
The Transcriptome and Proteome Networks of Malignant Tumours Reveal Atavistic Attractors of Polyploidy-Related Asexual Reproduction


 , and
, and 
Abstract
1. Introduction
2. Results
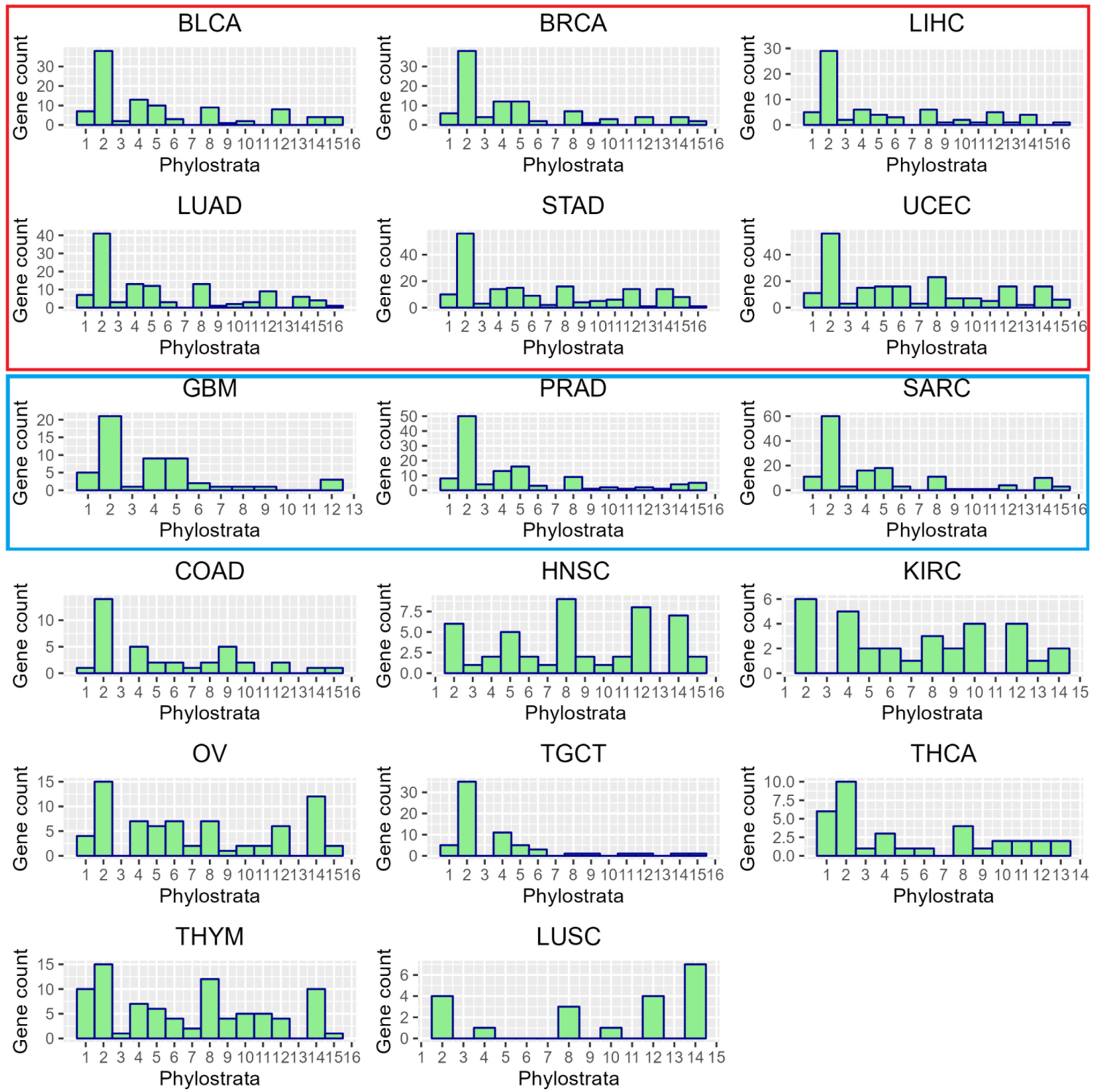
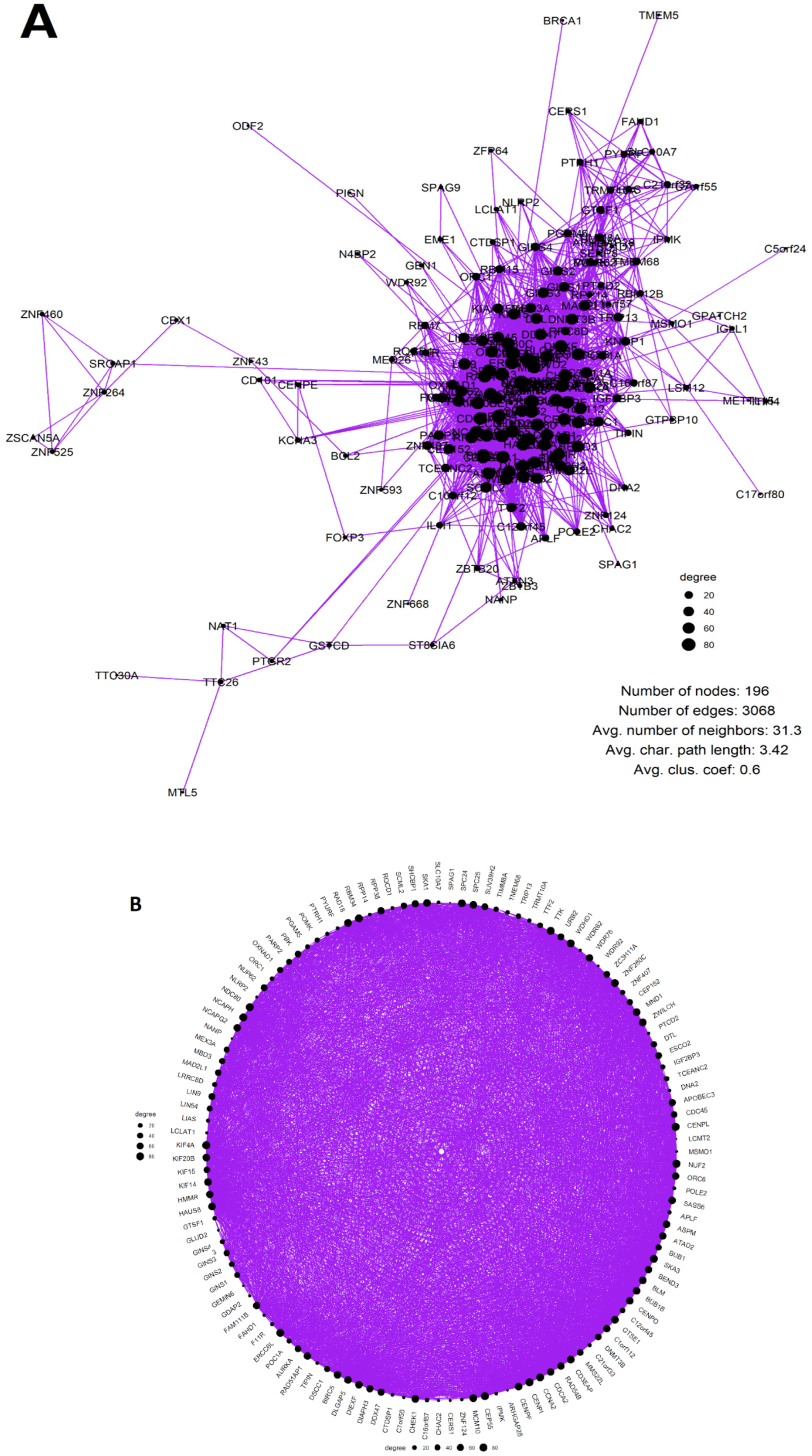
2.1. Whole-Genome Duplication Enriches GG Genes in TCGA Cancer Transcriptomes
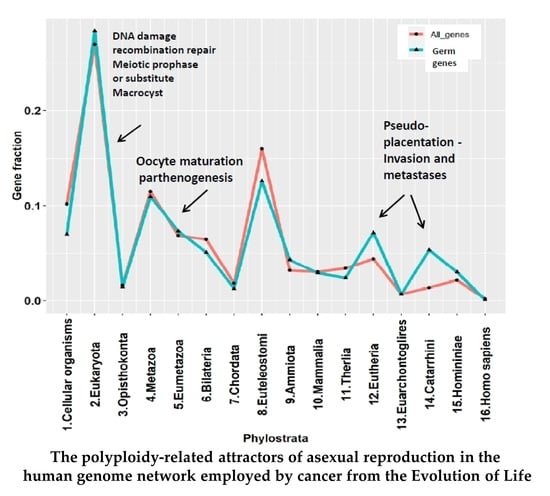
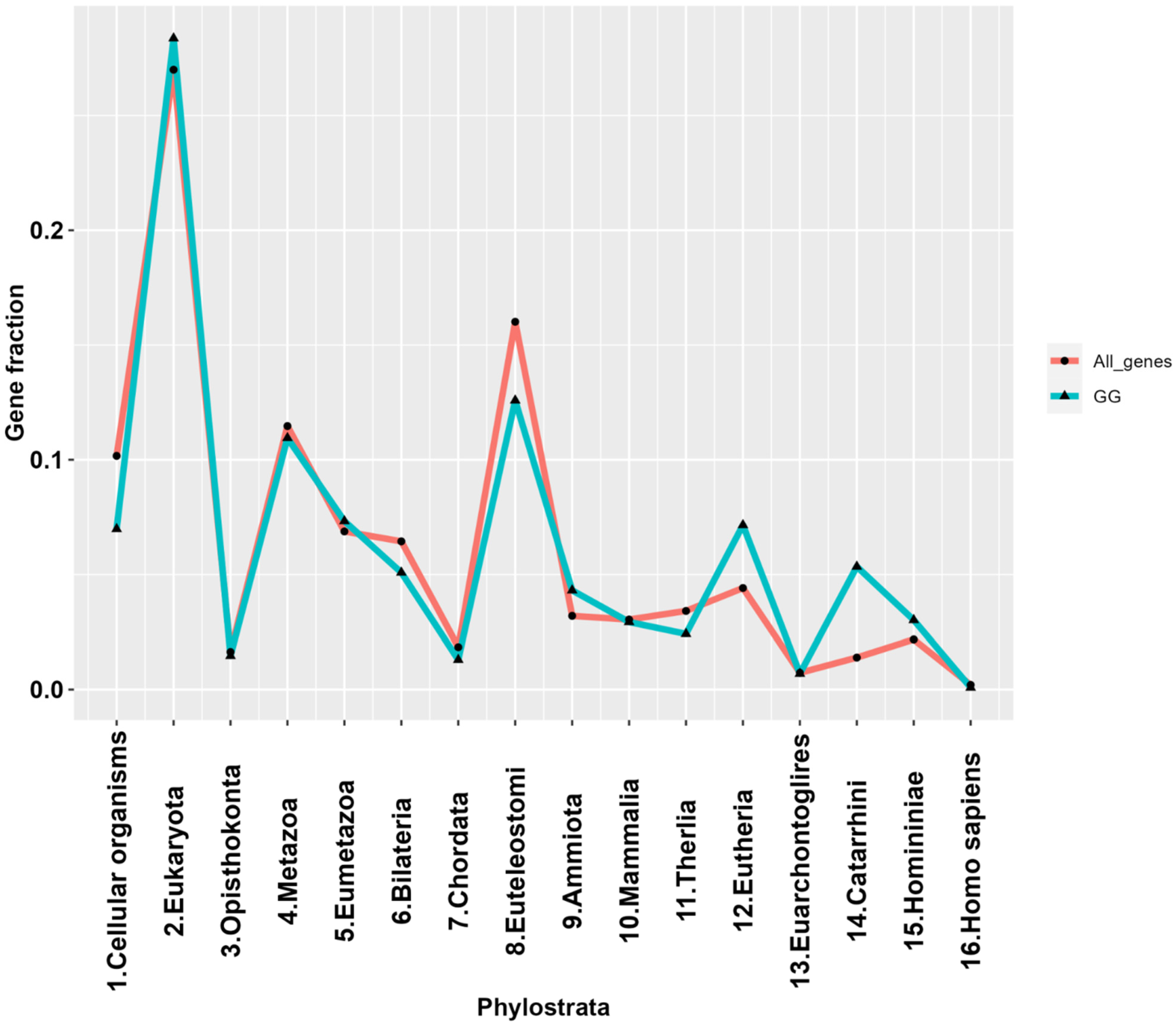
2.2. The Phylostratigraphic Distribution of GG Genes
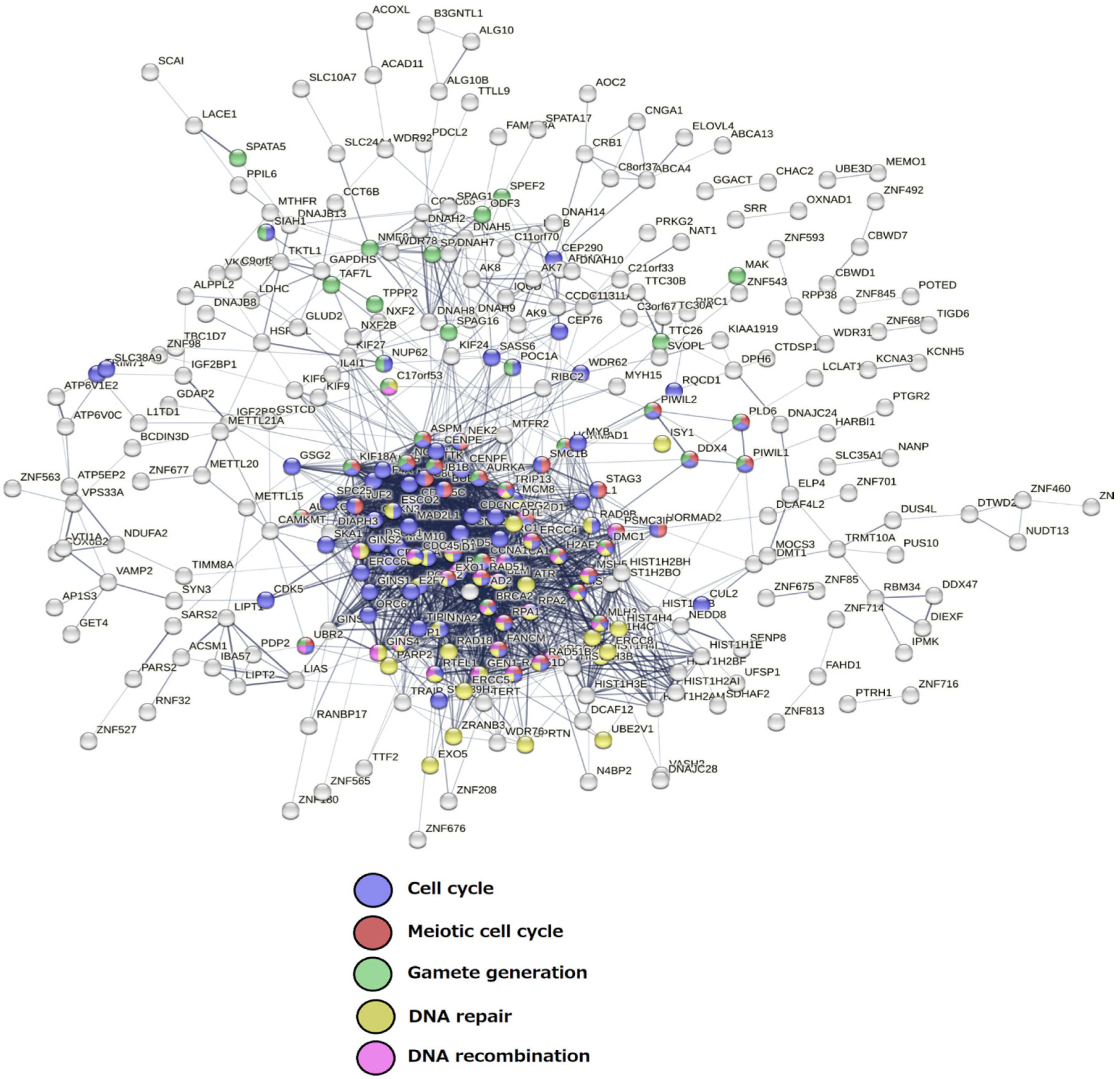
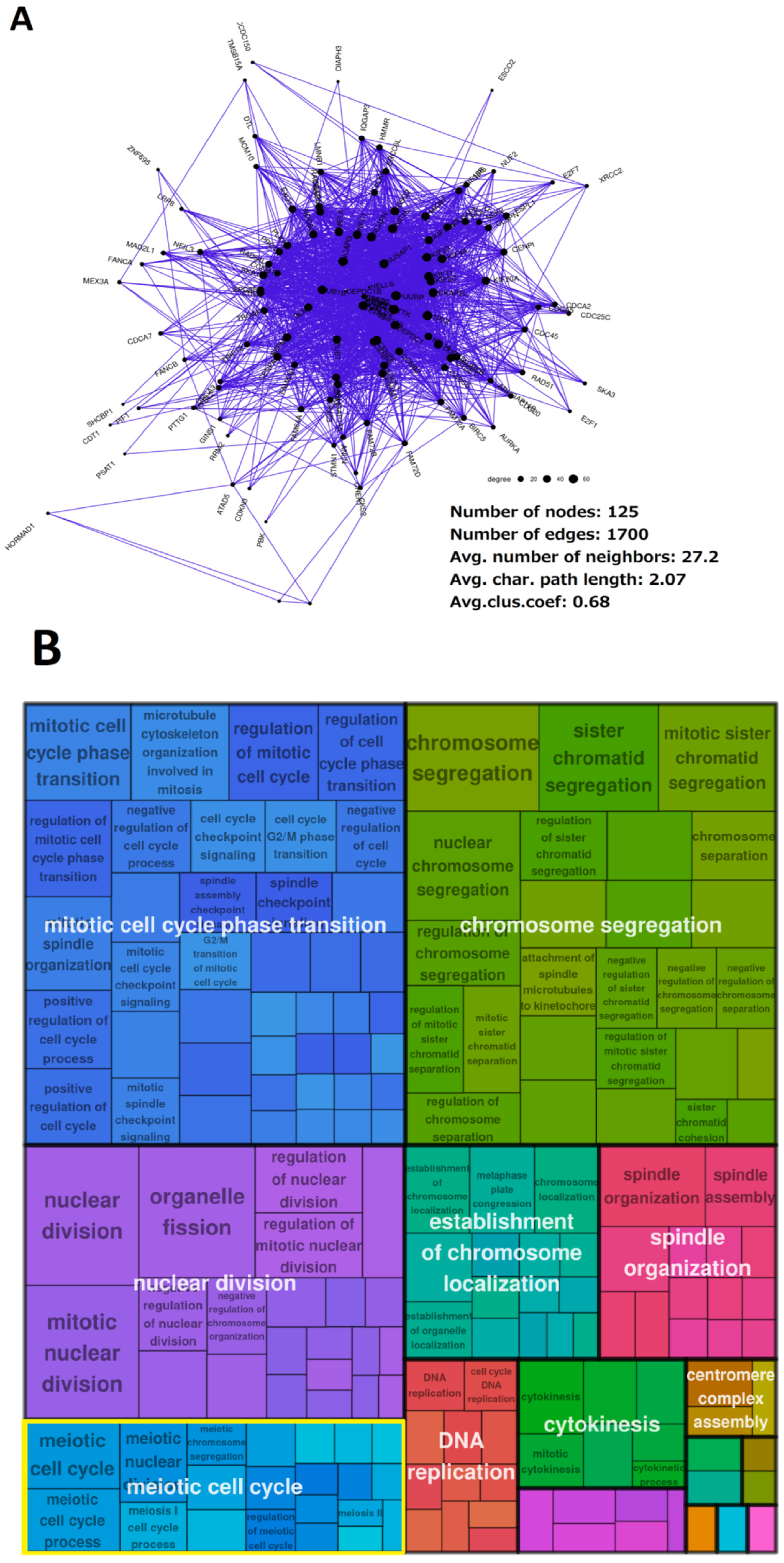
The Senescence Module of the Ploidy-Upregulated Gene Network
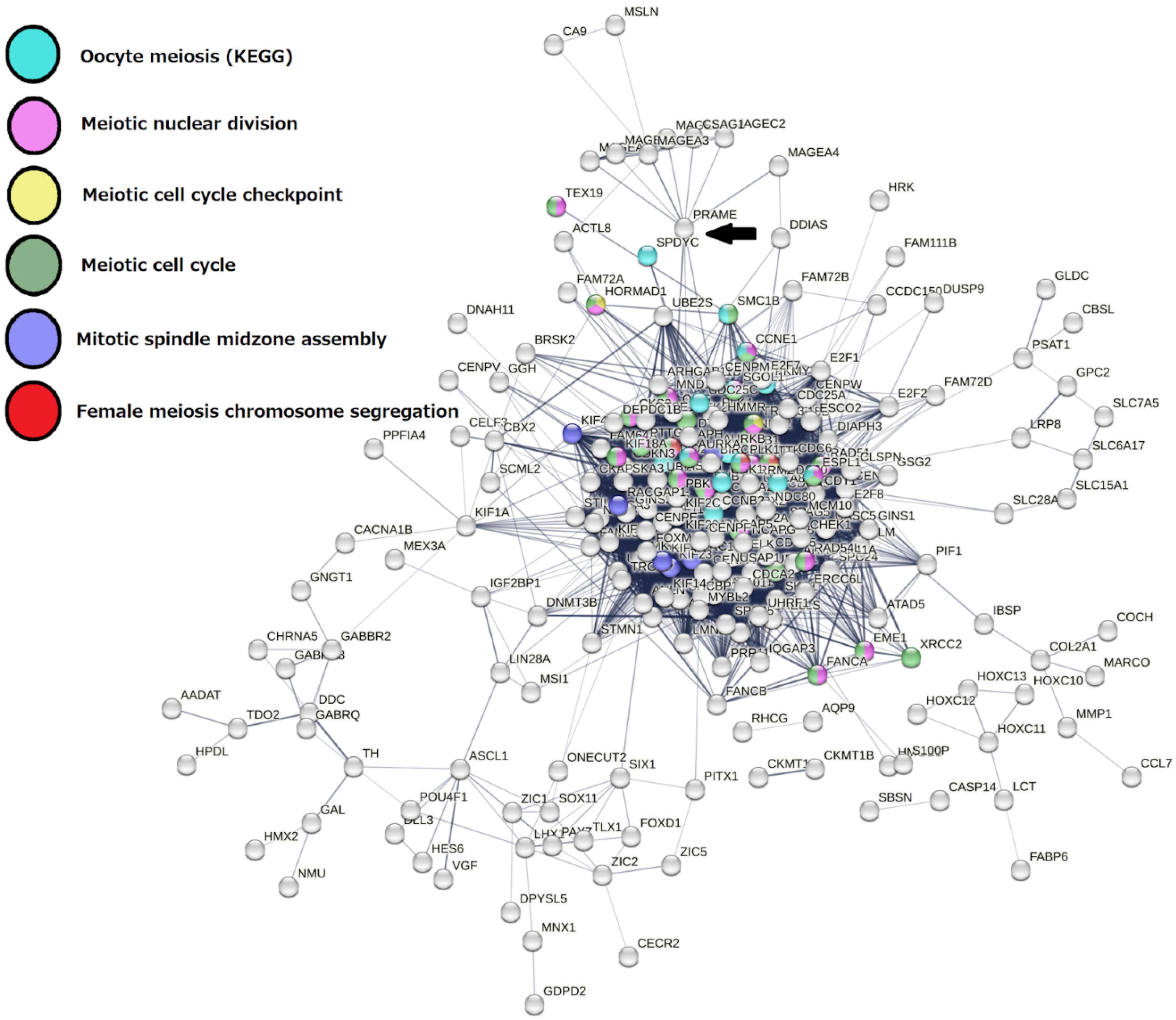
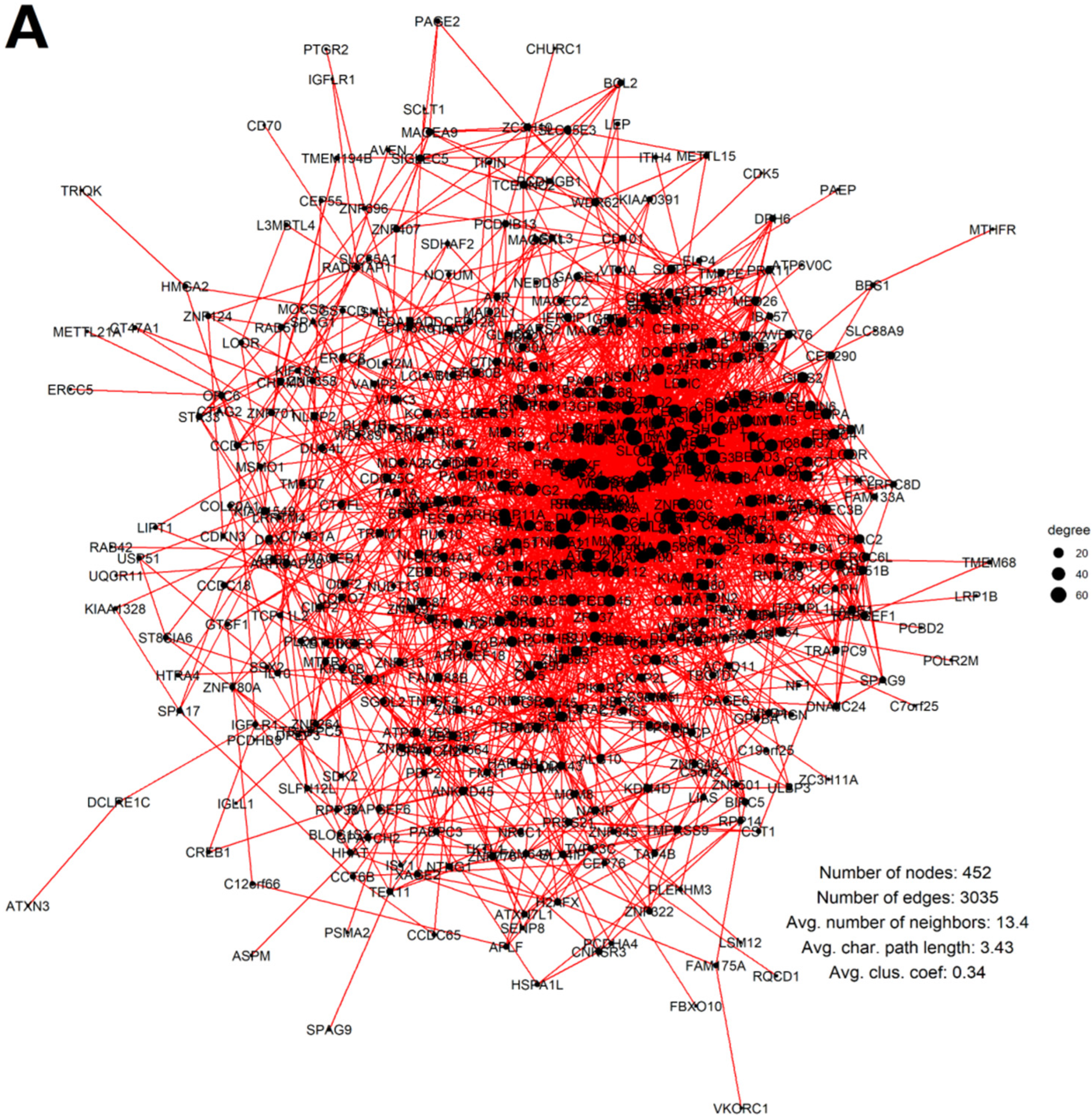
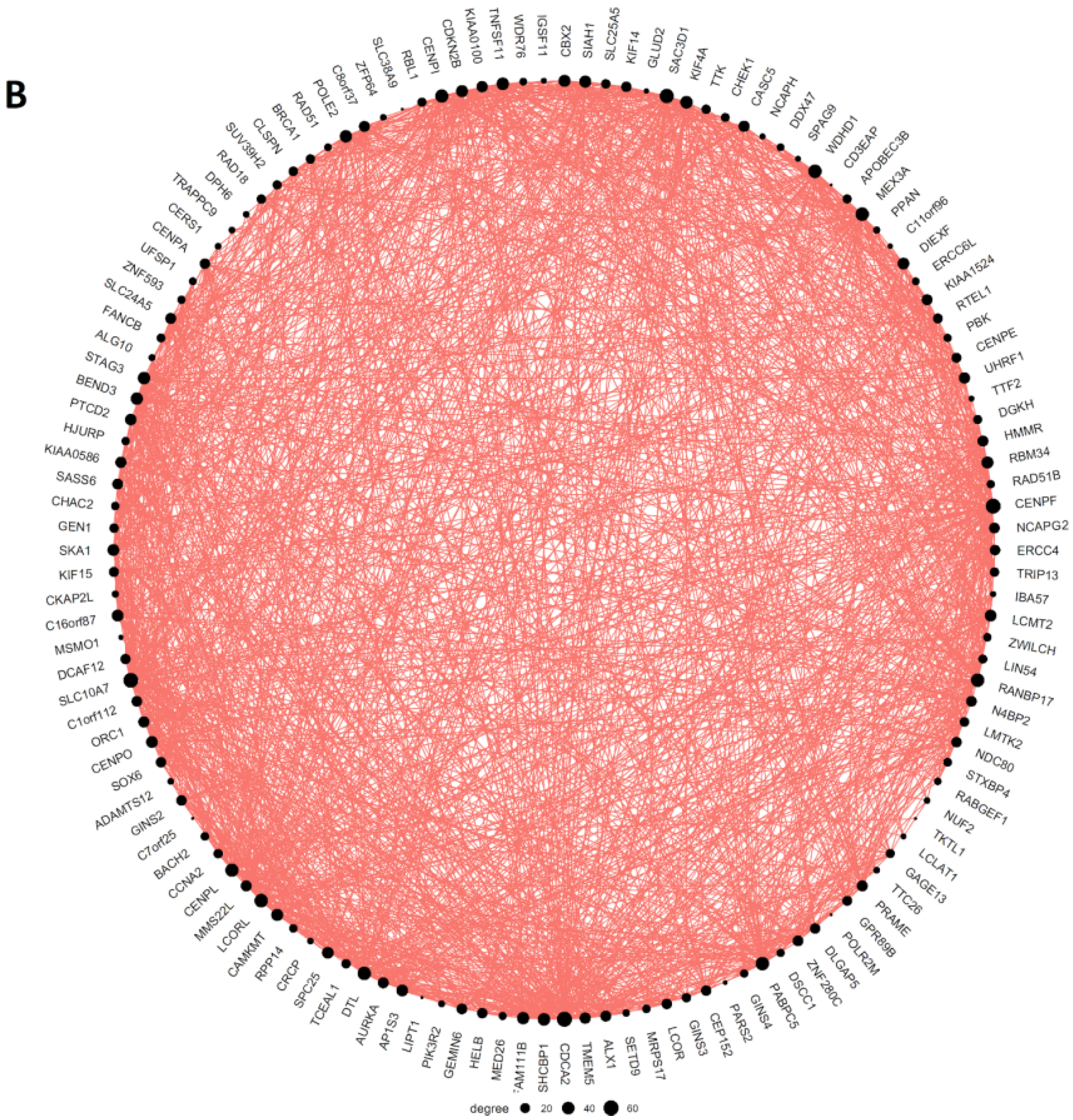
2.3. Malignant Melanoma (MM) and BRCA Proteome Analysis Reveal Robust Expression and Coexpression of GG Proteins
2.3.1. The Malignant Melanoma Proteome
2.3.2. The Breast Carcinoma Proteome
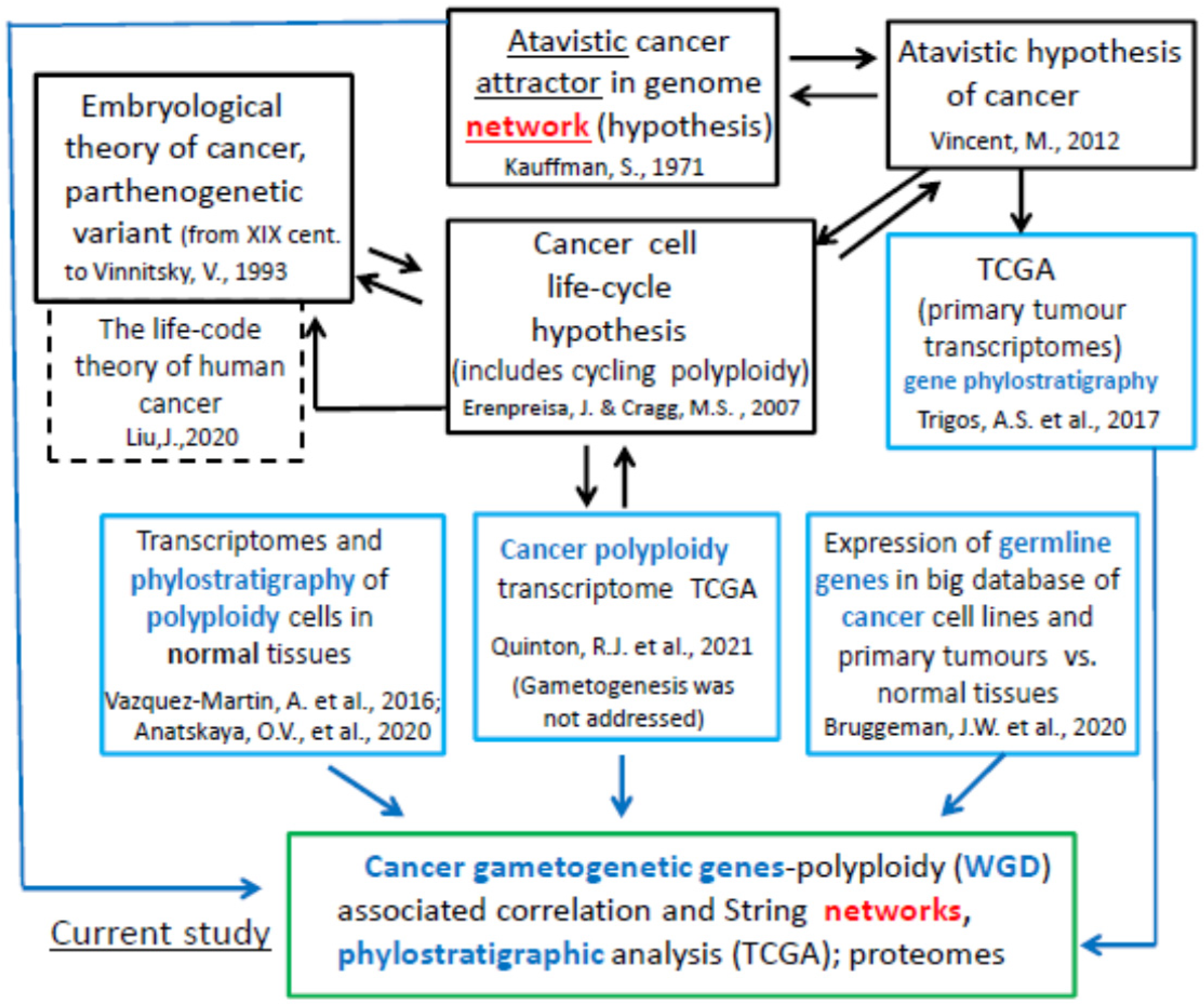
3. Discussion
4. Materials and Methods
4.1. TCGA Polyploid versus Diploid Tumour Transcriptome Comparison
4.2. Analysis of GG Protein Expression in the MM and BRCA Proteomes
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.; Lopez, A.D. Mortality by cause for eight regions of the world: Global Burden of Disease Study. Lancet 1997, 349, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Cancer overtakes CVD to become leading cause of death in high income countries. BMJ 2019, 366, l5368. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.-T. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Caballero, O.L.; Chen, Y.-T. Cancer/testis (CT) antigens: Potential targets for immunotherapy. Cancer Sci. 2009, 100, 2014–2021. [Google Scholar] [CrossRef] [PubMed]
- Old, L.J. Cancer/testis (CT) antigens—A new link between gametogenesis and cancer. Cancer Immun. 2001, 1, 1. [Google Scholar]
- Kalejs, M.; Erenpreisa, J. Cancer/testis antigens and gametogenesis: A review and “brain-storming” session. Cancer Cell Int. 2005, 5, 4. [Google Scholar] [CrossRef]
- Gantchev, J.; Martínez Villarreal, A.; Gunn, S.; Zetka, M.; Ødum, N.; Litvinov, I.V. The Ectopic Expression of meiCT Genes Promotes Meiomitosis and May Facilitate Carcinogenesis. Cell Cycle 2020, 19, 837–854. [Google Scholar] [CrossRef]
- McFarlane, R.J.; Wakeman, J.A. Meiosis-like Functions in Oncogenesis: A New View of Cancer. Cancer Res 2017, 77, 5712–5716. [Google Scholar] [CrossRef]
- Feichtinger, J.; McFarlane, R.J. Meiotic gene activation in somatic and germ cell tumours. Andrology 2019, 7, 415–427. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Horisawa-Takada, Y.; Inoue, E.; Tani, N.; Shibuya, H.; Fujimura, S.; Kariyazono, R.; Sakata, T.; Ohta, K.; Araki, K.; et al. Meiotic cohesins mediate initial loading of HORMAD1 to the chromosomes and coordinate SC formation during meiotic prophase. PLoS Genet. 2020, 16, e1009048. [Google Scholar] [CrossRef]
- Ianzini, F.; Kosmacek, E.A.; Nelson, E.S.; Napoli, E.; Erenpreisa, J.; Kalejs, M.; Mackey, M.A. Activation of Meiosis-Specific Genes Is Associated with Depolyploidization of Human Tumor Cells following Radiation-Induced Mitotic Catastrophe. Cancer Res. 2009, 69, 2296–2304. [Google Scholar] [CrossRef] [PubMed]
- Kalejs, M.; Ivanov, A.; Plakhins, G.; Cragg, M.S.; Emzinsh, D.; Illidge, T.M.; Erenpreisa, J. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer 2006, 6, 6. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S.; Salmina, K.; Hausmann, M.; Scherthan, H. The Role of Meiotic Cohesin REC8 in Chromosome Segregation in Gamma Irradiation-Induced Endopolyploid Tumour Cells. Exp. Cell Res. 2009, 315, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, J.W.; Irie, N.; Lodder, P.; Van Pelt, A.M.M.; Koster, J.; Hamer, G. Tumors Widely Express Hundreds of Embryonic Germline Genes. Cancers 2020, 12, 3812. [Google Scholar] [CrossRef] [PubMed]
- da Silva, V.L.; Fonseca, A.F.; Fonseca, M.; da Silva, T.E.; Coelho, A.C.; Kroll, J.E.; de Souza, J.E.S.; Stransky, B.; de Souza, G.A.; de Souza, S.J. Genome-wide identification of cancer/testis genes and their association with prognosis in a pan-cancer analysis. Oncotarget 2017, 8, 92966–92977. [Google Scholar] [CrossRef] [PubMed]
