

Identification of a Cardiac Glycoside Exhibiting Favorable Brain Bioavailability and Potency for Reducing Levels of the Cellular Prion Protein


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
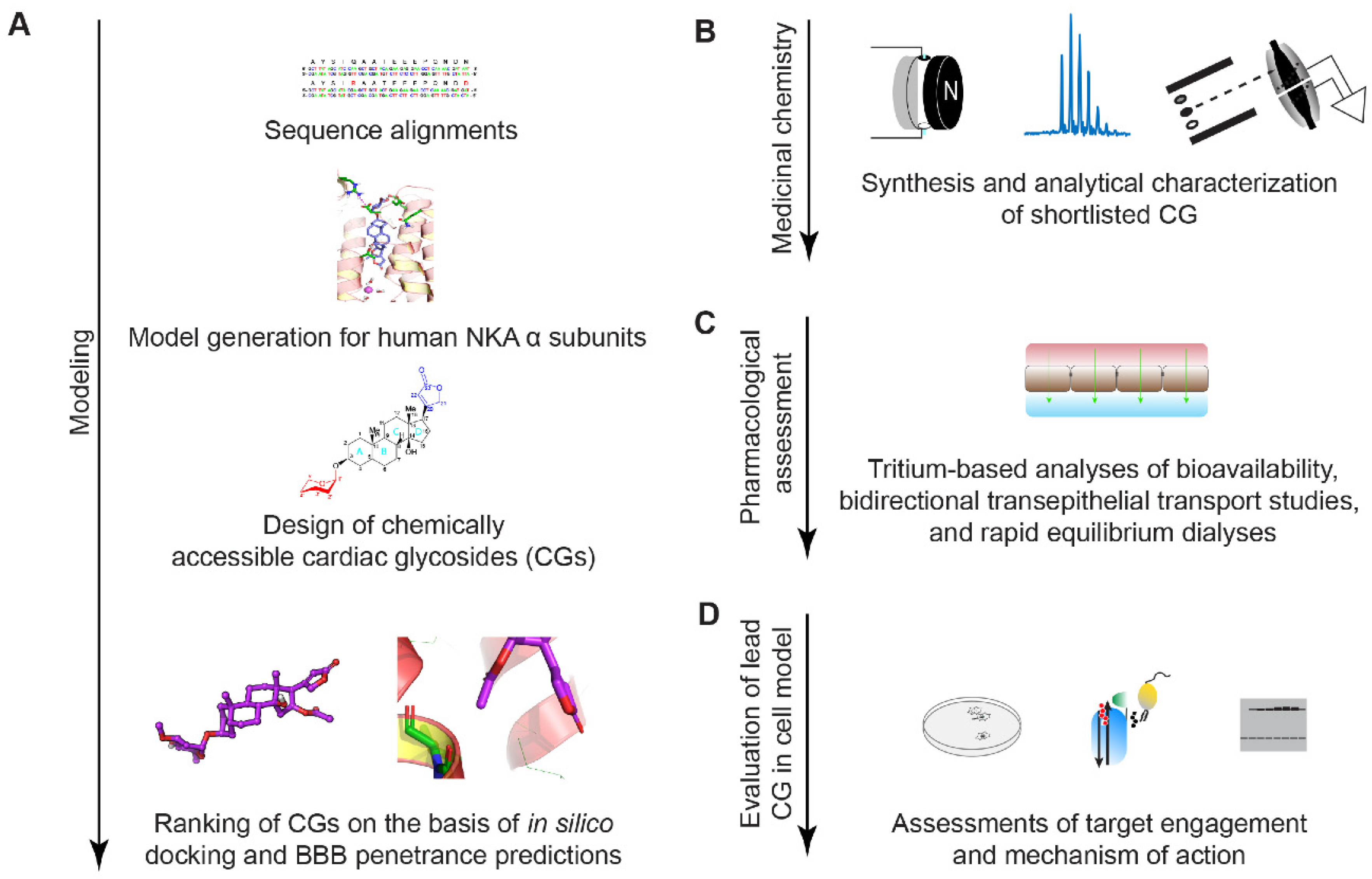
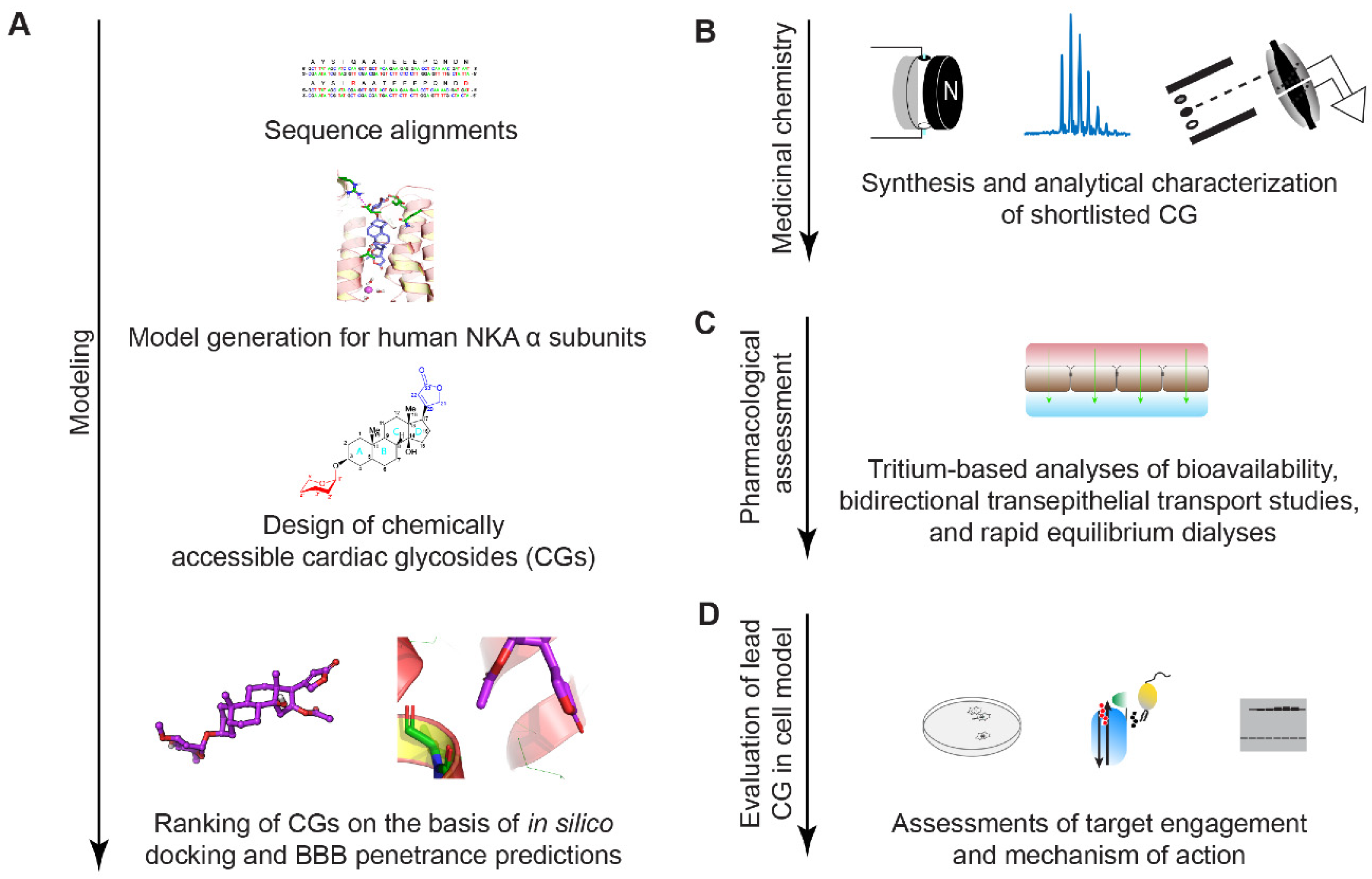
2.1. Study Design
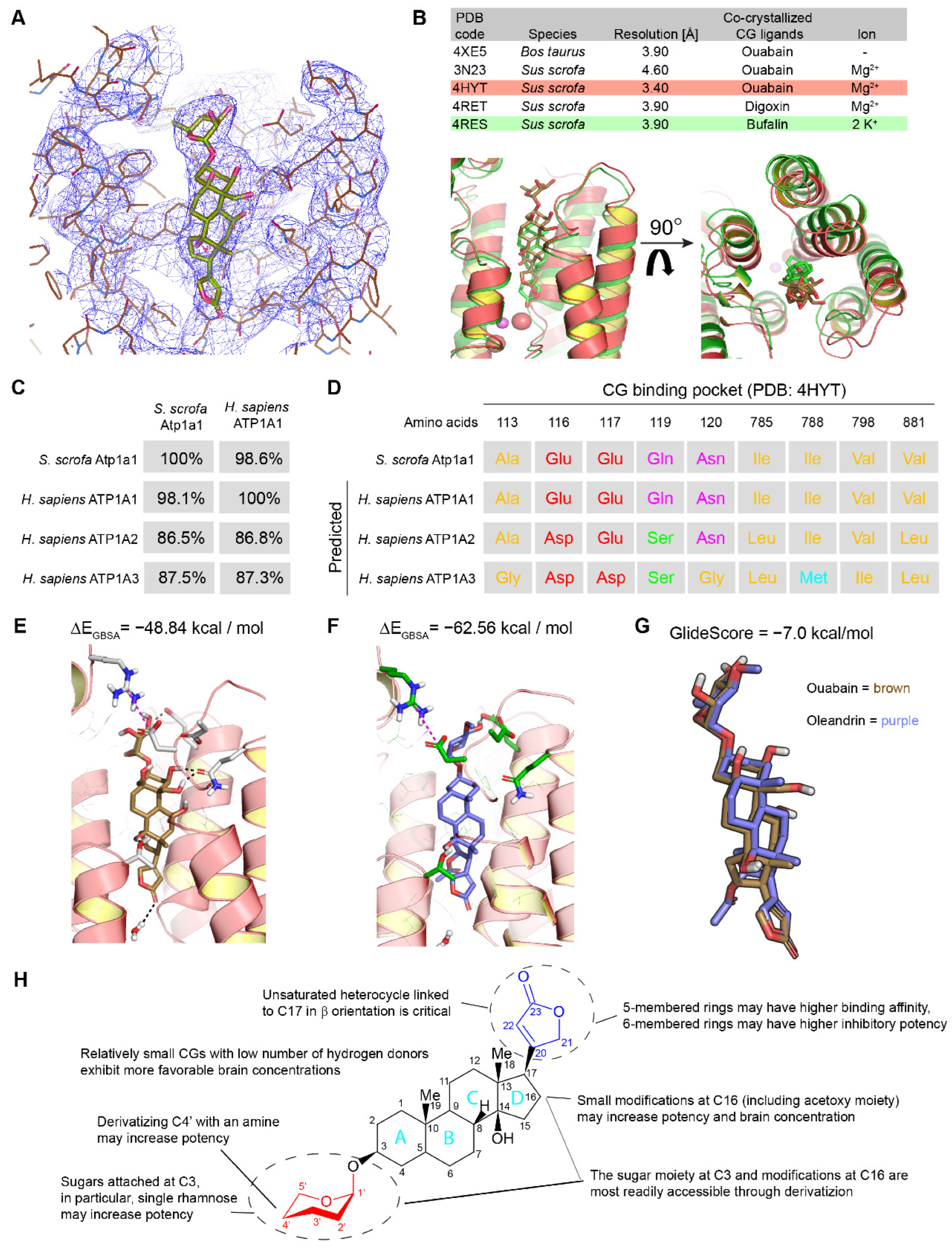
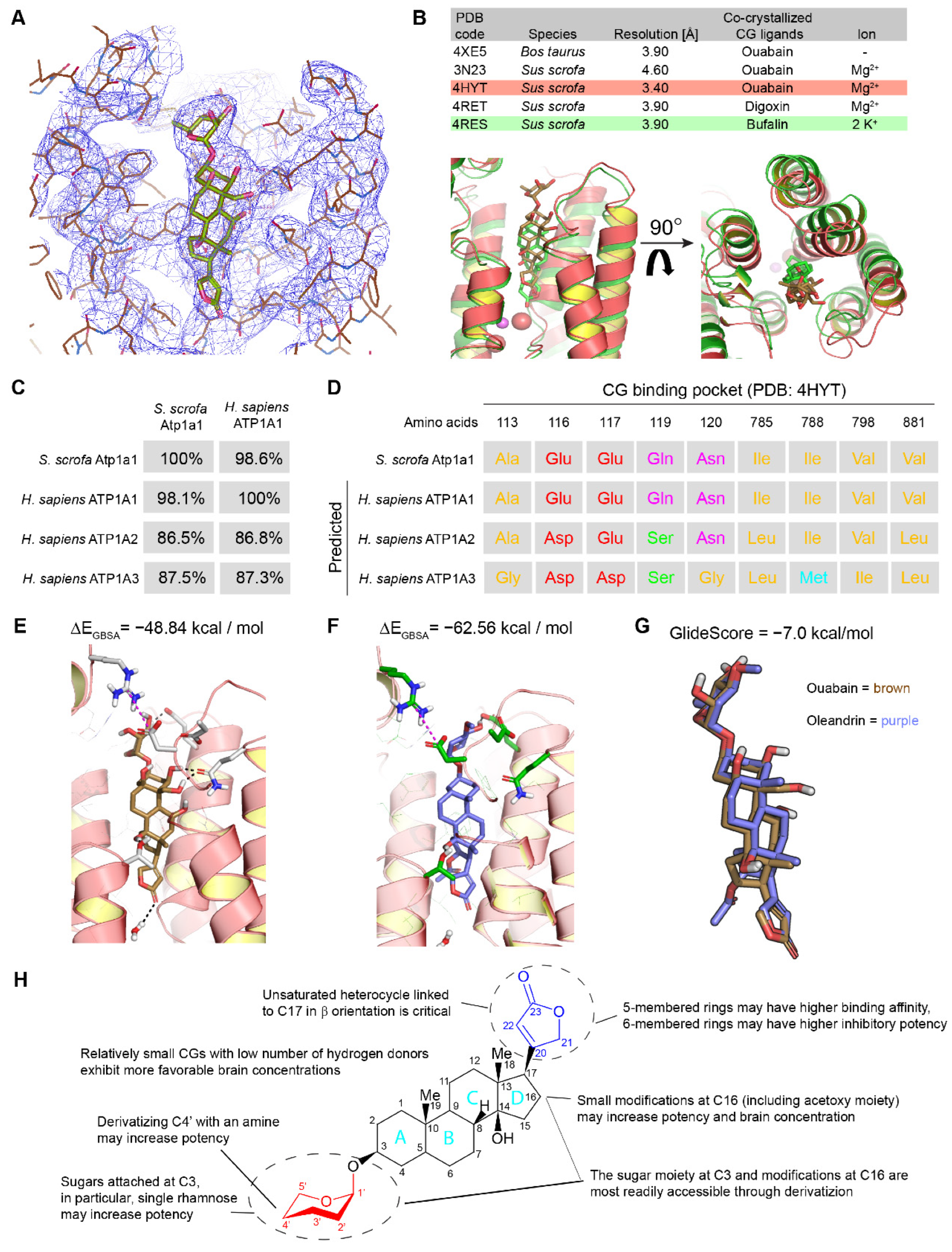
2.2. In Silico Modeling of Oleandrin Binding to Human NKA α Subunit
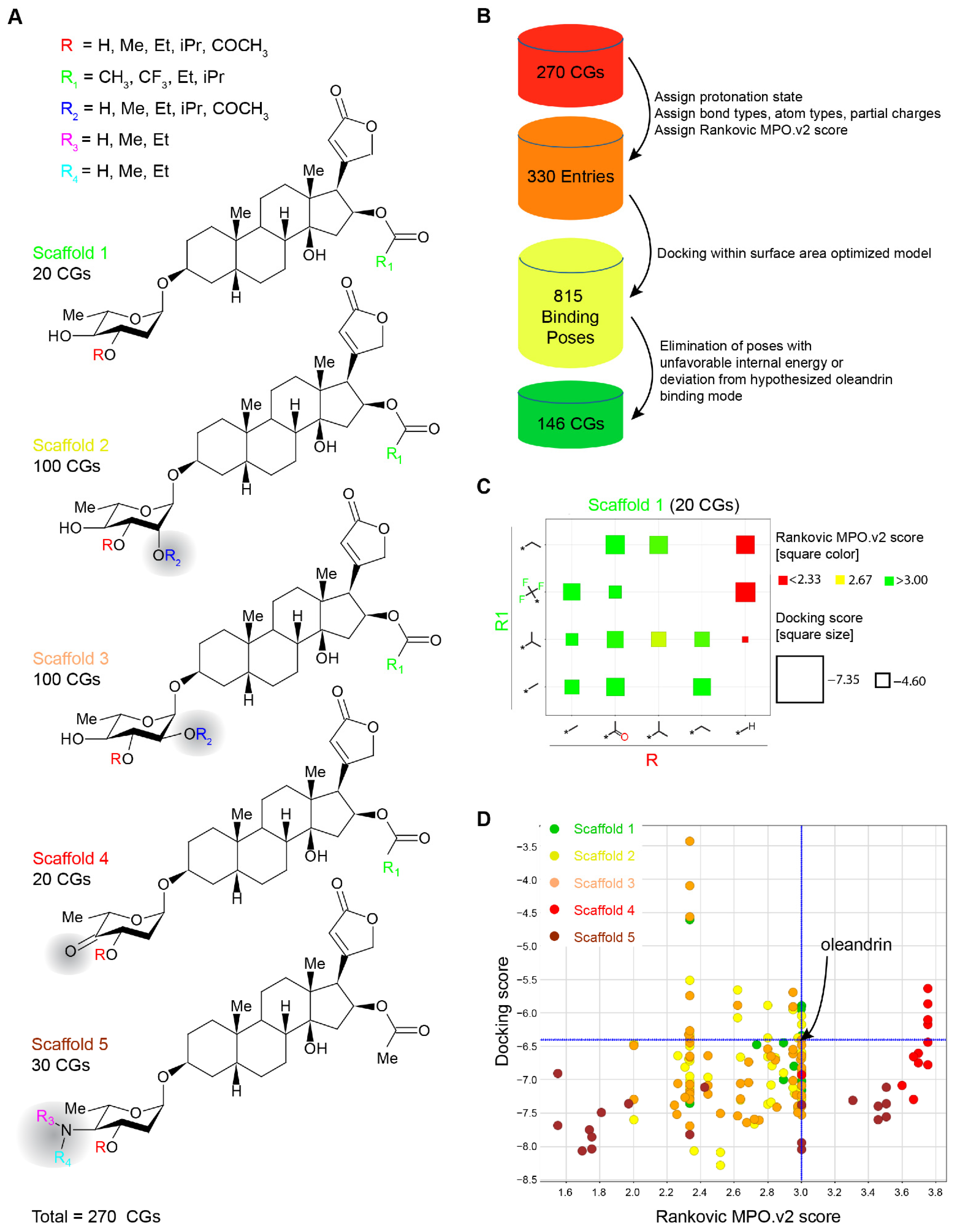
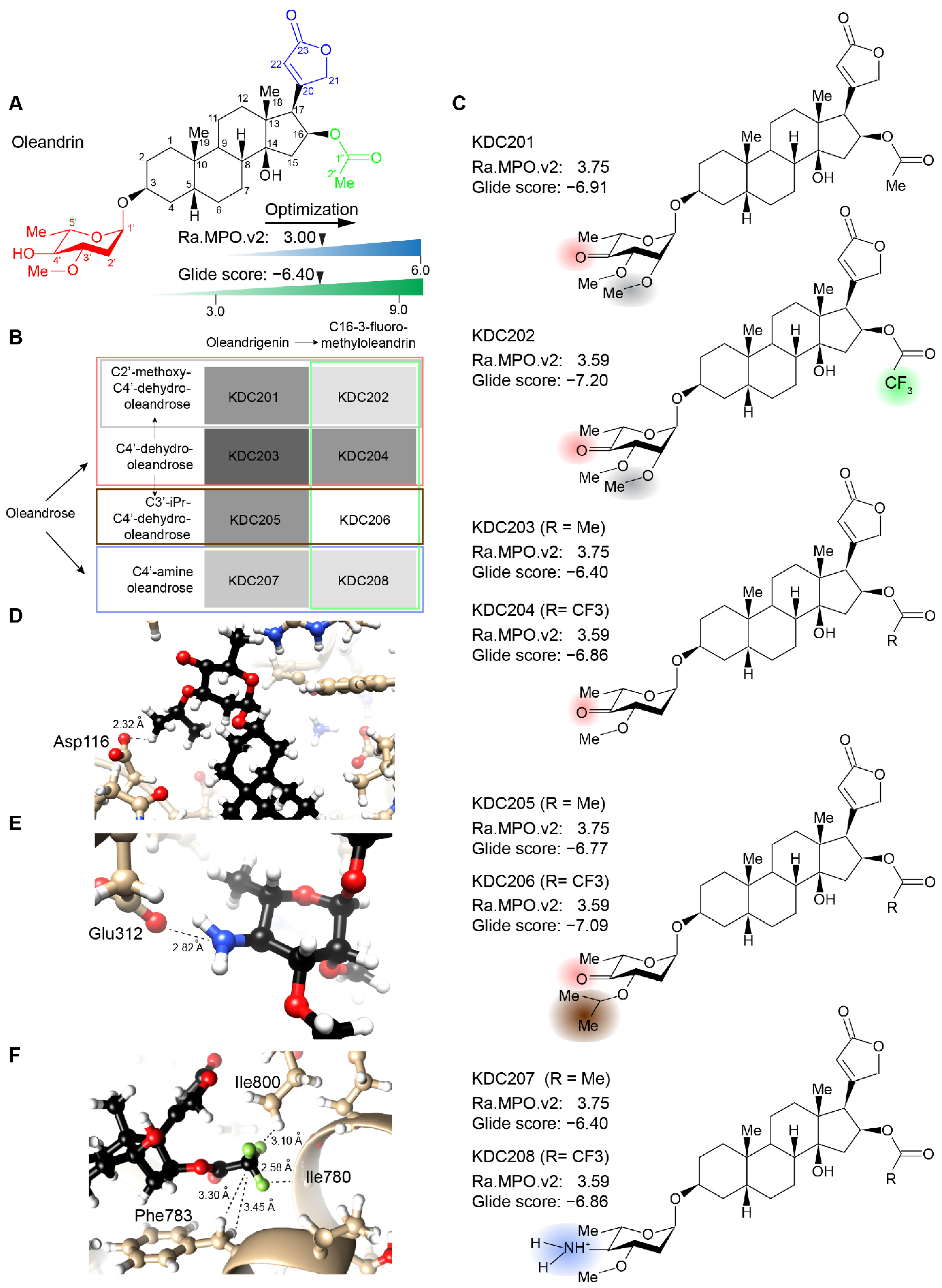
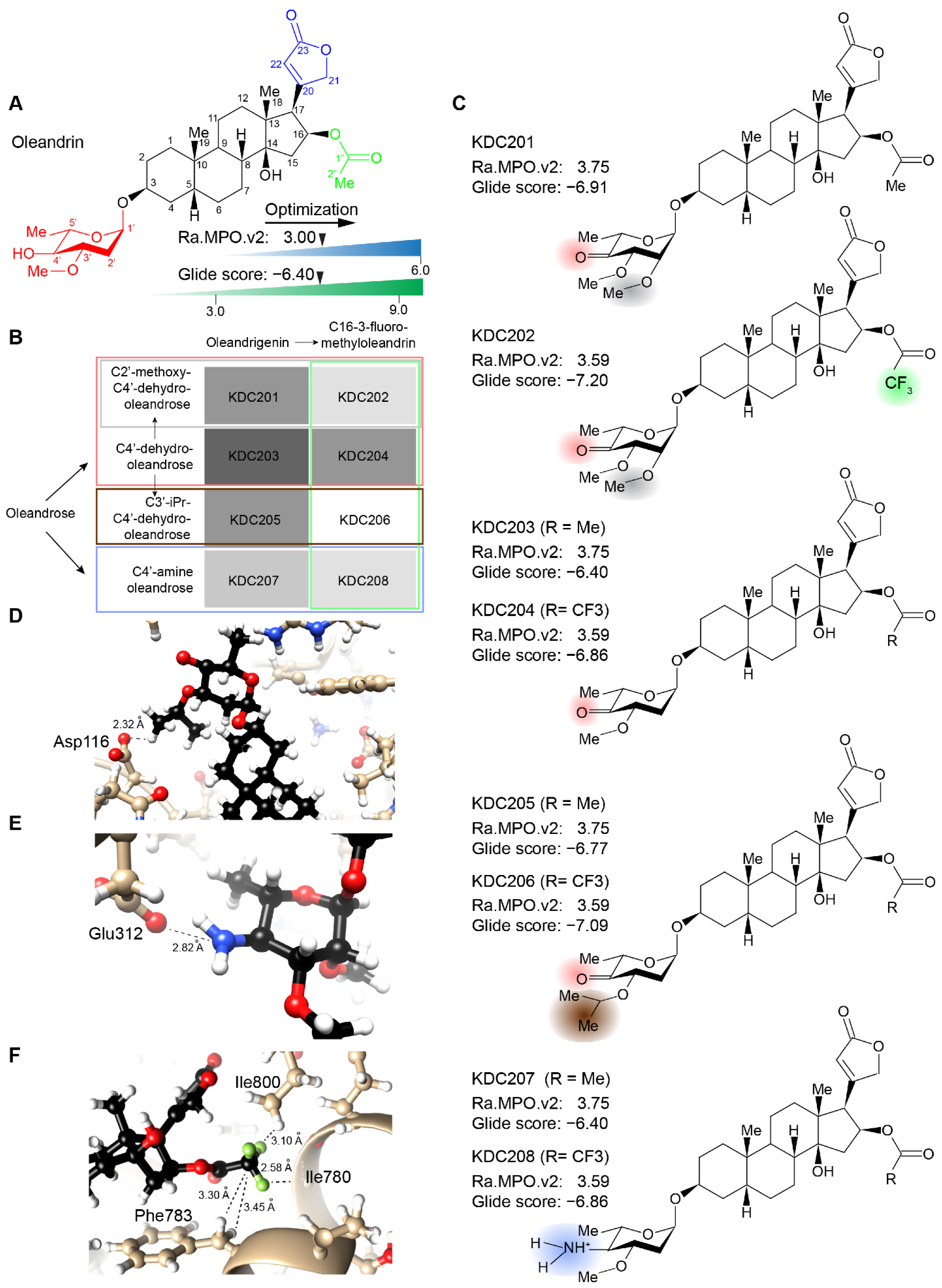
2.3. Evaluation of Chemically Accessible Oleandrin Derivatives Based on Binding Score and Brain Bioavailability
2.4. Synthesis of Lead Compound with Favorable Characteristics
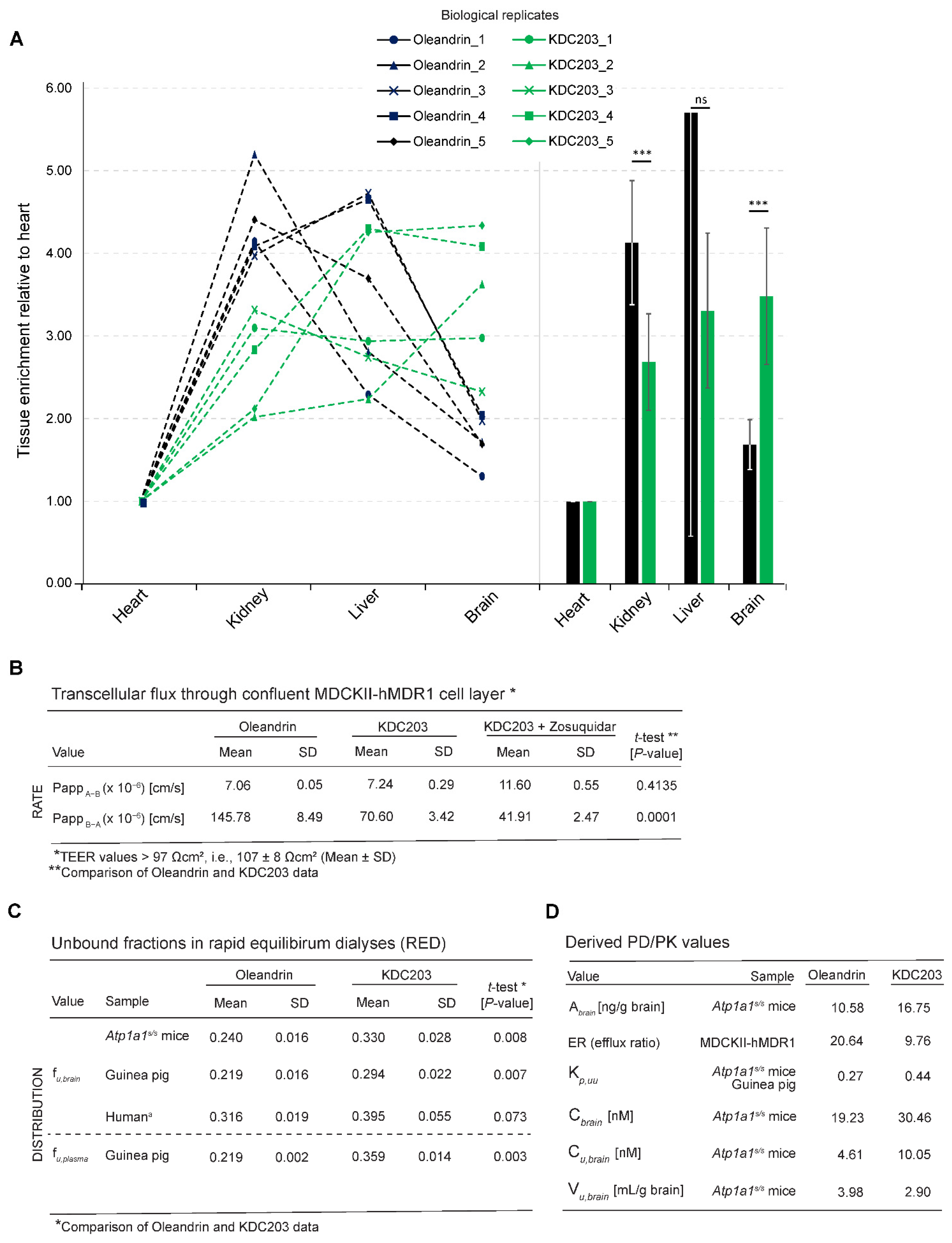
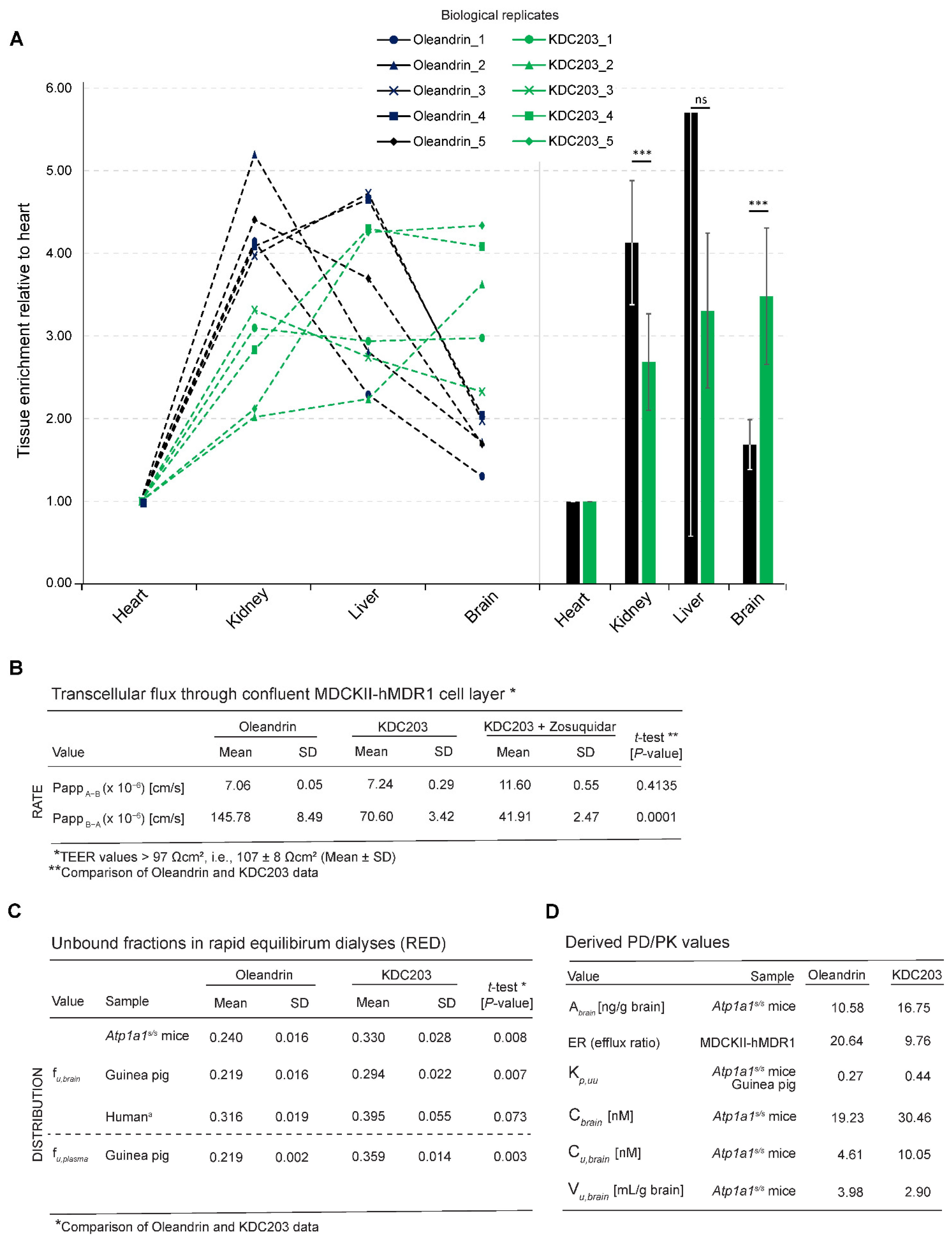
2.5. Analysis of Brain Bioavailability
2.6. Transepithelial Diffusion and Transport
2.7. Free Versus Bound Fraction
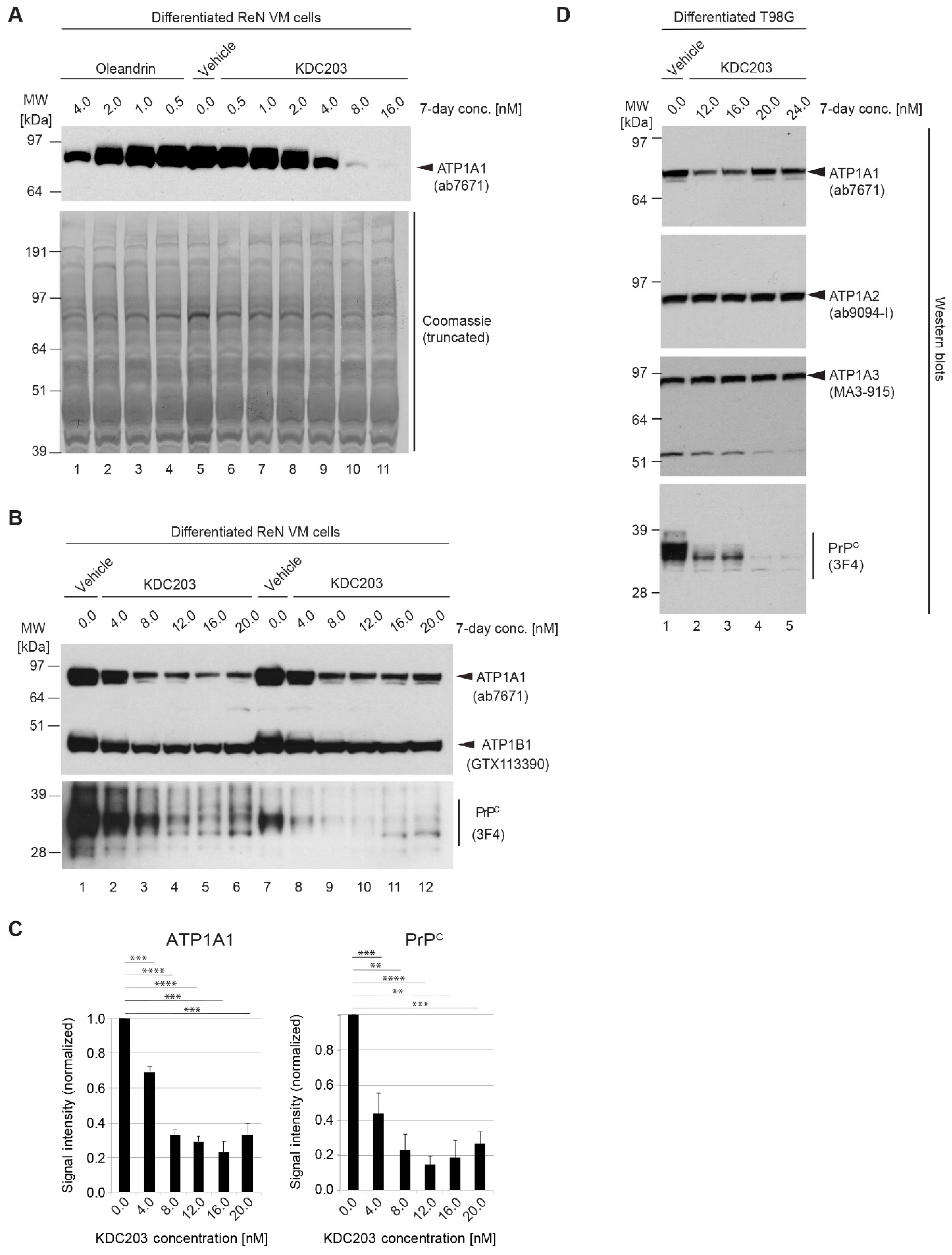
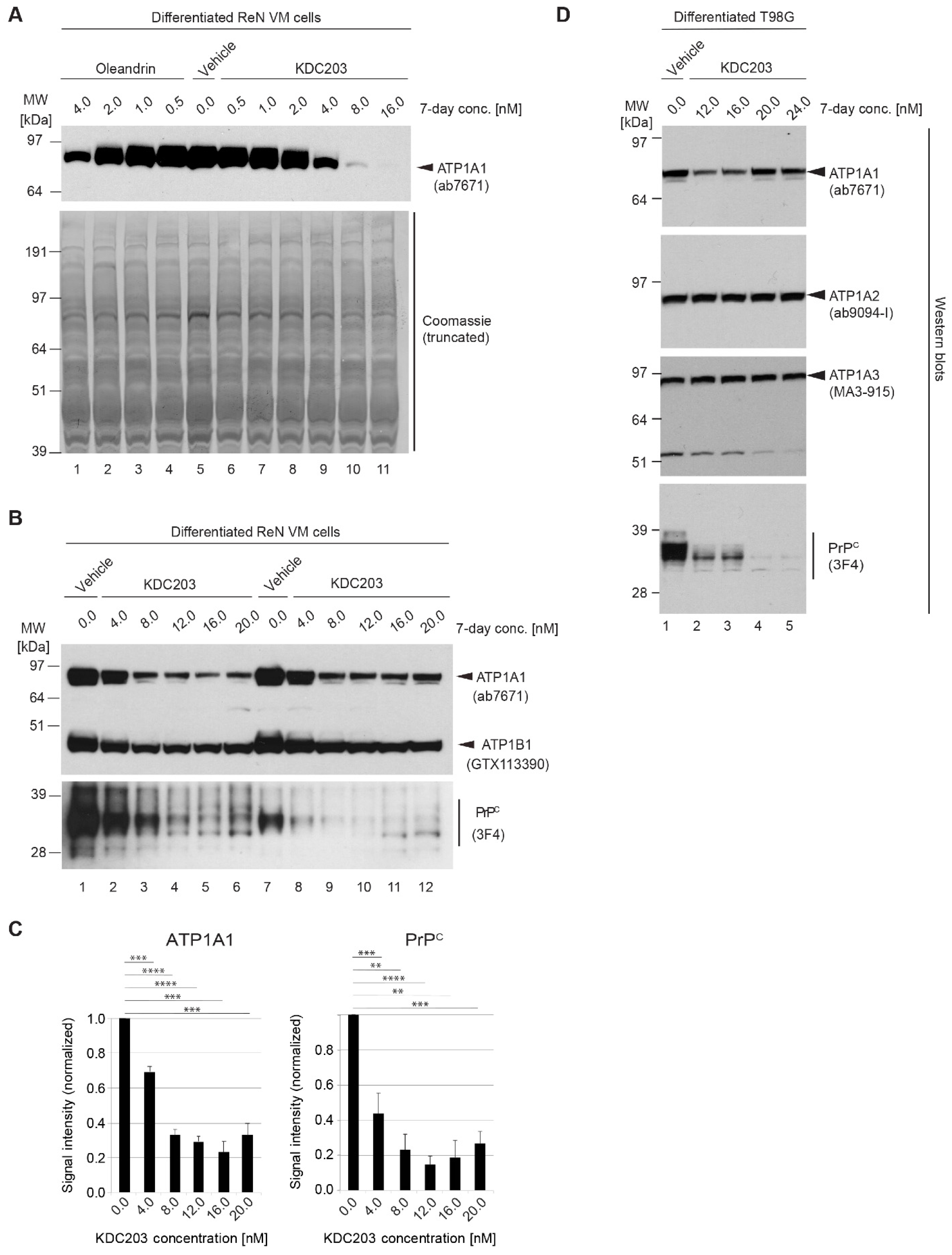
2.8. In Vitro Assessment of Potency in Differentiated ReN VM Cells
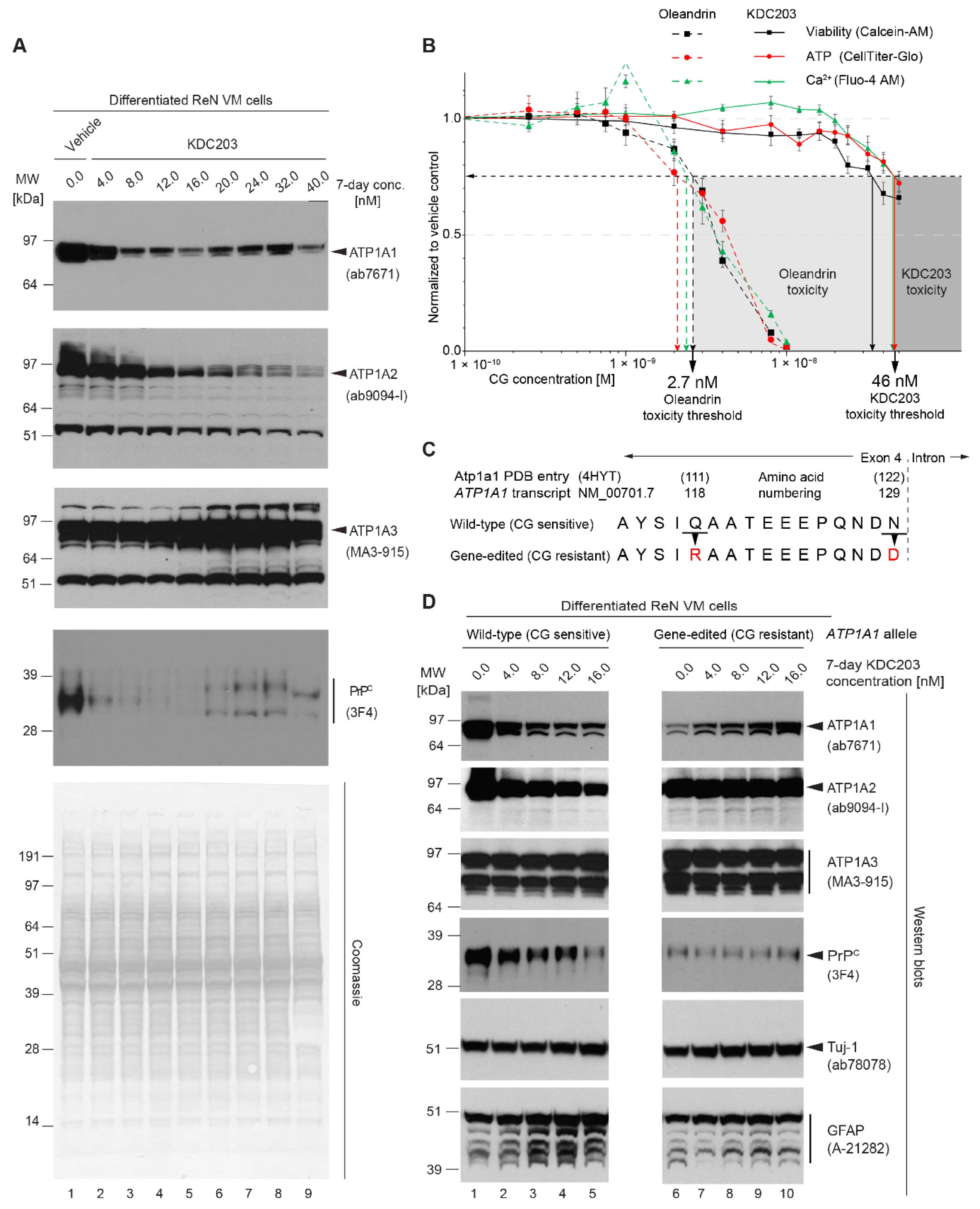
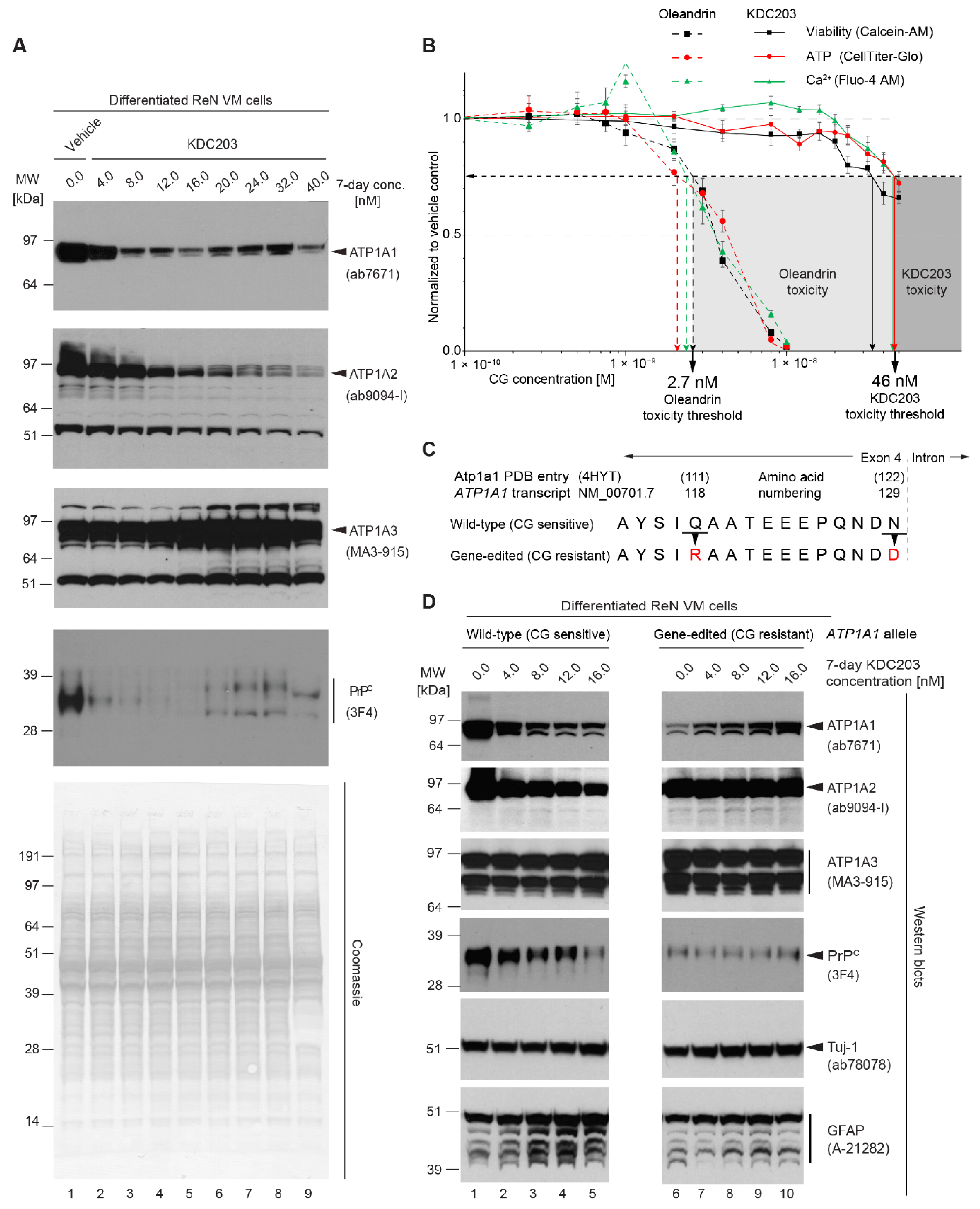
2.9. KDC203 Is Less Toxic Than Oleandrin
2.10. Mutagenesis of CG Binding Site within ATP1A1 Abolishes KDC203-Dependent PrPC Reduction
3. Discussion
4. Materials and Methods
4.1. Sequence Alignment and Identification of Residues Lining CG Binding Pocket
4.2. Structural Modeling and Assessment of Docking and BBB Penetrance Scores
4.3. Design of Chemically Accessible Oleandrin Derivatives
4.4. Synthesis of Shortlisted Oleandrin Derivative
4.5. Characterization of KDC203 by Mass Spectrometry
4.6. Animal Husbandry
4.7. Tritium-Based Comparison of Bioavailability of Oleandrin and KDC203
4.8. Bidirectional MDCK Assay
4.9. Rapid Equilibrium Dialysis
4.10. Treatment of ReN VM Cells with KDC203
4.11. Antibodies
4.12. Western Blot Analyses
4.13. Viability, Intracellular Ca2+, and ATP Content in Cells Exposed to KDC203 versus Oleandrin
4.14. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Richt, J.A.; Kasinathan, P.; Hamir, A.N.; Castilla, J.; Sathiyaseelan, T.; Vargas, F.; Sathiyaseelan, J.; Wu, H.; Matsushita, H.; Koster, J.; et al. Production of cattle lacking prion protein. Nat. Biotechnol. 2007, 25, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Austbø, L.; Tranulis, M.A.; Espenes, A.; Olsaker, I. Healthy goats naturally devoid of prion protein. Vet. Res. 2012, 43, 87. [Google Scholar] [CrossRef] [Green Version]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 8, 322ra9. [Google Scholar] [CrossRef] [Green Version]
