data to support manual and de novo sequencing of post-transcriptionally modified ribonucleic acids. Nucleic Acids Res. 2015, 43, e64. [Google Scholar] [CrossRef] [PubMed]
- Nyakas, A.; Blum, L.C.; Stucki, S.R.; Reymond, J.-L.; Schürch, S. OMA and OPA—Software-supported mass spectra analysis of native and modified nucleic acids. J. Am. Soc. Mass Spectrom. 2013, 24, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demelenne, A.; Nys, G.; Nix, C.; Fjeldsted, J.C.; Crommen, J.; Fillet, M. Separation of phosphorothioated oligonucleotide diastereomers using multiplexed drift tube ion mobility mass spectrometry. Anal. Chim. Acta 2022, 1191, 339297. [Google Scholar] [CrossRef] [PubMed]
- Stoll, D.R.; Shoykhet, K.; Petersen, P.; Buckenmaier, S. Active Solvent Modulation: A valve-based approach to improve separation compatibility in two-dimensional liquid chromatography. Anal. Chem. 2017, 89, 9260–9267. [Google Scholar] [CrossRef] [PubMed]
- Buckenmaier, S.; Petersson, P. Analysis of Peptide/Protein-Related Impurities Using the Integrated Solution of Bio 2D-LC/Q-TOF in Agilent MassHunter Software; 5994–4743EN; Agilent Technologies: Waldbronn, Germany, 2022. [Google Scholar]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stationary Phase | k | |||
|---|---|---|---|---|
| pH of 20 mM ammonium formate | ||||
| 4.5 | 5.5 | 6.3 | 7.5 | |
| CHOL | - | 16.62 ± 0.21 | 15.23 ± 0.16 | 13.09 ± 0.09 |
| AP | - | 13.22 ± 0.30 | 9.93 ± 0.02 | 7.15 ± 0.02 |
| Concentration of ammonium formate | ||||
| 10 mM | 20 mM | 40 mM | ||
| CHOL | 10.44 ± 0.11 | 15.23 ± 0.27 | 13.96 ± 0.20 | |
| AP | 8.08 ± 0.18 | 9.93 ± 0.13 | 9.12 ± 0.08 | |
| Type of salt | ||||
| 20 mM ammonium formate | 20 mM ammonium acetate | |||
| CHOL | 15.23 ± 0.14 | 10.98 ± 0.25 | ||
| AP | 9.93 ± 0.09 | 7.72 ± 0.13 | ||
| Type of organic solvent | ||||
| MeOH | ACN | |||
| CHOL | 15.23 ± 0.29 | 14.17 ± 0.22 | ||
| AP | 9.93 ± 0.018 | 8.18 ± 0.21 | ||
| Ion (m/z) | EIC Peak Area | |
|---|---|---|
| pH 6.3 | pH 7.5 | |
| Sense strand | ||
| 865.173 | 236,618 ± 1302 | 81,850 ± 591 |
| 1235.711 | 151,654 ± 973 | 68,514 ± 462 |
| 1155.188 | 66,958 ± 664 | 51,524 ± 349 |
| 1489.9089 | 87,257 ± 471 | 32,500 ± 588 |
| 1690.023 | 501,090 ± 1364 | 259,873 ± 1147 |
| Antisense strand | ||
| pH 6.3 | pH 7.5 | |
| 850.172 | 198,963 ± 1197 | 71,599 ± 759 |
| 902.147 | 201,337 ± 1372 | 98,004 ± 1024 |
| 1074.673 | 73,951± 1005 | 33,820 ± 1153 |
| 1247.197 | 89,620 ± 826 | 38,391 ± 944 |
| 1663.732 | 296,281 ± 1277 | 139,009 ± 1039 |
| m/z Values for the Most Abundant Ion | Charge | Impurity | Impurity Sequence | Deconvoluted Mass (Da) | Retention Time CHOL pH 6.3 (min) | Retention Time CHOL pH 7.5 (min) |
|---|---|---|---|---|---|---|
| ANTISENSE STRAND | ||||||
| 652.082 | −1 | 3′ N-19as | 5′ AU 3′—PO | 653.082 | 8.46 | 5.06 |
