
An In Vivo Definition of Brain Histamine Dynamics Reveals Critical Neuromodulatory Roles for This Elusive Messenger

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
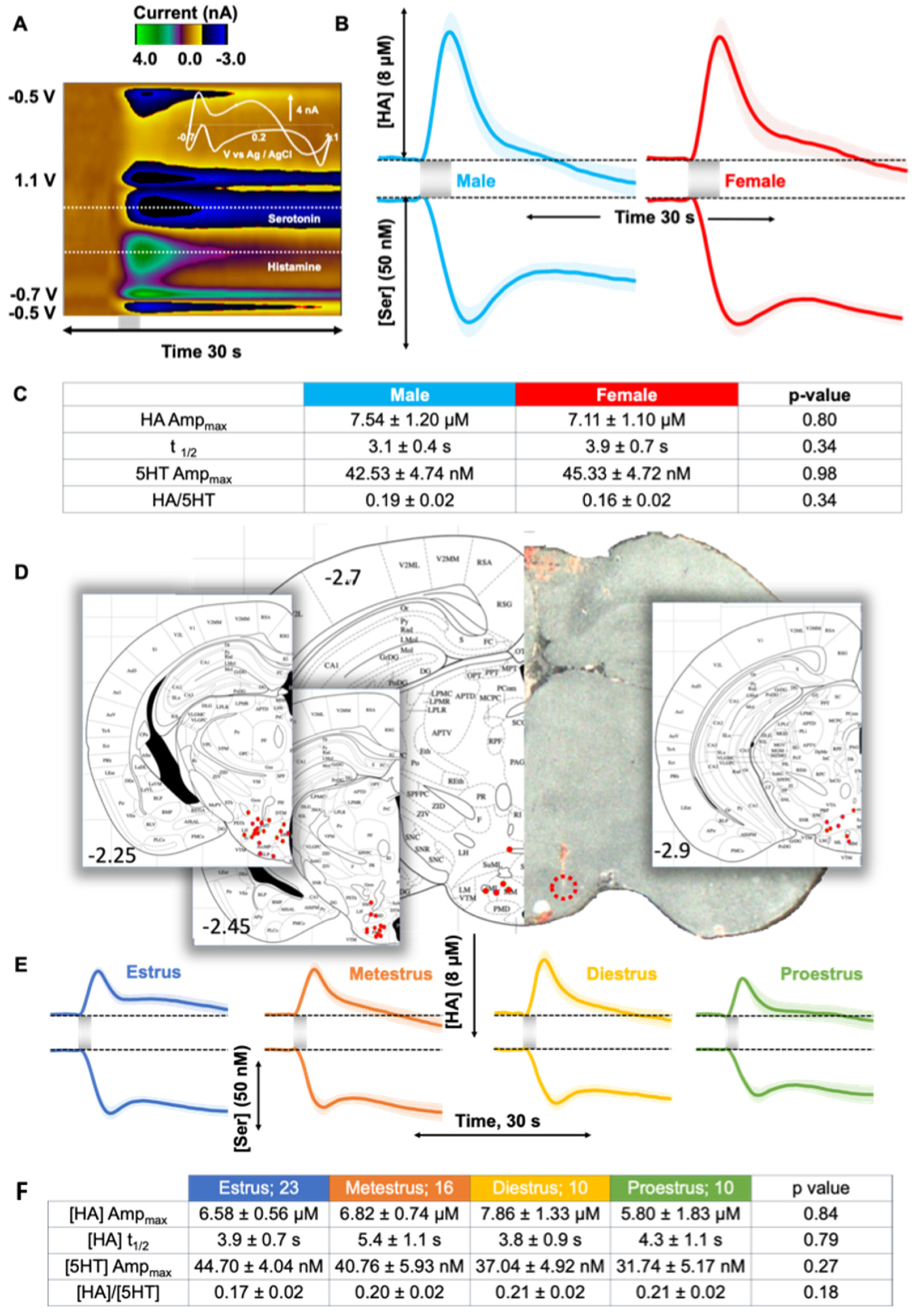
2.1. Evoked Histamine in Male and Female Mice
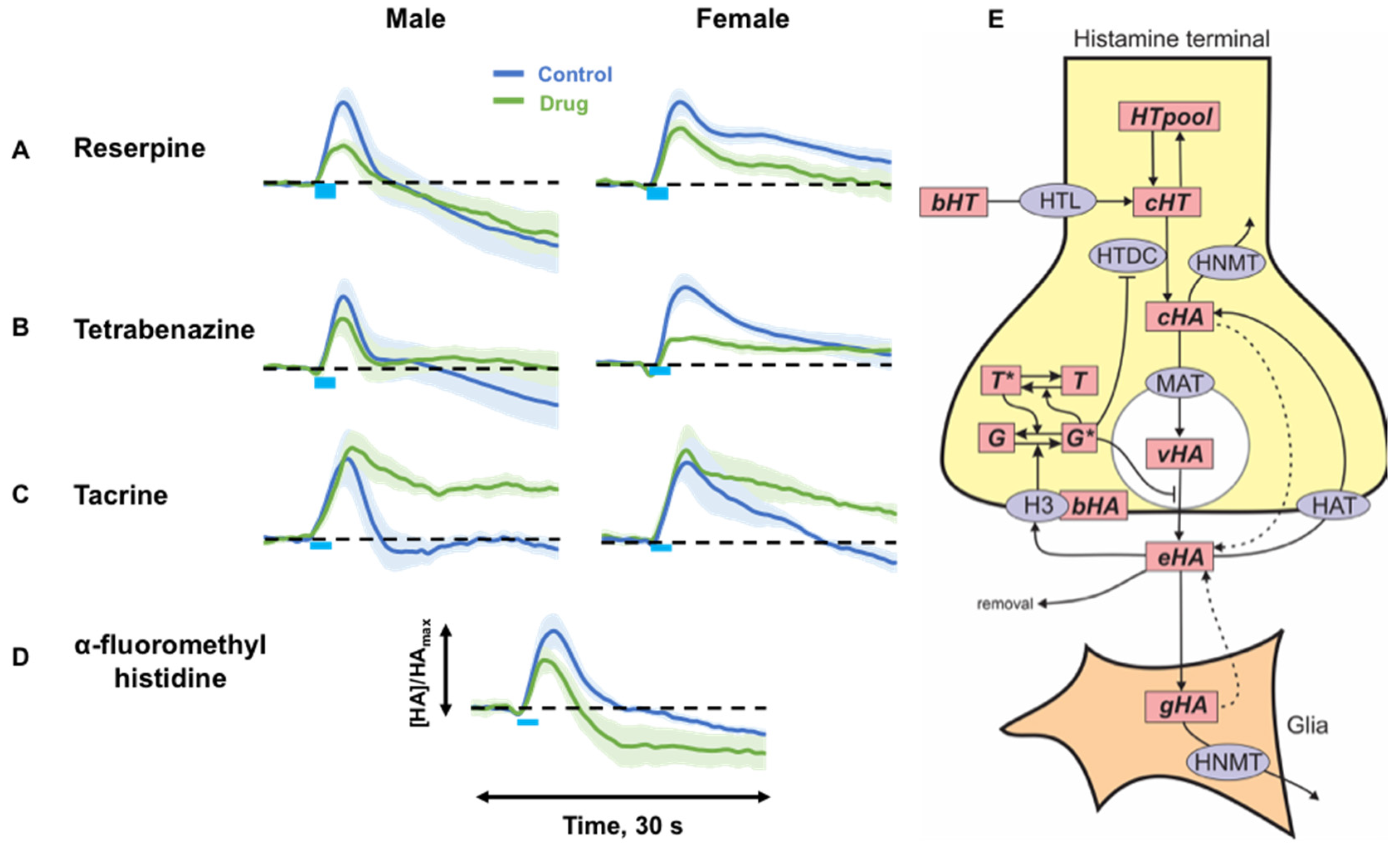
2.2. Targeting Histamine Packaging, Synthesis and Metabolism
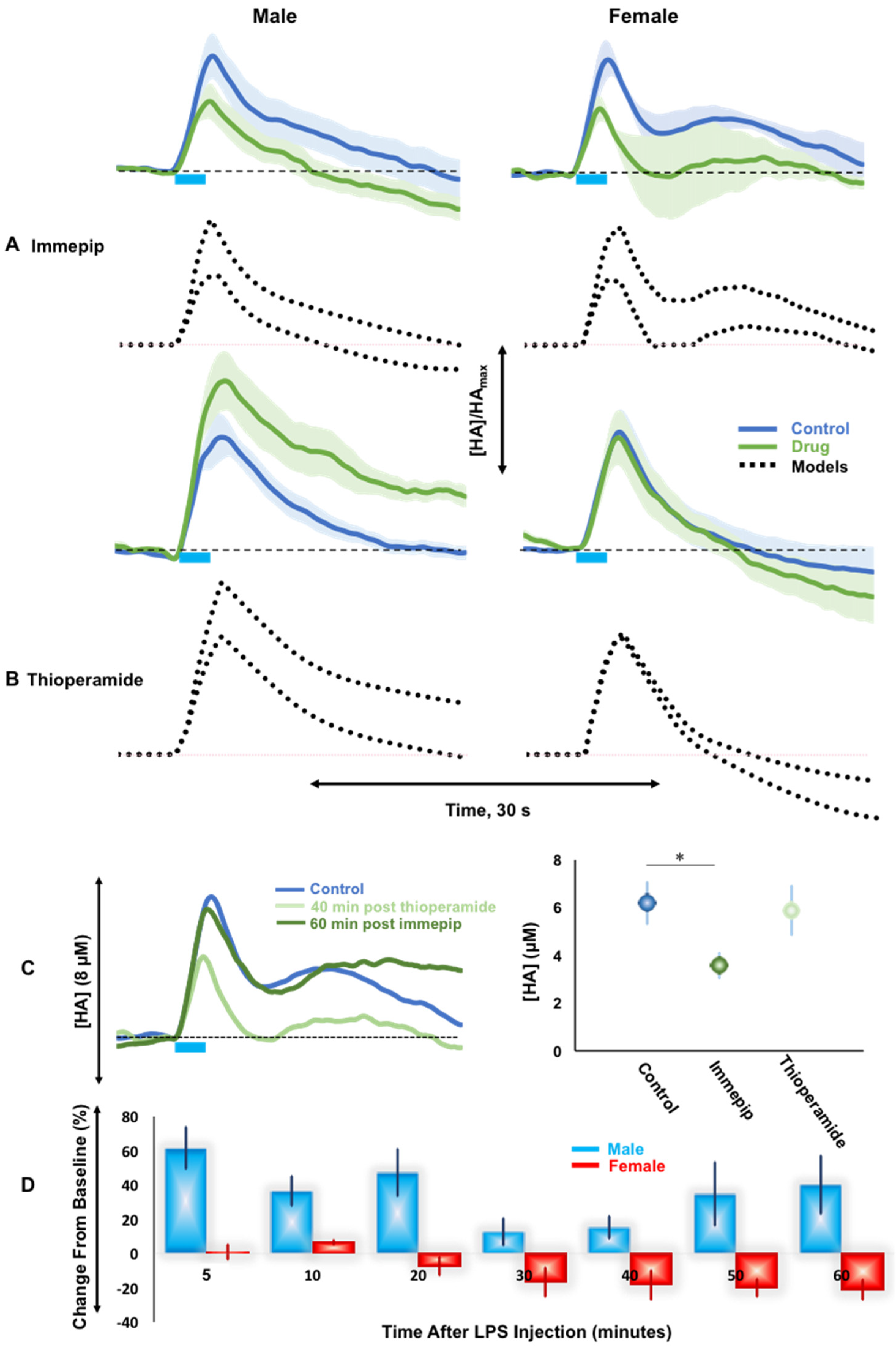
2.3. H3R Autoreceptor Control in Male and Female Mice
2.4. Histamine Post-Synaptic Receptor Pharmacology
3. Discussion
3.1. Control Evoked Histamine Does Not Vary between Sexes
3.2. Histamine Release Is Sensitive to Packaging, Synthesis and Metabolism
3.3. H3R Autoreceptors Differentiate Histaminergic Response in Male and Female Mice
3.4. H1R Antagonist Modulates Serotonin Levels
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Electrode Fabrication
4.3. Animals and Surgical Procedures
4.4. Data Collection and Analysis
4.5. Mathematical Modeling of the Experimental Data
4.6. Statistical Analyses
4.7. Post Experimental Histological Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ennis, M.; Truneh, A.; White, J.R.; Pearce, F.L. Inhibition of histamine secretion from mast cells. Nature 1981, 289, 186–187. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef] [PubMed]
- Jutel, M.; Watanabe, T.; Klunker, S.; Akdis, M.; Thomet, O.A.R.; Malolepszy, J.; Zak-Nejmark, T.; Koga, R.; Kobayashi, T.; Blaser, K.; et al. Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature 2001, 413, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.M.; Pires, J.; Esteves, M.; Baltazar, G.; Bernardino, L. Histamine: A new immunomodulatory player in the neuron-glia crosstalk. Front. Cell. Neurosci. 2014, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Niimi, M.; Yamamoto, Y.; Kawamura, T.; Morimoto-Ishizuka, T.; Sawada, M.; Takemori, H.; Yamatodani, A. Histamine production by cultured microglial cells of the mouse. Neurosci. Lett. 2001, 305, 181–184. [Google Scholar] [CrossRef]
- Panula, P.; Yang, H.Y.; Costa, E. Histamine-containing neurons in the rat hypothalamus. Proc. Natl. Acad. Sci. USA 1984, 81, 2572–2576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panula, P.; Pirvola, U.; Auvinen, S.; Airaksinen, M. Histamine-immunoreactive nerve fibers in the rat brain. Neuroscience 1989, 28, 585–610. [Google Scholar] [CrossRef]
- Watanabe, T.; Taguchi, Y.; Hayashi, H.; Tanaka, J.; Shiosaka, S.; Tohyama, M.; Kubota, H.; Terano, Y.; Wada, H. Evidence for the presence of a histaminergic neuron system in the rat brain: An immunohistochemical analysis. Neurosci. Lett. 1983, 39, 249–254. [Google Scholar] [CrossRef]