- Planells-Palop, V.; Hazazi, A.; Feichtinger, J.; Jezkova, J.; Thallinger, G.G.; Alsiwiehri, N.O.; Almutairi, M.; Parry, L.; Wakeman, J.; McFarlane, R.J. Human germ/stem cell-specific gene TEX19 influences cancer cell proliferation and cancer prognosis. Mol. Cancer 2017, 16, 84. [Google Scholar] [CrossRef]
- Nichols, B.A.; Oswald, N.W.; McMillan, E.A.; McGlynn, K.; Yan, J.; Kim, M.S.; Saha, J.; Mallipeddi, P.L.; LaDuke, S.A.; Villa-lobos, P.A.; et al. HORMAD1 Is a Negative Prognostic Indicator in Lung Adenocarcinoma and Specifies Resistance to Oxida-tive and Genotoxic Stress. Cancer Res. 2018, 78, 6196–6208. [Google Scholar] [CrossRef]
- Chung, J.-Y.; Kitano, H.; Takikita, M.; Cho, H.; Noh, K.H.; Kim, T.W.; Ylaya, K.; Hanaoka, J.; Fukuoka, J.; Hewitt, S.M. Syn-aptonemal Complex Protein 3 as a Novel Prognostic Marker in Early Stage Non-Small Cell Lung Cancer. Hum. Pathol. 2013, 44, 472–479. [Google Scholar] [CrossRef]
- Gu, E.A.Y.; Wang, C.; Zhu, R.; Yang, J.; Yuan, W.; Zhu, Y.; Zhou, Y.; Qin, N.; Shen, H.; Ma, H.; et al. The cancer-testis gene, MEIOB, sensitizes triple-negative breast cancer to PARP1 inhibitors by inducing homologous recombination deficiency. Cancer Biol. Med. 2021, 18, 74–87. [Google Scholar] [CrossRef]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef]
- Sheltzer, J.M.; Amon, A. The aneuploidy paradox: Costs and benefits of an incorrect karyotype. Trends Genet. 2011, 27, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Gerashchenko, B.I.; Salmina, K.; Eglitis, J.; Huna, A.; Grjunberga, V.; Erenpreisa, J. Disentangling the Aneuploidy and Senes-cence Paradoxes: A Study of Triploid Breast Cancers Non-Responsive to Neoadjuvant Therapy. Histochem. Cell Biol. 2016, 145, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Hammarlund, E.U.; Brown, J.S.; Amend, S.R.; Axelrod, R.M. Cancer recurrence and lethality are enabled by enhanced survival and reversible cell cycle arrest of polyaneuploid cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2020838118. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S. Cancer: A Matter of Life Cycle? Cell Biol. Int. 2007, 31, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, A.S. The asexual ploidy cycle and the origin of sex. Nature 1994, 370, 213–216. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic Catastrophe and Endomitosis in Tumour Cells: An Evolutionary Key to a Mo-lecular Solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Life-Cycle Features of Tumour Cells. In Evolutionary Biology from Concept to Application; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 61–71. ISBN 9783540789932. [Google Scholar]
- Niculescu, V.F. Developmental and Non Developmental Polyploidy in Xenic and Axenic Cultured Stem Cell Lines of Enta-moeba Invadens and E. Histolytica. Insights Stem Cells 2016, 2, 14. [Google Scholar]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of cancer: Survival at the brink. Review. Semin. Cancer Biol. 2022, 81, 119–131. [Google Scholar] [CrossRef]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef]
- Cohnheim, J. Vorlesungen über Allgemeine Pathologie: Ein Handbuch für Aertze und Studirende; A. Hirschwald: Berlin, Germany, 1882. [Google Scholar]
- Pierce, G.B.; Pantazis, C.G.; Caldwell, E.J.; Wells, R.S. Specificity of the control of tumor formation by the blastocyst. Cancer Res. 1982, 42, 1082–1087. [Google Scholar] [PubMed]
- Erenpreiss, J. Current Concepts of Malignant Growth. Part A: From a Normal Cell to Cancer; Zvaigzne: Riga, Latvia, 1993. [Google Scholar]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef] [PubMed]
- Bignold, L.P.; Coghlan, B.L.D.; Jersmann, H.P.A. David Paul von Hansemann: Contributions to Oncology: Context, Comments and Translations; Birkhauser Verlag: Basel, Switzerland, 2007. [Google Scholar]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “Virgin Birth”, Polyploidy, and the Origin of Cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Vinnitsky, V. Oncogerminative hypothesis of tumor formation. Med. Hypotheses 1993, 40, 19–27. [Google Scholar] [CrossRef]