| 979.118 | −1 | 3′ N-18as | 5′ AUG 3′—cyc | 980.118 | 10.76 | 8.68 |
| 850.170 | −1 | 5′ N-18as | 5′ CdTdT 3′—OH | 851.170 | 10.73 | 9.06 |
| 545.129 | −1 | 5′ N-19as | 5′ dTdT 3′—OH | 546.129 | 11.15 | 9.94 |
| 749.128 | −2 | 5′ N-16as | 5′ mUACdTdT 3′—OH | 1500.257 | 13.95 | 11.64 |
| 835.110 | −2 | 3′ N-16as | 5′ AUGGA 3′—PO | 1672.221 | 14.03 | 9.33 |
| 902.141 | −2 | 5′ N-15as | 5′ UmUACdTdT 3′—OH | 1806.282 | 14.06 | 11.56 |
| 1074.665 | −2 | 5′ N-14as | 5′ GCmUACdTdT 3′—OH | 2151.330 | 15.62 | 13.01 |
| 999.632 | −2 | 3′ N-15as | 5′ AUGGAA 3′—PO | 2001.264 | 16.11 | 10.73 |
| 1035.139 | −3 | 5′ N-11as | 5′ UUGGUmUACdTdT 3′—OH | 3108.418 | 16.35 | 13.6 |
| 831.122 | −3 | 5′ N-13as | 5′ GGUmUACdTdT 3′—OH | 2496.367 | 16.8 | 14.23 |
| 1136.820 | −3 | 5′ N-10as | 5′ CUUGGUmUACdTdT 3′—OH | 3413.459 | 17.04 | 14 |
| 1043.630 | −4 | 3′ N-8as | 5′ AUGGAAmUACUCUU 3′—PO | 4178.519 | 17.28 | 12.52 |
| 967.3734 | −4 | 3′ N-9as | 5′ AUGGAAmUAUUUU 3′—PO | 3873.496 | 17.28 | 12.51 |
| 928.867 | −4 | 5′ N-9as | 5′ UCUUGGUmUACdTdT 3′-OH | 3719.470 | 17.46 | 14.12 |
| 1005.132 | −4 | 5′ N-8as | 5′ CUCUUGGUmUACdTdT 3′—OH | 4024.528 | 17.46 | 13.78 |
| 882.455 | −3 | 3′ N-13as | 5′ AUGGAAmUA 3′—PO | 2650.364 | 17.66 | 12.56 |
| 1187.822 | −3 | 3′ N-10as | 5′ AUGGAAmUACUC 3′—PO | 3566.466 | 17.94 | 12.66 |
| 1167.402 | −4 | 5′ N-6as | 5′ mUACUCUUGGUmUACdTdT 3′—OH | 4673.609 | 18.21 | 14.16 |
| 984.135 | −3 | 3′ N-12as | 5′ AUGGAAmUAC 3′—PO | 2955.406 | 18.41 | 12.83 |
| 1129.892 | −4 | 3′ N-7as | 5′ AUGGAAmUACUCUUG 3′—PO | 4523.570 | 18.7 | 13.13 |
| 1249.665 | −4 | 5′ N-5as | 5′ AmUACUCUUGGCmUACdTdT 3′—OH | 5002.661 | 18.95 | 14.23 |
| 1216.152 | −4 | 3′ N-6as | 5′ AUGGAAmUACUCUUGG 3′—PO | 4868.606 | 19.04 | 13.55 |
| 1580.954 | −4 | 5′ N-1as | 5′ UGGAAmUACUCUUGGUmUACdTdT 3′—OH | 6327.814 | 19.16 | 15.28 |
| 1418.703 | −4 | 5′ N-3as | 5′ GAAmUACUCUUGGUmUACdTdT 3′—OH | 5676.783 | 19.29 | 15.46 |
| 1504.957 | −4 | 5′ N-2as | 5′ GGAAmUACUCUUGGUmUACdTdT 3′—OH | 6021.828 | 19.33 | 15.47 |
| 1531.187 | −4 | 3′ N-2as | 5′ AUGGAAmUACUCUUGGUmUAC 3′—PO | 6128.748 | 19.54 | 15.14 |
| 1454.931 | −4 | 3′ N-3as | 5′ AUGGAAmUACUCUUGGUmUA 3′—PO | 5823.723 | 19.74 | 14.92 |
| 1663.732 | −4 | Antisense | 5′ AUGGAAmUACUCUUGGUmUACdTdT 3′—OH | 6656.866 | 20.03 | 15.68 |
| SENSE STRAND | ||||||
| 1011.145 | −1 | 3′ N-18s | 5′ GmUA 3′—PO | 1012.130 | 11.77 | 7.49 |
| 865.169 | −1 | 5′ N-18s | 5′ mUdTdT 3′—OH | 866.169 | 13.58 | 11.94 |
| 1340.212 | −1 | 3′ N-17s | 5′GmUAA 3′—PO | 1341.212 | 14.49 | 9.42 |
| 915.667 | −2 | 5′ N-15s | 5′ mCmCAmUdTdT 3′—OH | 1833.337 | 15.12 | 12.44 |
| 756.134 | −2 | 5′ N-16s | 5′ mCAmUdTdT 3′—OH | 1514.268 | 15.18 | 12.94 |
| 1075.689 | −2 | 5′ N-14 OHs | 5′ mUmCmCAmUdTdT 3′—OH | 2153.353 | 16.18 | 13.22 |