- Giannoni, P.; Passani, B.; Nosi, D.; Chazot, P.L.; Shenton, F.C.; Medhurst, A.D.; Munari, L.; Blandina, P. Heterogeneity of histaminergic neurons in the tuberomammillary nucleus of the rat. Eur. J. Neurosci. 2009, 29, 2363–2374. [Google Scholar] [CrossRef]
- Haas, H.L.; Sergeeva, O.A.; Selbach, O. Histamine in the Nervous System. Physiol. Rev. 2008, 88, 1183–1241. [Google Scholar] [CrossRef]
- Nuutinen, S.; Panula, P. Histamine in Neurotransmission and Brain Diseases. Adv. Exp. Med. Biol. 2010, 709, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Panula, P. Histamine receptors, agonists, and antagonists in health and disease. Handb. Clin. Neurol. 2021, 180, 377–387. [Google Scholar] [CrossRef]
- Samaranayake, S.; Abdalla, A.; Robke, R.; Nijhout, H.F.; Reed, M.C.; Best, J.; Hashemi, P. A voltammetric and mathematical analysis of histaminergic modulation of serotonin in the mouse hypothalamus. J. Neurochem. 2016, 138, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varaschin, R.K.; Osterstock, G.; Ducrot, C.; Leino, S.; Bourque, M.-J.; Prado, M.A.; Prado, V.F.; Salminen, O.; Nuutinen, S.R.; Trudeau, L.-E. Histamine H (3) Receptors Decrease Dopamine Release in the Ventral Striatum by Reducing the Activity of Striatal Cholinergic Interneurons. Neuroscience 2018, 376, 188–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baux, G.; Fossier, P.; Trudeau, L.-E.; Tauc, L. Presynaptic receptors for FMRFamide, histamine and buccalin regulate acetylcholine release at a neuro-neuronal synapse of Aplysia by modulating N-type Ca2+ channels. J. Physiol. Paris 1992, 86, 3–13. [Google Scholar] [CrossRef]
- Vanhanen, J.K.; Nuutinen, S.; Tuominen, M.; Panula, P.; Kononoff, J. Histamine H3 Receptor Regulates Sensorimotor Gating and Dopaminergic Signaling in the Striatum. J. Pharmacol. Exp. Ther. 2016, 357, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Threlfell, S.; Cragg, S.J.; Kalló, I.; Turi, G.F.; Coen, C.W.; Greenfield, S.A. Histamine H3 Receptors Inhibit Serotonin Release in Substantia Nigra Pars Reticulata. J. Neurosci. 2004, 24, 8704–8710. [Google Scholar] [CrossRef] [Green Version]
- Carthy, E.; Ellender, T. Histamine, Neuroinflammation and Neurodevelopment: A Review. Front. Neurosci. 2021, 15, 680214. [Google Scholar] [CrossRef]
- Passani, M.B.; Blandina, P. Histamine receptors in the CNS as targets for therapeutic intervention. Trends Pharmacol. Sci. 2011, 32, 242–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.M.; Snyder, S.H. Isotopic Microassay of Histamine, Histidine, Histidine Decarboxylase and Histamine Methyltransferase in Brain Tissue. J. Neurochem. 1972, 19, 1343–1358. [Google Scholar] [CrossRef]