- Bueler, H.; Raeber, A.; Sailer, A.; Fischer, M.; Aguzzi, A.; Weissmann, C. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol. Med 1994, 1, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [Green Version]
- Mallucci, G.R.; White, M.D.; Farmer, M.; Dickinson, A.; Khatun, H.; Powell, A.D.; Brandner, S.; Jefferys, J.G.; Collinge, J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 2007, 53, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Silber, B.M.; Gever, J.R.; Rao, S.; Li, Z.; Renslo, A.R.; Widjaja, K.; Wong, C.; Giles, K.; Freyman, Y.; Elepano, M.; et al. Novel compounds lowering the cellular isoform of the human prion protein in cultured human cells. Bioorg. Med. Chem. 2014, 22, 1960–1972. [Google Scholar] [CrossRef]
- Karapetyan, Y.E.; Sferrazza, G.F.; Zhou, M.; Ottenberg, G.; Spicer, T.; Chase, P.; Fallahi, M.; Hodder, P.; Weissmann, C.; Lasmezas, C.I. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc. Natl. Acad. Sci. USA 2013, 110, 7044–7049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minikel, E.V.; Zhao, H.T.; Le, J.; O’Moore, J.; Pitstick, R.; Graffam, S.; Carlson, G.A.; Kavanaugh, M.P.; Kriz, J.; Kim, J.B.; et al. Prion protein lowering is a disease-modifying therapy across prion disease stages, strains and endpoints. Nucleic Acids Res. 2020, 48, 10615–10631. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef] [Green Version]
- Mazur, C.; Powers, B.; Zasadny, K.; Sullivan, J.M.; Dimant, H.; Kamme, F.; Hesterman, J.; Matson, J.; Oestergaard, M.; Seaman, M.; et al. Brain pharmacology of intrathecal antisense oligonucleotides revealed through multimodal imaging. JCI Insight 2019, 4, e129240. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Williams, D.; Mehrabian, M.; Arshad, H.; Eid, S.; Sackmann, C.; Zhao, W.; Wang, X.; Ghodrati, F.; Verkuyl, C.E.; Watts, J.C.; et al. The cellular prion protein interacts with and promotes the activity of Na,K-ATPases. PLoS ONE 2021, 16, e0258682. [Google Scholar] [CrossRef]