- Vinnitsky, V. The development of a malignant tumor is due to a desperate asexual self-cloning process in which cancer stem cells develop the ability to mimic the genetic program of germline cells. Intrinsically Disord. Proteins 2014, 2, e29997. [Google Scholar] [CrossRef][Green Version]
- Pierce, G.B. Carcinoma is to Embryology as Mutation is to Genetics. Am. Zool. 1985, 25, 707–712. [Google Scholar] [CrossRef]
- Liu, J. The “Life Code”: A Theory That Unifies the Human Life Cycle and the Origin of Human Tumors. Semin. Cancer Biol. 2020, 60, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Moein, S.; Adibi, R.; Meirelles, L.D.S.; Nardi, N.B.; Gheisari, Y. Cancer regeneration: Polyploid cells are the key drivers of tumor progression. Biochim. Biophys. Acta 2020, 1874, 188408. [Google Scholar] [CrossRef] [PubMed]
- Vainshelbaum, N.M.; Salmina, K.; Gerashchenko, B.I.; Lazovska, M.; Zayakin, P.; Cragg, M.S.; Pjanova, D.; Erenpreisa, J. Role of the Circadian Clock “Death-Loop” in the DNA Damage Response Underpinning Cancer Treatment Resistance. Cells 2022, 11, 880. [Google Scholar] [CrossRef]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid giant cancer cells: An emerging new field of cancer biology. Semin. Cancer Biol. 2022, 81, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lenormand, T.; Engelstädter, J.; Johnston, S.E.; Wijnker, E.; Haag, C.R. Evolutionary mysteries in meiosis. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20160001. [Google Scholar] [CrossRef]
- Mirzaghaderi, G.; Hörandl, E. The evolution of meiotic sex and its alternatives. Proc. R. Soc. B Boil. Sci. 2016, 283, 20161221. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of mitotic descendants by giant cells from irradiated burkitt’s lymphoma cell lines. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.-N. Two Types of Germ Cells, the Sexual Reproduction Cycle, and the Double-Ring Mode of Plant Developmental Pro-gram. Plant Signal. Behav. 2017, 12, e1320632. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Gordeeva, O. Cancer-testis antigens: Unique cancer stem cell biomarkers and targets for cancer therapy. Semin. Cancer Biol. 2018, 53, 75–89. [Google Scholar] [CrossRef]
- Miranda, A.; Hamilton, P.T.; Zhang, A.W.; Pattnaik, S.; Becht, E.; Mezheyeuski, A.; Bruun, J.; Micke, P.; de Reynies, A.; Nelson, B.H. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9020–9029. [Google Scholar] [CrossRef]
- Marcar, L.; Maclaine, N.J.; Hupp, T.R.; Meek, D.W. Mage-A Cancer/testis Antigens Inhibit p53 Function by Blocking Its Inter-action with Chromatin. Cancer Res. 2010, 70, 10362–10370. [Google Scholar] [CrossRef]
- Yang, B.; O’Herrin, S.M.; Wu, J.; Reagan-Shaw, S.; Ma, Y.; Bhat, K.M.; Gravekamp, C.; Setaluri, V.; Peters, N.; Hoffmann, F.M.; et al. MAGE-A, mMage-b, and MAGE-C Proteins Form Complexes with KAP1 and Suppress p53-Dependent Apoptosis in MAGE-Positive Cell Lines. Cancer Res 2007, 67, 9954–9962. [Google Scholar] [CrossRef]
- Ladelfa, M.F.; Peche, L.Y.; Toledo, M.F.; Laiseca, J.E.; Schneider, C.; Monte, M. Tumor-specific MAGE proteins as regulators of p53 function. Cancer Lett. 2012, 325, 11–17. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, Y.; Gong, J.; Rao, L.; Wu, Z.; Nie, T.; Shi, D.; Zhang, L. High expression of MAGE-A9 contributes to stemness and malignancy of human hepatocellular carcinoma. Int. J. Oncol. 2017, 52, 219–230. [Google Scholar] [CrossRef]
- Nettersheim, D.; Arndt, I.; Sharma, R.; Riesenberg, S.; Jostes, S.; Schneider, S.; Hölzel, M.; Kristiansen, G.; Schorle, H. The Cancer/testis-Antigen PRAME Supports the Pluripotency Network and Represses Somatic and Germ Cell Differentiation Pro-grams in Seminomas. Br. J. Cancer 2016, 115, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Al-Khadairi, G.; Decock, J. Cancer Testis Antigens and Immunotherapy: Where Do We Stand in the Targeting of PRAME? Cancers 2019, 11, 984. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Zhang, Z.; Wu, Y. PRAME Is a Potential Carcinogenic Biomarker That Correlates with Patient Prognosis and Tumor Immunity Based on Pan-Cancer Analysis. Ann. Clin. Lab. Sci. 2022, 52, 185–195. [Google Scholar] [PubMed]