- Ferretti, C. Hypothalamic Histamine Release in Normal and Stressed Rats Is Affected by Sex and Aging. Pharmacol. Biochem. Behav. 1998, 59, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Flik, G.; Folgering, J.H.A.; Cremers, T.I.H.F.; Westerink, B.H.C.; Dremencov, E. Interaction Between Brain Histamine and Serotonin, Norepinephrine, and Dopamine Systems: In Vivo Microdialysis and Electrophysiology Study. J. Mol. Neurosci. 2015, 56, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Oishi, R.; Nishibori, M.; Saeki, K. Characterization of Histamine Release from the Rat Hypothalamus as Measured by In Vivo Microdialysis. J. Neurochem. 1991, 56, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, S.; Abdalla, A.; Robke, R.; Wood, K.M.; Zeqja, A.; Hashemi, P. In vivo histamine voltammetry in the mouse premammillary nucleus. Analyst 2015, 140, 3759–3765. [Google Scholar] [CrossRef]
- Hersey, M.; Samaranayake, S.; Berger, S.N.; Tavakoli, N.; Mena, S.; Nijhout, H.F.; Reed, M.C.; Best, J.; Blakely, R.D.; Reagan, L.P.; et al. Inflammation-Induced Histamine Impairs the Capacity of Escitalopram to Increase Hippocampal Extracellular Serotonin. J. Neurosci. 2021, 41, 6564–6577. [Google Scholar] [CrossRef] [PubMed]
- Michael, D.; Travis, E.R.; Wightman, R.M. Color plots allow examination of all the cyclic voltammetry data simultaneously and provide an overview of chemical and temporal changes. Anal. Chem. 1998, 70, 586A–592A. [Google Scholar] [CrossRef]
- Saylor, R.A.; Hersey, M.; West, A.; Buchanan, A.M.; Berger, S.N.; Nijhout, H.F.; Reed, M.C.; Best, J.; Hashemi, P. In vivo hippocampal serotonin dynamics in male and female mice: Determining effects of acute escitalopram using fast scan cyclic voltammetry. Front. Neurosci. 2019, 13, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prell, G.D.; Khandelwal, J.K.; Burns, R.; LeWitt, P.A.; Green, J.P. Influence of age and gender on the levels of histamine metabolites and pros-methylimidazoleacetic acid in human cerebrospinal fluid. Arch. Gerontol. Geriatr. 1991, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Beery, A.K.; Zucker, I. Sex bias in neuroscience and biomedical research. Neurosci. Biobehav. Rev. 2011, 35, 565–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Cruz, S.; Mendoza-Rodríguez, Y.; Nava-Castro, K.E.; Yepez-Mulia, L.; Morales-Montor, J. Gender-Related Effects of Sex Steroids on Histamine Release and FcεRI Expression in Rat Peritoneal Mast Cells. J. Immunol. Res. 2015, 2015, 351829. [Google Scholar] [CrossRef] [PubMed]