- Mehrabian, M.; Wang, X.; Eid, S.; Yan, B.Q.; Grinberg, M.; Siegner, M.; Sackmann, C.; Sulman, M.; Zhao, W.; Williams, D.; et al. Cardiac glycoside-mediated turnover of Na, K-ATPases as a rational approach to reducing cell surface levels of the cellular prion protein. PLoS ONE 2022, 17, e0270915. [Google Scholar] [CrossRef]
- Yatime, L.; Buch-Pedersen, M.J.; Musgaard, M.; Morth, J.P.; Winther, A.M.L.; Pedersen, B.P.; Olesen, C.; Andersen, J.P.; Vilsen, B.; Schiott, B.; et al. P-type ATPases as drug targets: Tools for medicine and science. Biochim. Biophys. Acta. 2009, 1787, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Greeff, K. Cardiac Glycosides; Springer: Berlin/Heidelberg, Germany, 1981; Part, I. [Google Scholar]
- Schatzmann, H.J. Herzglykoside als Hemmstoffe für den aktiven Kalium- und Natriumtransport durch die Erythrocytenmembran. (Cardiac glycosides as inhibitors for the active potassium and sodium transport across the red cell membrane). Helv. Physiol. Pharm. Acta 1953, 11, 346–354. [Google Scholar]
- Khatri, H.R.; Bhattarai, B.; Kaplan, W.; Li, Z.; Long, M.J.C.; Aye, Y.; Nagorny, P. Modular total synthesis and cell-based anticancer activity evaluation of ouabagenin and other cardiotonic steroids with varying degrees of oxygenation. J. Am. Chem. Soc. 2019, 141, 4849–4860. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, T.; Dufrasne, F.; Kiss, R. Cardiotonic steroids-mediated targeting of the Na(+)/K(+)-ATPase to combat chemoresistant cancers. Curr. Med. Chem. 2012, 19, 627–646. [Google Scholar] [CrossRef] [PubMed]
- Menger, L.; Vacchelli, E.; Kepp, O.; Eggermont, A.; Tartour, E.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Cardiac glycosides and cancer therapy. Oncoimmunology 2013, 2, e23082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.K.T.; Portbury, S.; Thomas, M.B.; Barney, S.; Ricca, D.J.; Morris, D.L.; Warner, D.S.; Lo, D.C. Cardiac glycosides provide neuroprotection against ischemic stroke: Discovery by a brain slice-based compound screening platform. Proc. Natl. Acad. Sci. USA 2006, 103, 10461–10466. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, J.; Erdmann, E.; Rietbrock, N. Distribution of cardiac glycosides in heart and brain of dogs and their affinity to the (Na++K+)-ATPase. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1979, 307, 65–71. [Google Scholar] [CrossRef]