- Lindsey, S.F.; Byrnes, D.M.; Eller, M.S.; Rosa, A.M.; Dabas, N.; Escandon, J.; Grichnik, J.M. Potential Role of Meiosis Proteins in Melanoma Chromosomal Instability. J. Ski. Cancer 2013, 2013, 190109. [Google Scholar] [CrossRef] [PubMed]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [PubMed]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Somatic mutations in early metazoan genes disrupt regulatory links between unicellular and multicellular genes in cancer. eLife 2019, 8, 947. [Google Scholar] [CrossRef]
- Niculescu, V.F. Cancer genes and cancer stem cells in tumorigenesis: Evolutionary deep homology and controversies. Genes Dis. 2022, 9, 1234–1247. [Google Scholar] [CrossRef]
- Maciver, S.K.; Koutsogiannis, Z.; Valle, A.D.O.F.D. ‘Meiotic genes’ are constitutively expressed in an asexual amoeba and are not necessarily involved in sexual reproduction. Biol. Lett. 2019, 15, 20180871. [Google Scholar] [CrossRef]
- Lorenz, A.; Mpaulo, S.J. Gene conversion: A non-Mendelian process integral to meiotic recombination. Heredity 2022, 129, 56–63. [Google Scholar] [CrossRef]
- Kauffman, S. Differentiation of malignant to benign cells. J. Theor. Biol. 1971, 31, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ernberg, I.; Kauffman, S. Cancer Attractors: A Systems View of Tumors from a Gene Network Dynamics and De-velopmental Perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, T.; Tautz, D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E. Human transcriptome nexuses: Basic-eukaryotic and metazoan. Genomics 2010, 95, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M. Cancer: A de-Repression of a Default Survival Program Common to All Cells?: A Life-History Perspective on the Nature of Cancer. Bioessays 2012, 34, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Lineweaver, C.H.; Bussey, K.J.; Blackburn, A.C.; Davies, P.C.W. Cancer progression as a sequence of atavistic reversions. BioEssays 2021, 43, e2000305. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Klimovich, A.; Anokhin, B.; Anton-Erxleben, F.; Hamm, M.J.; Lange, C.; Bosch, T.C. Naturally occurring tumours in the basal metazoan Hydra. Nat. Commun. 2014, 5, 4222. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E.; Vainshelbaum, N.M.; Giuliani, A.; Erenpreisa, J. Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer. Int. J. Mol. Sci. 2020, 21, 8759. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Anatskaya, O.V. Growth of Biological Complexity from Prokaryotes to Hominids Reflected in the Human Genome. Int. J. Mol. Sci. 2021, 22, 11640. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic polyploidy is associated with the upregulation of c-MYC interacting genes and EMT-like signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Polyploidy and Myc Proto-Oncogenes Promote Stress Adaptation via Epigenetic Plasticity and Gene Regulatory Network Rewiring. Int. J. Mol. Sci. 2022, 23, 9691. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer Stem Cells as Key Drivers of Tumour Progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer Stem Cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. [Google Scholar] [CrossRef]
- Roeder, G.S.; Bailis, J.M. The Pachytene Checkpoint. Trends Genet. 2000, 16, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.V.; Hochwagen, A. The Meiotic Checkpoint Network: Step-by-Step through Meiotic Prophase. Cold Spring Harb. Perspect. Biol. 2014, 6, a016675. [Google Scholar] [CrossRef]
- Czarnecka-Herok, J.; Sliwinska, M.A.; Herok, M.; Targonska, A.; Strzeszewska-Potyrala, A.; Bojko, A.; Wolny, A.; Mosieniak, G.; Sikora, E. Therapy-Induced Senescent/Polyploid Cancer Cells Undergo Atypical Divisions Associated with Altered Expres-sion of Meiosis, Spermatogenesis and EMT Genes. Int. J. Mol. Sci. 2022, 23, 8288. [Google Scholar] [CrossRef]