- Schlicker, E.; Betz, R.; Göthert, M. Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1988, 337, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Puttonen, H.; Semenova, S.; Sundvik, M.; Panula, P. Storage of neural histamine and histaminergic neurotransmission is VMAT2 dependent in the zebrafish. Sci. Rep. 2017, 7, 3060. [Google Scholar] [CrossRef] [Green Version]
- Muroi, N.; Oishi, R.; Saeki, K. Effect of reserpine on histamine metabolism in the mouse brain. J. Pharmacol. Exp. Ther. 1991, 256, 967–972. Available online: http://jpet.aspetjournals.org/content/256/3/967.abstract (accessed on 1 September 2022). [PubMed]
- Garbarg, M.; Barbin, G.; Rodergas, E.; Schwartz, J.C. Inhibition of histamine synthesis in brain by α-fluoromethylhistidine, a new irreversible inhibitor: In vitro and in vivo studies. J. Neurochem. 1980, 35, 1045–1052. [Google Scholar] [CrossRef]
- Takehiko, W.; Atsushi, Y.; Kazutaka, M.; Hiroshi, W. Pharmacology of α fluoromethylhistidine, a specific inhibitor of histidine decarboxylase. Trends Pharmacol. Sci. 1990, 11, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Maeyama, K.; Watanabe, T.; Taguchi, Y.; Yamatodani, A.; Wada, H. Effect of α-fluoromethylhistidine, a suicide inhibitor of histidine decarboxylase, on histamine levels in mouse tissues. Biochem. Pharmacol. 1982, 31, 2367–2370. [Google Scholar] [CrossRef] [PubMed]
- Taraschenko, O.; Barnes, W.; Herrick-Davis, K.; Yakoyama, Y.; Boyd, D.; Hough, L. Actions of tacrine and galanthamine on histamine-N-methytransferase. Methods Find. Exp. Clin. Pharmacol. 2005, 27, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Cumming, P.; Vincent, S.R. Inhibition of histamine-N-methyltransferase (HNMT) by fragments of 9-amino-1,2,3,4-tetrahydroacridine (tacrine) and by β-carbolines. Biochem. Pharmacol. 1992, 44, 989–999. [Google Scholar] [CrossRef]
- Farlow, M.; Gracon, S.I.; Hershey, L.A.; Lewis, K.W.; Sadowsky, C.H.; Dolan-Ureno, J. A controlled trial of tacrine in Alzheimer’s disease. The Tacrine Study Group. JAMA 1992, 268, 2523–2529. [Google Scholar] [CrossRef]
- Eagger, S.; Levy, R.; Sahakian, B. Tacrine in Alzheimer’s disease. Lancet 1991, 337, 989–992. [Google Scholar] [CrossRef]
- Prast, H.; Tran, M.; Fischer, H.; Kraus, M.; Lamberti, C.; Grass, K.; Philippu, A. Histaminergic neurons modulate acetylcholine release in the ventral striatum: Role of H3 histamine receptors. Naunyn. Schmiedebergs. Arch. Pharmacol. 1999, 360, 558–564. [Google Scholar] [CrossRef]