- Dutta, S.; Marks, B.H.; Schoener, E.P. Accumulation of radioactive cardiac glycosides by various brain regions in relation to the dysrhythmogenic effect. Br. J. Pharmacol. 1977, 59, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Elmaci, I.; Alturfan, E.E.; Cengiz, S.; Ozpinar, A.; Altinoz, M.A. Neuroprotective and tumoricidal activities of cardiac glycosides. Could oleandrin be a new weapon against stroke and glioblastoma? Int. J. Neurosci. 2018, 128, 865–877. [Google Scholar] [CrossRef]
- Dunn, D.E.; He, D.N.; Yang, P.; Johansen, M.; Newman, R.A.; Lo, D.C. In vitro and in vivo neuroprotective activity of the cardiac glycoside oleandrin from Nerium oleander in brain slice-based stroke models. J. Neurochem. 2011, 119, 805–814. [Google Scholar] [CrossRef]
- Flasch, H.; Heinz, N. Concentration of cardiac glycosides in the heart and brain (author’s translation). Arzneim. Forsch. 1976, 26, 1213–1216. [Google Scholar]
- Ni, D.; Madden, T.L.; Johansen, M.; Felix, E.; Ho, D.H.; Newman, R.A. Murine pharmacokinetics and metabolism of oleandrin, a cytotoxic component of Nerium oleander. J. Exp. Ther. Oncol. 2002, 2, 278–285. [Google Scholar] [CrossRef]
- Botelho, A.F.M.; Miranda, A.L.S.; Freitas, T.G.; Milani, P.F.; Barreto, T.; Cruz, J.S.; Melo, M.M. Comparative Cardiotoxicity of Low Doses of Digoxin, Ouabain, and Oleandrin. Cardiovasc. Toxicol. 2020, 20, 539–547. [Google Scholar] [CrossRef]
- Mayer, U.; Wagenaar, E.; Beijnen, J.H.; Smit, J.W.; Meijer, D.K.; van Asperen, J.; Borst, P.; Schinkel, A.H. Substantial excretion of digoxin via the intestinal mucosa and prevention of long-term digoxin accumulation in the brain by the mdr 1a P-glycoprotein. Br. J. Pharmacol. 1996, 119, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Turan, N.; Akgun-Dar, K.; Kuruca, S.E.; Kilicaslan-Ayna, T.; Seyhan, V.G.; Atasever, B.; Mericli, F.; Carin, M. Cytotoxic effects of leaf, stem and root extracts of Nerium oleander on leukemia cell lines and role of the p-glycoprotein in this effect. J. Exp. Ther. Oncol. 2006, 6, 31–38. [Google Scholar] [PubMed]
- Zhou, C.; Yu, F.; Zeng, P.; Zhang, T.; Huang, H.; Chen, W.; Wu, B. Circadian sensitivity to the cardiac glycoside oleandrin is associated with diurnal intestinal P-glycoprotein expression. Biochem. Pharmacol. 2019, 169, 113622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Reddy, M.S.; Phoenix, S.; Deslongchamps, P. Total synthesis of ouabagenin and ouabain. Angew. Chem. 2008, 47, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.S.; Zhang, H.; Phoenix, S.; Deslongchamps, P. Total synthesis of ouabagenin and ouabain. Chem. Asian J. 2009, 4, 725–741. [Google Scholar] [CrossRef]