- Berdieva, M.; Demin, S.; Goodkov, A. Amoeba proteus and ploidy cycles: From simple model to complex issues. Protistology 2019, 13, 166–173. [Google Scholar] [CrossRef]
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 2021, 590, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Yu, X.; Seitz, E.M.; Kowalczykowski, S.C.; Egelman, E.H. Archaeal RadA Protein Binds DNA as Both Helical Fila-ments and Octameric Rings. J. Mol. Biol. 2001, 314, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Sauvageau, S.; Masson, J.-Y. Exploring the multiple facets of the meiotic recombinase Dmc1. BioEssays 2004, 26, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.-H.; Choi, Y.; Erdin, S.U.; Yatsenko, S.A.; Kloc, M.; Yang, F.; Wang, P.J.; Meistrich, M.L.; Rajkovic, A. Hormad1 Muta-tion Disrupts Synaptonemal Complex Formation, Recombination, and Chromosome Segregation in Mammalian Meiosis. PLoS Genet. 2010, 6, e1001190. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. MOS, aneuploidy and the ploidy cycle of cancer cells. Oncogene 2010, 29, 5447–5451. [Google Scholar] [CrossRef]
- Yamamoto, A.; Hiraoka, Y. Monopolar spindle attachment of sister chromatids is ensured by two distinct mechanisms at the first meiotic division in fission yeast. EMBO J. 2003, 22, 2284–2296. [Google Scholar] [CrossRef]
- Jungbluth, A.A.; Silva, A.W.; Iversen, K.; Frosina, D.; Zaidi, B.; Coplan, K.; Eastlake-Wade, S.K.; Castelli, S.B.; Spagnoli, G.C.; Old, L.J.; et al. Expression of cancer-testis (CT) antigens in placenta. Cancer Immun. 2007, 7, 15. [Google Scholar]
- Old, L.J. Cancer is a somatic cell pregnancy. Cancer Immun. 2007, 7, 19. [Google Scholar]
- Kulkarni, P.; Shiraishi, T.; Rajagopalan, K.; Kim, R.; Mooney, S.M.; Getzenberg, R.H. Cancer/testis antigens and urological malignancies. Nat. Rev. Urol. 2012, 9, 386–396. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’Ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Arcaro, A. Targeting the insulin-like growth factor-1 receptor in human cancer. Front. Pharmacol. 2013, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Git, A.; Allison, R.; Perdiguero, E.; Nebreda, A.R.; Houliston, E.; Standart, N. Vg1RBP phosphorylation by Erk2 MAP kinase correlates with the cortical release of Vg1 mRNA during meiotic maturation of Xenopus oocytes. RNA 2009, 15, 1121–1133. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andrabi, S.M.; Ganai, N.A. Long Non-Coding RNA: From Disease Biomarkers to Targeted Therapeutics; Cambridge Scholars Publishing: Cambridge, UK, 2020; ISBN 9781527559103. [Google Scholar]
- Hammer, N.A.; Hansen, T.O.; Byskov, A.G.; Rajpert-De Meyts, E.; Grøndahl, M.L.; Bredkjaer, H.E.; Wewer, U.M.; Christiansen, J.; Nielsen, F.C. Expression of IGF-II mRNA-Binding Proteins (IMPs) in Gonads and Testicular Cancer. Reproduction 2005, 130, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, Z.; Mao, S.; Chen, W.; Zeng, R.; Zhou, S.; Liu, J. Effect of hypoxia on hypoxia inducible factor-1α, insulin-like growth factor I and vascular endothelial growth factor expression in hepatocellular carcinoma HepG2 cells. Oncol. Lett. 2015, 9, 1142–1148. [Google Scholar] [CrossRef]
- Betancourt, L.H.; Gil, J.; Kim, Y.; Doma, V.; Çakır, U.; Sanchez, A.; Murillo, J.R.; Kuras, M.; Parada, I.P.; Sugihara, Y.; et al. The human melanoma proteome atlas—Defining the molecular pathology. Clin. Transl. Med. 2021, 11, e473. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cyto-scape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Johansson, H.J.; Consortia Oslo Breast Cancer Research Consortium (OSBREAC); Socciarelli, F.; Vacanti, N.M.; Haugen, M.H.; Zhu, Y.; Siavelis, I.; Fernandez, A.; Aure, M.R.; Sennblad, B.; et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat. Commun. 2019, 10, 1600. [Google Scholar] [CrossRef]
- Pjanova, D.; Vainshelbaum, M.N.; Salmina, K.; Erenpreisa, J. The Role of the Meiotic Component in Reproduction of B-RAF-Mutated Melanoma: A Review and “brainstorming” Session. In Melanoma; IntechOpen: London, UK, 2021; ISBN 9781838808785. [Google Scholar]