- Fantozzi, R.; Moroni, F.; Masini, E.; Blandina, P.; Mannaioni, P.F. Modulation of the spontaneous histamine release by adrenergic and cholinergic drugs. Agents Actions 1978, 8, 347–358. [Google Scholar] [CrossRef]
- Parst, H.; Fischer, H.P.; Parst, M.; Philippu, A. In vivo modulation of histamine release by autoreceptors and muscarinic acetylcholine receptors in the rat anterior hypothalamus. Naunyn. Schmiedebergs. Arch. Pharmacol. 1994, 350, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Ghi, P.; Orsetti, M.; Gamalero, S.R.; Ferretti, C. Sex differences in memory performance in the object recognition test. Possible role of histamine receptors. Pharmacol. Biochem. Behav 1999, 64, 761–766. [Google Scholar] [CrossRef]
- Roof, R.L.; Hall, E.D. Gender Differences in Acute CNS Trauma and Stroke: Neuroprotective Effects of Estrogen and Progesterone. J. Neurotrauma 2000, 17, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; Von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R.; et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Miyazawa, K.W.; Pinho-Ribeiro, F.A.; Zarpelon, A.C.; Staurengo-Ferrari, L.; Silva, R.L.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q.; Casagrande, R.; Verri, W.A. Vinpocetine reduces lipopolysaccharide-induced inflammatory pain and neutrophil recruitment in mice by targeting oxidative stress, cytokines and NF-κB. Chem. Biol. Interact. 2015, 237, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Calil, I.L.; Zarpelon, A.C.; Guerrero, A.T.G.; Alves-Filho, J.C.; Ferreira, S.H.; Cunha, F.Q.; Cunha, T.M.; Verri, W.A. Lipopolysaccharide induces inflammatory hyperalgesia triggering a TLR4/MyD88-dependent cytokine cascade in the mice paw. PLoS ONE 2014, 9, e90013. [Google Scholar] [CrossRef]
- Watkins, L.; Wiertelak, E.; Goehler, L.; Mooney-Heiberger, K.; Martinez, J.; Furness, L.; Smith, K.; Maier, S. Neurocircuitry of illness-induced hyperalgesia. Brain Res. 1994, 639, 283–299. [Google Scholar] [CrossRef]
- Meseguer, V.; Alpizar, Y.A.; Luis, E.; Tajada, S.; Denlinger, B.; Fajardo, O.; Manenschijn, J.-A.; Fernández-Pena, C.; Talavera, A.; Kichko, T.; et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat. Commun. 2014, 5, 3125. [Google Scholar] [CrossRef] [PubMed]
- Mobarakeh, J.I.; Sakurada, S.; Katsuyama, S.; Kutsuwa, M.; Kuramasu, A.; Lin, Z.Y.; Watanabe, T.; Hashimoto, Y.; Yanai, K. Role of histamine H1 receptor in pain perception: A study of the receptor gene knockout mice. Eur. J. Pharmacol. 2000, 391, 81–89. [Google Scholar] [CrossRef]
- Obara, I.; Telezhkin, V.; AlRashdi, I.; Chazot, P. Histamine, histamine receptors, and neuropathic pain relief. Br. J. Pharmacol. 2020, 177, 580–599. [Google Scholar] [CrossRef]