- Renata, H.; Zhou, Q.; Baran, P.S. Strategic redox relay enables a scalable synthesis of ouabagenin, a bioactive cardenolide. Science 2013, 339, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Mukai, K.; Kasuya, S.; Nakagawa, Y.; Urabe, D.; Inoue, M. A convergent total synthesis of ouabagenin. Chem. Sci. 2015, 6, 3383–3387. [Google Scholar] [CrossRef] [Green Version]
- Yatime, L.; Laursen, M.; Morth, J.P.; Esmann, M.; Nissen, P.; Fedosova, N.U. Structural insights into the high affinity binding of cardiotonic steroids to the Na+,K+-ATPase. J. Struct. Biol. 2011, 174, 296–306. [Google Scholar] [CrossRef]
- Laursen, M.; Yatime, L.; Nissen, P.; Fedosova, N.U. Crystal structure of the high-affinity Na+K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Proc. Natl. Acad. Sci. USA 2013, 110, 10958–10963. [Google Scholar] [CrossRef] [Green Version]
- Miles, A.J.; Fedosova, N.U.; Hoffmann, S.V.; Wallace, B.A.; Esmann, M. Stabilisation of Na,K-ATPase structure by the cardiotonic steroid ouabain. Biochem. Biophys. Res. Commun. 2013, 435, 300–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, H.; Shinoda, T.; Cornelius, F.; Toyoshima, C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. USA 2009, 106, 13742–13747. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laursen, M.; Gregersen, J.L.; Yatime, L.; Nissen, P.; Fedosova, N.U. Structures and characterization of digoxin- and bufalin-bound Na+,K+-ATPase compared with the ouabain-bound complex. Proc. Natl. Acad. Sci. USA 2015, 112, 1755–1760. [Google Scholar] [CrossRef] [Green Version]
- Rankovic, Z. CNS Physicochemical Property Space Shaped by a Diverse Set of Molecules with Experimentally Determined Exposure in the Mouse Brain. J. Med. Chem. 2017, 60, 5943–5954. [Google Scholar] [CrossRef]
- Chen, H.; Lei, M.; Ma, B.; Liu, M.; Guo, D.; Liu, X.; Hu, L. Synthesis and cytotoxicity evaluation of 4′-amino-4′-dehydroxyloleandrin derivatives. Fitoterapia 2016, 113, 85–90. [Google Scholar] [CrossRef]