- Erenpreisa, J.; Cragg, M.S.; Anisimov, A.P.; Illidge, T.M. Tumor Cell Embryonality and the Ploidy Number 32n: Is It a Developmental Checkpoint? Cell Cycle 2011, 10, 1873–1874. [Google Scholar] [CrossRef][Green Version]
- Bernstein, H.; Bernstein, C.; Michod, R.E. Meiosis as an Evolutionary Adaptation for DNA Repair. DNA Repair 2011, 11, 357. [Google Scholar] [CrossRef]
- Skejo, J.; Garg, S.G.; Gould, S.B.; Hendriksen, M.; Tria, F.D.K.; Bremer, N.; Franjević, D.; Blackstone, N.W.; Martin, W.F. Evidence for a Syncytial Origin of Eukaryotes from Ancestral State Reconstruction. Genome Biol. Evol. 2021, 13, evab096. [Google Scholar] [CrossRef] [PubMed]
- Muller, H. The relation of recombination to mutational advance. Mutat. Res. Mol. Mech. Mutagen. 1964, 1, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M. Recombination and loss of complementation: A more than two-fold cost for parthenogenesis. J. Evol. Biol. 2004, 17, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Archetti, M. Inverted meiosis and the evolution of sex by loss of complementation. J. Evol. Biol. 2019, 33, 460–467. [Google Scholar] [CrossRef]
- Archetti, M. Polyploidy as an Adaptation against Loss of Heterozygosity in Cancer. Int. J. Mol. Sci. 2022, 23, 8528. [Google Scholar] [CrossRef]
- Soppa, J. Ploidy and gene conversion in Archaea. Biochem. Soc. Trans. 2011, 39, 150–154. [Google Scholar] [CrossRef]
- Hofstatter, P.G.; Brown, M.; Lahr, D.J.G. Comparative Genomics Supports Sex and Meiosis in Diverse Amoebozoa. Genome Biol. Evol. 2018, 10, 3118–3128. [Google Scholar] [CrossRef]
- Nielsen, A.Y.; Gjerstorff, M.F. Ectopic Expression of Testis Germ Cell Proteins in Cancer and Its Potential Role in Genomic Instability. Int. J. Mol. Sci. 2016, 17, 890. [Google Scholar] [CrossRef]
- Rivera, M.; Wu, Q.; Hamerlik, P.; Hjelmeland, A.B.; Bao, S.; Rich, J.N. Acquisition of meiotic DNA repair regulators maintain genome stability in glioblastoma. Cell Death Dis. 2015, 6, e1732. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-Induced Polyploidy Shifts Somatic Cells towards a pro-Tumourogenic Unicellular Gene Transcription Network. Cancer Hypotheses 2018, 1, 1–20. [Google Scholar]
- Walen, K.H. Epigenetic Enabled Normal Human Cells, Lead to First Cell’s Unique Division System, Driving Tumorigenesis Evolution. J. Cancer Ther. 2022, 13, 48–69. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Ivanov, A.; Wheatley, S.P.; Kosmacek, E.A.; Ianzini, F.; Anisimov, A.P.; Mackey, M.; Davis, P.J.; Plakhins, G.; Illidge, T.M. Endopolyploidy in irradiated p53-deficient tumour cell lines: Persistence of cell division activity in giant cells expressing Aurora-B kinase. Cell Biol. Int. 2008, 32, 1044–1056. [Google Scholar] [CrossRef]
- Salmina, K.; Gerashchenko, B.I.; Hausmann, M.; Vainshelbaum, N.M.; Zayakin, P.; Erenpreiss, J.; Freivalds, T.; Cragg, M.S.; Erenpreisa, J. When Three Isn’t a Crowd: A Digyny Concept for Treatment-Resistant, Near-Triploid Human Cancers. Genes 2019, 10, 551. [Google Scholar] [CrossRef] [PubMed]
- Adl, S.M.; Simpson, A.G.B.; Lane, C.E.; Lukes, J.; Bass, D.; Bowser, S.S.; Brown, M.; Burki, F.; Dunthorn, M.; Hampl, V.; et al. The Revised Classification of Eukaryotes. J. Eukaryot. Microbiol. 2012, 59, 429–514. [Google Scholar] [CrossRef] [PubMed]
- Raĭkov, I.B. The Protozoan Nucleus, Morphology and Evolution; Springer: Berlin/Heidelberg, Germany, 1982; ISBN 9780387816784. [Google Scholar]
- Davies, P. Exposing cancer’s deep evolutionary roots. Phys. World 2013, 26, 37–40. [Google Scholar] [CrossRef]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A Novel Type of Cell Division in Cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of Cancer Stem-like Cells through the For-mation of Polyploid Giant Cancer Cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Ivanov, A.; Cragg, M.; Selivanova, G.; Illidge, T. Nuclear envelope-limited chromatin sheets are part of mitotic death. Histochem. Cell Biol. 2002, 117, 243–255. [Google Scholar] [CrossRef]