- Mogil, J.S.; Wilson, S.G.; Chesler, E.J.; Rankin, A.L.; Nemmani, K.V.S.; Lariviere, W.R.; Groce, M.K.; Wallace, M.R.; Kaplan, L.; Staud, R.; et al. The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc. Natl. Acad. Sci. USA 2003, 100, 4867–4872. [Google Scholar] [CrossRef] [Green Version]
- Wiesenfeld-Hallin, Z. Sex differences in pain perception. Gend. Med. 2005, 2, 137–145. [Google Scholar] [CrossRef]
- Sorge, R.E.; Mapplebeck, J.C.S.; Rosen, S.; Beggs, S.; Taves, S.; Alexander, J.K.; Martin, L.J.; Austin, J.-S.; Sotocinal, S.G.; Chen, D.; et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 2015, 18, 1081–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, P.M.; Dubal, D.B.; Wilson, M.E.; Rau, S.W.; Böttner, M. Minireview: Neuroprotective Effects of Estrogen—New Insights into Mechanisms of Action. Endocrinology 2001, 142, 969–973. [Google Scholar] [CrossRef]
- Gotoh, K.; Masaki, T.; Chiba, S.; Higuchi, K.; Kakuma, T.; Shimizu, H.; Mori, M.; Sakata, T.; Yoshimatsu, H. Hypothalamic neuronal histamine signaling in the estrogen deficiency-induced obesity. J. Neurochem. 2009, 110, 1796–1805. [Google Scholar] [CrossRef]
- Mori, H.; Matsuda, K.-I.; Yamawaki, M.; Kawata, M. Estrogenic regulation of histamine receptor subtype H1 expression in the ventromedial nucleus of the hypothalamus in female rats. PLoS ONE 2014, 9, e96232. [Google Scholar] [CrossRef]
- Baker, A.E.; Brautigam, V.M.; Watters, J.J. Estrogen Modulates Microglial Inflammatory Mediator Production via Interactions with Estrogen Receptor β. Endocrinology 2004, 145, 5021–5032. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.; Khalili, H.; Pergolizzi, R.G.; Michael, S.D.; Chang, M.-D.Y. Estradiol Down-Regulates LPS-Induced Cytokine Production and NFkB Activation in Murine Macrophages. Am. J. Reprod. Immunol. 1997, 38, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Vegeto, E.; Bonincontro, C.; Pollio, G.; Sala, A.; Viappiani, S.; Nardi, F.; Brusadelli, A.; Viviani, B.; Ciana, P.; Maggi, A. Estrogen Prevents the Lipopolysaccharide-Induced Inflammatory Response in Microglia. J. Neurosci. 2001, 21, 1809–1818. [Google Scholar] [CrossRef] [Green Version]
- Lei, B.; Mace, B.; Dawson, H.N.; Warner, D.S.; Laskowitz, D.T.; James, M.L. Anti-Inflammatory Effects of Progesterone in Lipopolysaccharide-Stimulated BV-2 Microglia. PLoS ONE 2014, 9, e103969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paton, D.M.; Webster, D.R.; Paton, D.M. Clinical Pharmacokinetics of H1-Receptor Antagonists (The Antihistamines). Clin. Pharmacokinet. 1985, 10, 477–497. [Google Scholar] [CrossRef] [PubMed]