- Repke, K.; Est, M.; Portius, H.J. On the cause of species differences in digitalis sensitivity. Biochem. Pharmacol. 1965, 14, 1785–1802. [Google Scholar] [CrossRef]
- Wallick, E.T.; Pitts, B.J.; Lane, L.K.; Schwartz, A. A kinetic comparison of cardiac glycoside interactions with Na+,K+-ATPases from skeletal and cardiac muscle and from kidney. Arch. Biochem. Biophys. 1980, 202, 442–449. [Google Scholar] [CrossRef]
- Gupta, R.S.; Chopra, A.; Stetsko, D.K. Cellular basis for the species differences in sensitivity to cardiac glycosides (digitalis). J. Cell. Physiol. 1986, 127, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Price, E.M.; Rice, D.A.; Lingrel, J.B. Structure-function studies of Na,K-ATPase. Site-directed mutagenesis of the border residues from the H1-H2 extracellular domain of the alpha subunit. J. Biol. Chem. 1990, 265, 6638–6641. [Google Scholar] [CrossRef] [PubMed]
- Dostanic-Larson, I.; Van Huysse, J.W.; Lorenz, J.N.; Lingrel, J.B. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc. Natl. Acad. Sci USA 2005, 102, 15845–15850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dostanic, I.; Jel, J.S.; Lorenz, J.N.; Lingrel, J.B. The alpha 1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J. Biol. Chem. 2004, 279, 54053–54061. [Google Scholar] [CrossRef]
- Hellinger, É.; Veszelka, S.; Tóth, A.E.; Walter, F.; Kittel, Á.; Bakk, M.L.; Tihanyi, K.; Hada, V.; Nakagawa, S.; Dinh Ha Duy, T.; et al. Comparison of brain capillary endothelial cell-based and epithelial (MDCK-MDR1, Caco-2, and VB-Caco-2) cell-based surrogate blood–brain barrier penetration models. Eur. J. Pharm. Biopharm. 2012, 82, 340–351. [Google Scholar] [CrossRef]
- Wang, Q.; Rager, J.D.; Weinstein, K.; Kardos, P.S.; Dobson, G.L.; Li, J.; Hidalgo, I.J. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood-brain barrier. Int. J. Pharm. 2005, 288, 349–359. [Google Scholar] [CrossRef]
- Donato, R.; Miljan, E.A.; Hines, S.J.; Aouabdi, S.; Pollock, K.; Patel, S.; Edwards, F.A.; Sinden, J.D. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci. 2007, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Williams, D.; Muller, I.; Lemieux, M.; Dukart, R.; Maia, I.B.L.; Wang, H.; Woerman, A.L.; Schmitt-Ulms, G. Tau interactome analyses in CRISPR-Cas9 engineered neuronal cells reveal ATPase-dependent binding of wild-type but not P301L Tau to non-muscle myosins. Sci. Rep. 2019, 9, 16238. [Google Scholar] [CrossRef] [Green Version]