- Westbrook, V.A.; Schoppee, P.D.; Diekman, A.B.; Klotz, K.L.; Allietta, M.; Hogan, K.T.; Slingluff, C.L.; Patterson, J.W.; Frierson, H.F.; Irvin, W.P.; et al. Genomic Organization, Incidence, and Localization of the SPAN-X Family of Cancer-Testis Antigens in Melanoma Tumors and Cell Lines. Clin. Cancer Res. 2004, 10, 101–112. [Google Scholar] [CrossRef][Green Version]
- Lazar, I.; Fabre, B.; Feng, Y.; Khateb, A.; Turko, P.; Gomez, J.M.M.; Frederick, D.T.; Levesque, M.P.; Feld, L.; Zhang, G.; et al. SPANX Control of Lamin A/C Modulates Nuclear Architecture and Promotes Melanoma Growth. Mol. Cancer Res. 2020, 18, 1560–1573. [Google Scholar] [CrossRef]
- Kouprina, N.; Noskov, V.N.; Pavlicek, A.; Collins, N.K.; Bortz, P.D.S.; Ottolenghi, C.; Loukinov, D.; Goldsmith, P.; Risinger, J.I.; Kim, J.-H.; et al. Evolutionary Diversification of SPANX-N Sperm Protein Gene Structure and Expression. PLoS ONE 2007, 2, e359. [Google Scholar] [CrossRef] [PubMed]
- Zybina, T. Genome Modifications Involved in Developmental Programs of the Placental Trophoblast. In Cytogenetics-Classical and Molecular Strategies for Analysing Heredity Material; IntechOpen: London, UK, 2021; ISBN 9781839689413. [Google Scholar]
- Kshitiz; Afzal, J.; Maziarz, J.D.; Hamidzadeh, A.; Liang, C.; Erkenbrack, E.M.; Kim, H.N.; Haeger, J.-D.; Pfarrer, C.; Ho-ang, T.; et al. Evolution of Placental Invasion and Cancer Metastasis Are Causally Linked. Nat. Ecol. Evol. 2019, 3, 1743–1753. [Google Scholar]
- Costanzo, V.; Bardelli, A.; Siena, S.; Abrignani, S. Exploring the links between cancer and placenta development. Open Biol. 2018, 8, 180081. [Google Scholar] [CrossRef] [PubMed]
- Lala, P.K.; Nandi, P.; Hadi, A.; Halari, C. A Crossroad between Placental and Tumor Biology: What Have We Learnt? Placenta 2021, 116, 12–30. [Google Scholar] [CrossRef]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, A.B.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef]
- Almeida, L.G.; Sakabe, N.J.; Deoliveira, A.R.; Silva, M.C.C.; Mundstein, A.S.; Cohen, T.; Chen, Y.-T.; Chua, R.; Gurung, S.; Gnjatic, S.; et al. CTdatabase: A knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2008, 37, D816–D819. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, D.; Ali, A.; Xu, B.; Liu, W.; Wen, J.; Zhang, H.; Shi, Q.; Zhang, Y. MeiosisOnline: A Manually Curated Database for Tracking and Predicting Genes Associated With Meiosis. Front. Cell Dev. Biol. 2021, 9, 673073. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein–protein Networks, and Functional Characterization of User-Uploaded Gene/measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Rahman, M.; Jackson, L.K.; Johnson, W.; Li, D.Y.; Bild, A.H.; Piccolo, S.R. Alternative preprocessing of RNA-Sequencing data in The Cancer Genome Atlas leads to improved analysis results. Bioinformatics 2015, 31, 3666–3672. [Google Scholar] [CrossRef]
- Censi, F.; Giuliani, A.; Bartolini, P.; Calcagnini, G. A Multiscale Graph Theoretical Approach to Gene Regulation Net-works: A Case Study in Atrial Fibrillation. IEEE Trans. Biomed. Eng. 2011, 58, 2943–2946. [Google Scholar] [CrossRef] [PubMed]
- Gustavsen, J.A.; Pai, S.; Isserlin, R.; Demchak, B.; Pico, A.R. RCy3: Network Biology Using Cytoscape from within R. F1000Research 2019, 8, 1774. [Google Scholar] [CrossRef] [PubMed]
- Settle, B.; Otasek, D.; Morris, J.H.; Demchak, B. aMatReader: Importing adjacency matrices via Cytoscape Automation. F1000Research 2018, 7, 823. [Google Scholar] [CrossRef]
- Sayols, S. Rrvgo: A Bioconductor Package to Reduce and Visualize Gene Ontology Terms. Available online: https://ssayols.github.io/rrvgo/ (accessed on 1 September 2022).
- Csardi, N. The Igraph Software Package for Complex Network Research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Pedersen, T.L. Ggraph: An Implementation of Grammar of Graphics for Graphs and Networks, R Package Version 1.0. 2; R Foundation: Vienna, Austria, 2018. [Google Scholar]
- Pratt, D.; Chen, J.; Welker, D.; Rivas, R.; Pillich, R.; Rynkov, V.; Ono, K.; Miello, C.; Hicks, L.; Szalma, S.; et al. NDEx, the Network Data Exchange. Cell Syst. 2015, 1, 302–305. [Google Scholar] [CrossRef]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar] [CrossRef]