- Bell, N.J.; Hunt, R.H. Role of gastric acid suppression in the treatment of gastro-oesophageal reflux disease. Gut 1992, 33, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Oishi, R.; Adachi, N.; Saeki, K. Nα-Methylhistamine inhibits intestinal transit in mice by central histamine H1 receptor activation. Eur. J. Pharmacol. 1993, 237, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Benoit-Marand, M.; Borrelli, E.; Gonon, F. Inhibition of Dopamine Release Via Presynaptic D2 Receptors: Time Course and Functional Characteristics In Vivo. J. Neurosci. 2001, 21, 9134–9141. [Google Scholar] [CrossRef] [Green Version]
- Garris, P.; Budygin, E.; Phillips, P.; Venton, B.; Robinson, D.; Bergstrom, B.; Rebec, G.; Wightman, R. A role for presynaptic mechanisms in the actions of nomifensine and haloperidol. Neuroscience 2003, 118, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Bamford, N.S.; Wightman, R.M.; Sulzer, D. Dopamine’s Effects on Corticostriatal Synapses during Reward-Based Behaviors. Neuron 2018, 97, 494–510. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.E.; Stevens, D.R.; Haas, H.L. The physiology of brain histamine. Prog. Neurobiol. 2001, 63, 637–672. [Google Scholar] [CrossRef] [PubMed]
- Best, J.; Nijhout, H.F.; Samaranayake, S.; Hashemi, P.; Reed, M. A mathematical model for histamine synthesis, release, and control in varicosities. Theor. Biol. Med. Model. 2017, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Suvarna, Y.; Maity, N.; Shivamurthy, M.C. Emerging Trends in Retrograde Signaling. Mol. Neurobiol. 2015, 53, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Prast, H.; Lamberti, C.; Fischer, H.; Tran, M.H.; Philippu, A. Nitric oxide influences the release of histamine and glutamate in the rat hypothalamus. Naunyn. Schmiedebergs. Arch. Pharmacol. 1996, 354, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Kreitzer, A. Retrograde signaling by endocannabinoids. Curr. Opin. Neurobiol. 2002, 12, 324–330. [Google Scholar] [CrossRef]
- Hughes, A.R.; Lin, A.; Hendrickson, R.G.; on behalf of Toxicology Investigator’s Consortium (ToxIC). Clinical and patient characteristics associated with severe outcome in diphenhydramine toxicity. Clin. Toxicol. 2021, 59, 918–925. [Google Scholar] [CrossRef]
- FDA. FDA Warns about Serious Problems with High Doses of the Allergy Medicine Diphenhydramine (Benadryl). 2020. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-warns-about-serious-problems-high-doses-allergy-medicine-diphenhydramine-benadryl (accessed on 1 September 2022).