- Price, E.M.; Lingrel, J.B. Structure-function relationships in the Na,K-ATPase alpha subunit: Site-directed mutagenesis of glutamine-111 to arginine and asparagine-122 to aspartic acid generates a ouabain-resistant enzyme. Biochemistry 1988, 27, 8400–8408. [Google Scholar] [CrossRef]
- Unal, I.; Caliskan-Ak, E.; Ustundag, U.V.; Ates, P.S.; Alturfan, A.A.; Altinoz, M.A.; Elmaci, I.; Emekli-Alturfan, E. Neuroprotective Effects of Mitoquinone and Oleandrin on Parkinson’s Disease Model in Zebrafish. Int. J. Neurosci. 2019, 130, 574–582. [Google Scholar] [CrossRef]
- Van Kanegan, M.J.; Dunn, D.E.; Kaltenbach, L.S.; Shah, B.; He, D.N.; McCoy, D.D.; Yang, P.; Peng, J.; Shen, L.; Du, L.; et al. Dual activities of the anti-cancer drug candidate PBI-05204 provide neuroprotection in brain slice models for neurodegenerative diseases and stroke. Sci. Rep. 2016, 6, 25626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, R. 4′-Dehydro-Oleandrin and Pharmaceutical Composition Thereof. U.S. Patent No. 3,898,331, 5 August 1975. [Google Scholar]
- Reichel, A. Addressing central nervous system (CNS) penetration in drug discovery: Basics and implications of the evolving new concept. Chem. Biodiv. 2009, 6, 2030–2049. [Google Scholar] [CrossRef] [PubMed]
- Fridén, M.; Bergström, F.; Wan, H.; Rehngren, M.; Ahlin, G.; Hammarlund-Udenaes, M.; Bredberg, U. Measurement of unbound drug exposure in brain: Modeling of pH partitioning explains diverging results between the brain slice and brain homogenate methods. Drug Metab. Dispos. 2011, 39, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, X.; Zhou, Y.; Hou, J.; Long, H.; Zhang, Z.; Lei, M.; Wu, W. Synthesis of oleandrin derivatives and their cytotoxic activity. Steroids 2020, 159, 108650. [Google Scholar] [CrossRef]
- Czernecki, S.; Georgoulis, C.; Stevens, C.L.; Vijayakumaran, K. Pyridinium dichromate oxidation. Modifications enhancing its synthetic utility. Tetrahedron Lett. 1985, 26, 1699–1702. [Google Scholar] [CrossRef]