- Dujon, A.M.; Aktipis, A.; Alix-Panabières, C.; Amend, S.R.; Boddy, A.M.; Brown, J.S.; Capp, J.; DeGregori, J.; Ewald, P.; Gatenby, R.; et al. Identifying key questions in the ecology and evolution of cancer. Evol. Appl. 2020, 14, 877–892. [Google Scholar] [CrossRef]
- Gyurko, D.M.; Veres, D.; Modos, D.; Lenti, K.; Korcsmáros, T.; Csermely, P. Adaptation and learning of molecular networks as a description of cancer development at the systems-level: Potential use in anti-cancer therapies. Semin. Cancer Biol. 2013, 23, 262–269. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA Cancer Type | % of WGD+ Samples | Number of Ploidy-Upregulated Genes | Upregulated Genes Enriched with GG (p < 0.05) | Number of Ploidy-Downregulated Genes | Downregulated Genes Enriched with GG (p < 0.05) |
|---|---|---|---|---|---|
| ACC | 53.7% | 84 | FALSE | 443 | FALSE |
| BLCA | 60.2% | 424 | TRUE | 1000 | FALSE |
| BRCA | 44.0% | 277 | TRUE | 1395 | FALSE |
| CESC | 33.7% | 16 | FALSE | 157 | FALSE |
| CHOL | 22.2% | 1 | FALSE | 0 | FALSE |
| COAD | 40.0% | 431 | FALSE | 641 | FALSE |
| ESCA | 55.4% | 0 | FALSE | 1 | FALSE |
| GBM | 17.6% | 164 | TRUE | 850 | FALSE |
| HNSC | 39.4% | 309 | FALSE | 867 | FALSE |
| KICH | 15.4% | 164 | FALSE | 204 | FALSE |
| KIRC | 22.2% | 305 | FALSE | 658 | FALSE |
| KIRP | 7.3% | 36 | FALSE | 5 | FALSE |
| LIHC | 35.0% | 435 | TRUE | 894 | FALSE |
| LUAD | 57.1% | 431 | TRUE | 1445 | FALSE |
| LUSC | 54.5% | 97 | TRUE | 1106 | FALSE |
| MESO | 22.6% | 0 | FALSE | 4 | FALSE |
| OV | 57.6% | 400 | TRUE | 1374 | FALSE |
| PAAD | 29.1% | 11 | FALSE | 1083 | FALSE |
| PCPG | 21.4% | 3 | FALSE | 6 | FALSE |
| PRAD | 11.6% | 427 | TRUE | 610 | FALSE |
| READ | 52.3% | 0 | FALSE | 4 | FALSE |
| SARC | 47.4% | 573 | TRUE | 1040 | FALSE |
| STAD | 41.3% | 1047 | TRUE | 928 | FALSE |
| TGCT | 95.3% | 549 | FALSE | 523 | FALSE |
| THCA | 9.0% | 686 | FALSE | 1138 | FALSE |
| THYM | 20.0% | 1562 | FALSE | 1046 | FALSE |
| UCEC | 22.3% | 1671 | FALSE | 1691 | FALSE |
| UCS | 82.8% | 1 | FALSE | 1 | FALSE |
| UVM | 6.3% | 13 | FALSE | 69 | FALSE |
| Tumour Type | Number of Samples (WGD+, >0.5 Purity) | Number of Upregulated Genes (pAdj < 0.05; logFC > 0.5) Expressed in High-Purity Samples | Percent of Upregulated Genes in the Giant Component of Network | Average Clustering Coefficient of Network | Meiotic GO/KEGG Modules Enriched in Coexpression Network (Y/N) | Female-Specific Meiotic GO/KEGG Modules Enriched in Coexpression Network (Y/N) |
|---|---|---|---|---|---|---|
| BRCA | 277 | 220 | 56.8% | 0.68 | Y | Y |
| LUAD | 103 | 340 | 52.9% | 0.74 | Y | Y |
| LIHC | 91 | 290 | 94.5% | 0.61 | Y | Y |
| STAD | 82 | 756 | 58.6% | 0.47 | Y | Y |
| BLCA | 142 | 348 | 54.9% | 0.5 | Y | Y |
| UCEC | 97 | 1217 | 48.5% | 0.56 | Y | Y |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vainshelbaum, N.M.; Giuliani, A.; Salmina, K.; Pjanova, D.; Erenpreisa, J. The Transcriptome and Proteome Networks of Malignant Tumours Reveal Atavistic Attractors of Polyploidy-Related Asexual Reproduction. Int. J. Mol. Sci. 2022, 23, 14930. https://doi.org/10.3390/ijms232314930
Vainshelbaum NM, Giuliani A, Salmina K, Pjanova D, Erenpreisa J. The Transcriptome and Proteome Networks of Malignant Tumours Reveal Atavistic Attractors of Polyploidy-Related Asexual Reproduction. International Journal of Molecular Sciences. 2022; 23(23):14930. https://doi.org/10.3390/ijms232314930
Chicago/Turabian StyleVainshelbaum, Ninel M., Alessandro Giuliani, Kristine Salmina, Dace Pjanova, and Jekaterina Erenpreisa. 2022. "The Transcriptome and Proteome Networks of Malignant Tumours Reveal Atavistic Attractors of Polyploidy-Related Asexual Reproduction" International Journal of Molecular Sciences 23, no. 23: 14930. https://doi.org/10.3390/ijms232314930
APA StyleVainshelbaum, N. M., Giuliani, A., Salmina, K., Pjanova, D., & Erenpreisa, J. (2022). The Transcriptome and Proteome Networks of Malignant Tumours Reveal Atavistic Attractors of Polyploidy-Related Asexual Reproduction. International Journal of Molecular Sciences, 23(23), 14930. https://doi.org/10.3390/ijms232314930