- Liu, H.; Farley, J.M. Effects of first and second generation antihistamines on muscarinic induced mucus gland cell ion transport. BMC Pharmacol. 2005, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemi, P.; Dankoski, E.C.; Petrovic, J.; Keithley, R.B.; Wightman, R.M. Voltammetric Detection of 5-Hydroxytryptamine Release in the Rat Brain. Anal. Chem. 2009, 81, 9462–9471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paxinos, G.; Franklin, K.B.J. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates; Academic press: Cambridge, MA, USA, 2019. [Google Scholar]
- Caligioni, C.S. Assessing Reproductive Status/Stages in Mice. Curr. Protoc. Neurosci. 2009, 48, A.4I.1–A.4I.8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalla, A.; Atcherley, C.W.; Pathirathna, P.; Samaranayake, S.; Qiang, B.; Peña, E.; Morgan, S.L.; Heien, M.L.; Hashemi, P. In Vivo Ambient Serotonin Measurements at Carbon-Fiber Microelectrodes. Anal. Chem. 2017, 89, 9703–9711. [Google Scholar] [CrossRef] [Green Version]
- Mena, S.; Dietsch, S.; Berger, S.N.; Witt, C.E.; Hashemi, P. Novel, User-Friendly Experimental and Analysis Strategies for Fast Voltammetry: 1. The Analysis Kid for FSCV. ACS Meas. Sci. Au 2021, 1, 11–19. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berger, S.N.; Baumberger, B.; Samaranayake, S.; Hersey, M.; Mena, S.; Bain, I.; Duncan, W.; Reed, M.C.; Nijhout, H.F.; Best, J.; et al. An In Vivo Definition of Brain Histamine Dynamics Reveals Critical Neuromodulatory Roles for This Elusive Messenger. Int. J. Mol. Sci. 2022, 23, 14862. https://doi.org/10.3390/ijms232314862
Berger SN, Baumberger B, Samaranayake S, Hersey M, Mena S, Bain I, Duncan W, Reed MC, Nijhout HF, Best J, et al. An In Vivo Definition of Brain Histamine Dynamics Reveals Critical Neuromodulatory Roles for This Elusive Messenger. International Journal of Molecular Sciences. 2022; 23(23):14862. https://doi.org/10.3390/ijms232314862
Chicago/Turabian StyleBerger, Shane N., Beatrice Baumberger, Srimal Samaranayake, Melinda Hersey, Sergio Mena, Ian Bain, William Duncan, Michael C. Reed, H. Frederik Nijhout, Janet Best, and et al. 2022. "An In Vivo Definition of Brain Histamine Dynamics Reveals Critical Neuromodulatory Roles for This Elusive Messenger" International Journal of Molecular Sciences 23, no. 23: 14862. https://doi.org/10.3390/ijms232314862
APA StyleBerger, S. N., Baumberger, B., Samaranayake, S., Hersey, M., Mena, S., Bain, I., Duncan, W., Reed, M. C., Nijhout, H. F., Best, J., & Hashemi, P. (2022). An In Vivo Definition of Brain Histamine Dynamics Reveals Critical Neuromodulatory Roles for This Elusive Messenger. International Journal of Molecular Sciences, 23(23), 14862. https://doi.org/10.3390/ijms232314862