- Gozalpour, E.; Wilmer, M.J.; Bilos, A.; Masereeuw, R.; Russel, F.G.M.; Koenderink, J.B. Heterogeneous transport of digitalis-like compounds by P-glycoprotein in vesicular and cellular assays. Toxicol. Vitro 2016, 32, 138–145. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Dukes, J.D.; Whitley, P.; Chalmers, A.D. The MDCK variety pack: Choosing the right strain. BMC Cell Biol. 2011, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Muiznieks, L.D.; Ghosh, P.; Williams, D.; Solarski, M.; Fang, A.; Ruiz-Riquelme, A.; Pomes, R.; Watts, J.C.; Chakrabartty, A.; et al. Somatostatin binds to the human amyloid beta peptide and favors the formation of distinct oligomers. eLife 2017, 6, e28401. [Google Scholar] [CrossRef]
- Gustafsson, S.; Lindstrom, V.; Ingelsson, M.; Hammarlund-Udenaes, M.; Syvanen, S. Intact blood-brain barrier transport of small molecular drugs in animal models of amyloid beta and alpha-synuclein pathology. Neuropharmacol 2018, 128, 482–491. [Google Scholar] [CrossRef]
- Kalvass, J.C.; Maurer, T.S. Influence of nonspecific brain and plasma binding on CNS exposure: Implications for rational drug discovery. Biopharm. Drug. Dispos. 2002, 23, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Henary, H.; Falchook, G.S.; Naing, A.; Fu, S.; Moulder, S.; Wheler, J.J.; Tsimberidou, A.; Durand, J.B.; Khan, R.; et al. First-in-human study of pbi-05204, an oleander-derived inhibitor of akt, fgf-2, nf-kappaBeta and p70s6k, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 1204–1212. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eid, S.; Zerbes, T.; Williams, D.; Wang, X.; Sackmann, C.; Meier, S.; Dulin, N.O.; Nagorny, P.; Schmitt-Ulms, G. Identification of a Cardiac Glycoside Exhibiting Favorable Brain Bioavailability and Potency for Reducing Levels of the Cellular Prion Protein. Int. J. Mol. Sci. 2022, 23, 14823. https://doi.org/10.3390/ijms232314823
Eid S, Zerbes T, Williams D, Wang X, Sackmann C, Meier S, Dulin NO, Nagorny P, Schmitt-Ulms G. Identification of a Cardiac Glycoside Exhibiting Favorable Brain Bioavailability and Potency for Reducing Levels of the Cellular Prion Protein. International Journal of Molecular Sciences. 2022; 23(23):14823. https://doi.org/10.3390/ijms232314823
Chicago/Turabian StyleEid, Shehab, Thomas Zerbes, Declan Williams, Xinzhu Wang, Chris Sackmann, Sammy Meier, Nickolai O. Dulin, Pavel Nagorny, and Gerold Schmitt-Ulms. 2022. "Identification of a Cardiac Glycoside Exhibiting Favorable Brain Bioavailability and Potency for Reducing Levels of the Cellular Prion Protein" International Journal of Molecular Sciences 23, no. 23: 14823. https://doi.org/10.3390/ijms232314823