| 1115.676 | −2 | 5′ N-14 POs | 5′ mUmCmCAmUdTdT 3′—PO | 2233.352 | 16.67 | 16.62 |
| 1235.714 | −2 | 5′ N-13s | 5′ mUmUmCmCAmUdTdT 3′—OH | 2473.396 | 17.21 | 14.12 |
| 1153.181 | −2 | 3′ N-14s | 5′ GmUAAmCmCA 3′—PO | 2308.362 | 17.92 | 15.7 |
| 1395.741 | −2 | 5′ N-12 (A-mU)s | 5′ mUmUmUmCmCAmUdTdT 3′—OH | 2793.442 | 18.26 | 14.9 |
| 933.150 | −3 | 5′ N-12s | 5′ AmUmUmCmCAmUdTdT 3′—OH | 2802.450 | 18.65 | 15.33 |
| 1039.831 | −3 | 5′ N-11s | 5′ mUAmUmUmCmCAmUdTdT 3′—OH | 3122.492 | 18.79 | 15.48 |
| 1317.703 | −2 | 3′ N-13s | 5′ GmUAAmCmCAA 3′—Np | 2637.407 | 18.87 | 12.17 |
| 1154.857 | −3 | 5′ N-10s | 5′ GmUAmUmUmCmCAmUdTdT 3′—OH | 3467.530 | 19.14 | 15.7 |
| 1264.544 | −3 | 5′ N-9s | 5′ AGmUAmUmUmCmCAmUdTdT 3′—OH | 3796.590 | 20.31 | 16.95 |
| 1489.237 | −3 | 5′ N-7s | 5′ AGAGmUAmUmUmCmCAmUdTdT 3′—OH | 4470.688 | 20.4 | 16.63 |
| 1598.928 | −3 | 5′ N-6s | 5′ AAGAGmUAmUmUmCmCAmUdTdT 3′—OH | 4799.718 | 20.58 | 16.96 |
| 1103.173 | −3 | 3′ N-11s | 5′ GmUAAmCmCAAGA 3′—PO | 3311.495 | 20.62 | 13.39 |
| 1217.849 | −3 | 3′ N-10s | 5′ GmUAAmCmCAAGAG 3′—PO | 3656.547 | 20.69 | 13.59 |
| 1358.973 | −4 | 5′ N-4s | 5′ mCmCAAGAGmUAmUmUmCmCAmUdTdT 3′—OH | 5437.841 | 20.68 | 16.58 |
| 1603.513 | −4 | 5′ N-1s | 5′ mUAAmCmCAAGAGmUAmUmUmCmCAmUdTdT 3′—OH | 6416.030 | 21.08 | 17.07 |
| 1690.023 | −5 | Sense (guide strand) | 5′ GmUAAmCmCAAGAGmUAmUmUmCmCAmUdTdT 3′—OH | 6761.087 | 21.8 | 17.17 |
| 1441.236 | −4 | 5′ N-3s | 5′ AmCmCAAGAGmUAmUmUmCmCAmUdTdT 3′—OH | 5766.896 | 22.71 | 17.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Studzińska, S.; Li, F.; Szumski, M.; Buszewski, B.; Lämmerhofer, M. Cholesterol Stationary Phase in the Separation and Identification of siRNA Impurities by Two-Dimensional Liquid Chromatography-Mass Spectrometry. Int. J. Mol. Sci. 2022, 23, 14960. https://doi.org/10.3390/ijms232314960
Studzińska S, Li F, Szumski M, Buszewski B, Lämmerhofer M. Cholesterol Stationary Phase in the Separation and Identification of siRNA Impurities by Two-Dimensional Liquid Chromatography-Mass Spectrometry. International Journal of Molecular Sciences. 2022; 23(23):14960. https://doi.org/10.3390/ijms232314960
Chicago/Turabian StyleStudzińska, Sylwia, Feiyang Li, Michał Szumski, Bogusław Buszewski, and Michael Lämmerhofer. 2022. "Cholesterol Stationary Phase in the Separation and Identification of siRNA Impurities by Two-Dimensional Liquid Chromatography-Mass Spectrometry" International Journal of Molecular Sciences 23, no. 23: 14960. https://doi.org/10.3390/ijms232314960
APA StyleStudzińska, S., Li, F., Szumski, M., Buszewski, B., & Lämmerhofer, M. (2022). Cholesterol Stationary Phase in the Separation and Identification of siRNA Impurities by Two-Dimensional Liquid Chromatography-Mass Spectrometry. International Journal of Molecular Sciences, 23(23), 14960. https://doi.org/10.3390/ijms232314960